

Abstract
Aflatoxin contamination, caused by fungal pathogen Aspergillus flavus, is a major quality and health problem delimiting the trade and consumption of groundnut (Arachis hypogaea L.) worldwide. RNA-seq approach was deployed to understand the host-pathogen interaction by identifying differentially expressed genes (DEGs) for resistance to in-vitro seed colonization (IVSC) at four critical stages after inoculation in J 11 (resistant) and JL 24 (susceptible) genotypes of groundnut. About 1,344.04 million sequencing reads have been generated from sixteen libraries representing four stages in control and infected conditions. About 64% and 67% of quality filtered reads (1,148.09 million) were mapped onto A (A. duranensis) and B (A. ipaёnsis) subgenomes of groundnut respectively. About 101 million unaligned reads each from J 11 and JL 24 were used to map onto A. flavus genome. As a result, 4,445 DEGs including defense-related genes like senescence-associated proteins, resveratrol synthase, 9s-lipoxygenase, pathogenesis-related proteins were identified. In A. flavus, about 578 DEGs coding for growth and development of fungus, aflatoxin biosynthesis, binding, transport, and signaling were identified in compatible interaction. Besides identifying candidate genes for IVSC resistance in groundnut, the study identified the genes involved in host-pathogen cross-talks and markers that can be used in breeding resistant varieties.
Introduction
Aflatoxins are a group of carcinogenic mycotoxins produced mainly by Aspergillus flavus and A. parasiticus. They are considered to be a threat to human health, global food safety, and security1, 2. Maize (Zea mays L.) and groundnut (Arachis hypogaea L.) are the most susceptible crops to aflatoxin contamination and serve as the main source of aflatoxin exposure for humans. The aflatoxins were reported as growth retardants in children3 and immune-suppressors leading to AIDS4, 5. Aflatoxin B1 produced by A. flavus and A. parasiticus in groundnut is considered to be the major cause of liver cancer along with the chronic infection with hepatitis B and C viruses and acute aflatoxicosis6.
Groundnuts tend to be colonized and contaminated by Aspergillus spp. at pre-harvest, during harvest, post-harvest drying, in storage and also during transport covering the whole value chain7, 8. Use of cultivars resistant to seed invasion by Aspergillus spp. is one of the possible means of reducing aflatoxin contamination during groundnut storage with no extra input cost for the farmers. Development of aflatoxin resistant groundnut varieties has been a challenging task for breeders due to lack of availability of reliable resistance sources, poor understanding of plant-fungus interactions and large environmental influence. Three types of resistance mechanisms namely in-vitro seed colonization resistance (IVSC), resistance to pre-harvest aflatoxin contamination (PAC) and resistance to aflatoxin production in seeds have been reported in groundnut. However, the aflatoxin is produced only in the cotyledons of groundnut kernels after fungal infection9. Sources of resistance to these mechanisms were identified independently7, 10, 11. Therefore, there is a need to understand the molecular mechanisms of resistance to aflatoxin contamination.
The differential transcriptome analysis based on next-generation sequencing (NGS), can provide insights to understand resistance mechanisms and offers the possibilities of identifying novel genes and splice variants12, 13. Few reports on expression profiles are available for selected tissues of plant growth and development in groundnut using either microarray14–16 or transcriptome sequencing17–22. Transcriptome studies have been carried out for biotic stresses like late leaf spot23, 24 and bacterial wilt25. In addition, transcriptome studies related to Aspergillus spp. infection26, 27 under drought conditions28 have also been carried out using microarray technology. Very recently, transcript profiling of resistant and susceptible groundnut post-harvest seeds has been undertaken in response A. flavus infection29. However, none of the above studies related to aflatoxin contamination provided the decisive insights on genomic control and solution for their deployment in developing improved varieties.
As aflatoxin is produced preferentially in the seed and IVSC is the first step towards fungal infection, it is considered as the most important stage during aflatoxin contamination. Identifying molecular mechanisms and candidate genes associated with IVSC resistance could lead to a breakthrough in controlling fungal colonization and aflatoxin contamination in groundnut. Furthermore, the availability of genome sequence information of diploid progenitors of tetraploid cultivated groundnut30, 31 and advances in next generation sequencing (NGS) technologies provide tools to understand the genomic control for such complex traits32, 33. The present study, therefore, aims identification of candidate genes and enhancing understanding of resistance mechanisms for IVSC using RNA-seq approach in groundnut.
Results
Microscopic observation and aflatoxin estimation
Observation of the seed coat of the control and infected seeds using the florescence microscope showed considerably low mycelial growth at first day after inoculation (1DAI) in both the genotypes (J 11 and JL 24). The infected samples of JL 24 began to show higher mycelial growth than that of J 11 at 2DAI and even more at 3DAI. Sporulation was observed in JL 24 at 7DAI as compared to no or very less number of spores in J 11. There was a clear inhibition of fungal colonization on groundnut seeds in J 11 compared to JL 24. In control conditions, both the genotypes showed uniform germination while after infection, JL 24 failed to germinate due to heavy fungal invasion and colonization. Highest toxin production (1,826 µg aflatoxin /kg seed) was observed at 3DAI in the case of JL 24 (Fig. 1).
Figure 1.

Phenotypic observations for seeds of J 11 and JL 24 during in-vitro seed colonization by Aspergillus flavus at different time points along with microscopic observation and aflatoxin estimation. The figure shows the microscopic observations of seed coat after fungal inoculation in J 11 (resistant) and JL 24 (susceptible) that clearly shows the presence of mycelium at Day 3 and sporulation at Day 7 after inoculation.
Transcriptome sequencing and gene expression analysis
The paired-end (2 × 100 bp) sequencing of sixteen samples generated 268.81 Gb of data comprising of 1,344.04 million raw reads with an average of 84 million reads per sample (Table 1) and was submitted to National Centre for Biotechnology Information (NCBI) with BioProject ID PRJNA355201 and Sequence Read Archive SRP094387. After the stringent quality check, a total of 1,064.21 million reads along with the high quality (QC ≥ 30) 83.88 million singleton were used for mapping onto A (A. duranensis) and B (A. ipaёnsis) subgenomes of groundnut. Percent mapping was slightly higher on the B subgenome (770.84 million reads; 67.14% of quality filtered) compared to the A subgenome (737.75 million reads; 64.26% of quality filtered) (Table 1). Interestingly, about 74–77% of the reads were mapped to exonic region, 7% to intronic region and about 11% to inter-genic regions of both A and B subgenomes (Supplementary Table 1). A total of 19,569 and 20,299 genes expressed during infection could be mapped to A and B subgenomes, respectively. Out of which 19,398 and 20,227 genes were mapped on the pseudomolecules of A and B subgenomes, rest of 171 and 72 genes were mapped onto scaffolds, hence do not have assigned coordinate information.
Table 1.
Summary of sequencing data generated and mapped on A and B subgenomes of groundnut.
| Library information | Total reads (millions) | High quality reads (millions, >Q30 filtered) | High quality singletons (millions, >Q30 filtered) | Number of reads (millions) uniquely mapped on A subgenome | Number of reads (millions) uniquely mapped on B subgenome |
|---|---|---|---|---|---|
| J 11_Control_1DAI | 100.20 | 79.69 | 6.02 | 63.17 | 63.50 |
| J 11_Control_2DAI | 90.06 | 71.10 | 5.59 | 52.88 | 53.06 |
| J 11_Control_3DAI | 88.01 | 69.53 | 5.45 | 51.97 | 52.19 |
| J 11_Control_7DAI | 90.68 | 67.66 | 6.76 | 42.72 | 47.02 |
| J 11_Infected_1DAI | 73.82 | 57.92 | 4.69 | 43.00 | 44.37 |
| J 11_Infected_2DAI | 77.79 | 56.41 | 6.35 | 28.15 | 32.68 |
| J 11_Infected_3DAI | 75.07 | 60.19 | 4.49 | 41.01 | 42.59 |
| J 11_Infected_7DAI | 75.02 | 59.07 | 4.80 | 38.00 | 40.55 |
| JL 24_Control_1DAI | 85.28 | 72.09 | 5.36 | 51.74 | 54.27 |
| JL 24_Control_2DAI | 89.96 | 73.30 | 4.92 | 55.72 | 56.53 |
| JL 24_Control_3DAI | 89.73 | 70.04 | 4.99 | 50.50 | 51.52 |
| JL 24_Control_7DAI | 86.77 | 67.54 | 5.35 | 43.72 | 47.48 |
| JL 24_Infected_1DAI | 85.61 | 67.05 | 5.59 | 44.12 | 47.64 |
| JL 24_Infected_2DAI | 89.88 | 75.60 | 4.77 | 47.06 | 51.05 |
| JL 24_Infected_3DAI | 69.52 | 56.68 | 3.86 | 43.47 | 43.42 |
| JL 24_Infected_7DAI | 76.62 | 60.33 | 4.89 | 40.53 | 42.96 |
| Total | 1344.04 | 1064.21 | 83.88 | 737.75 | 770.84 |
The unaligned reads i.e. 101.4 and 101.3 million reads from J 11 and JL 24 genotypes were assembled and used to map onto Aspergillus genome. An average of 4.45% and 5% of reads of infected samples of J 11 and JL 24 genotypes were mapped onto the fungal genome. The study indicated marginally higher mapping in JL 24, as compared to J 11 genotype. As expected the reads from control samples did not have any alignment on fungal genome indicating the sanity of the experiment.
Differentially expressed genes (DEGs) during Aspergillus infection
From differential expression analysis, a total 4,445 DEGs were identified in different combinations across four stages (Supplementary Table 2). A total of 1,194 DEGs were identified when J 11 was compared with its control counterparts. Similarly, 930 DEGs were observed when infected JL 24 was compared with the control samples and 995 DEGs were identified when infected J 11 samples were compared with infected JL 24. The highest number of DEGs were observed during 1DAI (1,283), followed by 7DAI (1,141), 3DAI (1,061) and 2DAI (960). Upon comparing infected samples of J 11 with JL 24, the number of upregulated DEGs (640) were higher than down-regulated genes (355) in both the subgenomes. Among all the DEGs, most abundant genes expressed included senescence-associated proteins, resveratrol synthase, seed linoleate 9s-lipoxygenases (9s-LOX), Mlp like protein 43, pathogenesis-related (PR) proteins, peroxidases, glutathione-S-transferase, chalcone synthase, defensins, chitinases, etc. The relative abundance of expression of DEGs at 2DAI were higher as compared to other days (Fig. 2a).
Figure 2.
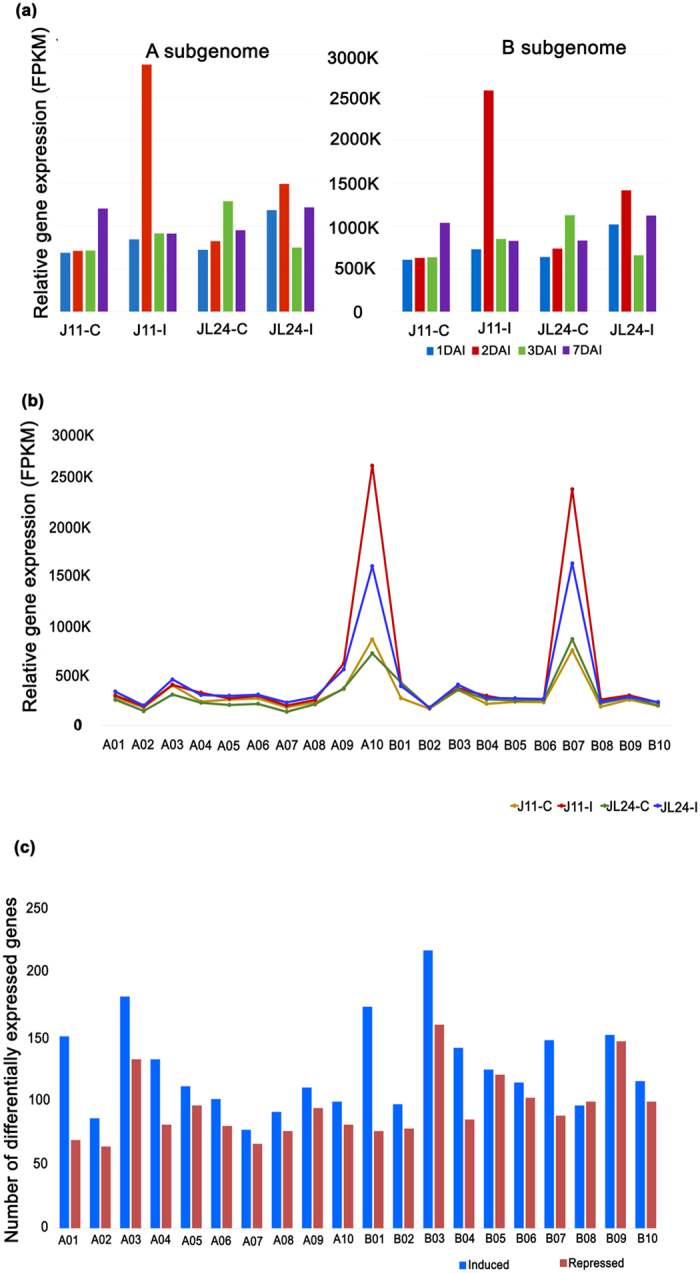
Gene expression during in-vitro seed colonization of Aspergillus flavus in groundnut. (a) Temporal distribution of relative abundance in expressed genes at different stages of fungal infection. The relative expression is found to be higher in infected samples of J 11 (J11-I) as compared to their control counter parts (J11-C) at 2nd day after inoculation (2DAI). The expression level of infected samples of JL 24 (JL24-I) was higher as compared to JL24-C. (b) Relative abundance of all the genes expressed during in-vitro seed colonization across subgenomes of groundnut progenitors. The relative abundance in terms of FPKM values were mapped onto A10 and B07 of A and B subgenomes, respectively. (c) Number of differentially expressed genes-DEGs (induced and repressed) during in-vitro seed colonization. The highest number of DEGs were mapped onto A03 and B03 of A and B subgenomes, respectively.
The relative expression of all DEGs across different stages in control and infected samples of J 11 and JL 24 are presented in Supplementary Table 3. Senescence-associated proteins were found to be highly expressed at 1DAI in both J 11 and JL 24, though the level of expression was higher in JL 24. The expression of resveratrol synthase was found to be pathogen-induced as in control samples there was no significant level of expression of this gene. The level of expression was higher at 3DAI in J 11 as compared to its control counterpart. Interestingly, no significant difference was found when infected samples of J 11 and JL 24 were compared. The 9s-LOX were found be induced at 1DAI and flared up at 7DAI in J 11, whereas, its expression was repressed from 1DAI through 7DAI in JL 24. Similarly, Mlp-like protein 43 was repressed in infected samples as compared to control during initial days and its expression shot up in case of J 11 at 7DAI. PR proteins were found highly expressed after infection in case of J 11, though there was a considerable expression of these genes in JL 24 samples as well. Among the PR proteins, PR-10 was found to be in abundant during Aspergillus infection and expression was more at 3DAI. However, PR-like proteins were found to be accumulated in uninfected samples as well.
Peroxidases were another class of enzymes that were induced during infection in case of J 11 as compared to JL 24 and lignin-forming anionic peroxidases and cationic peroxidases were found in the initial period of infection i.e., 1DAI and 2DAI and other peroxidase classes like peroxidase 3, 10, 15, 16, 21, 44 etc. were found to be highly expressed at 7DAI in case of J 11. Glutathione-S-transferases (GST) were found to be induced in case of J 11 and clear repression was found in the case of JL 24 at 3DAI. Moderate expression was found during initiation of pathogenesis (1DAI and 2DAI) and limited expression was found during 7DAI in J 11. Similarly, chalcone synthases (CHS) and isomerases and isoflavone reductases were also found to be induced during infection in J 11 and repressed in JL 24 at 3DAI. Chitinase enzymes were found to be upregulated in J 11 at 7DAI and downregulated in JL 24 at 2DAI. Similarly, defensin enzymes and Phenylalanine ammonia lyase (PAL) related transmembrane protein-9 were found to be highly expressed in the case of J 11 at 3DAI as compared to their controls. The expression of heat shock proteins was also found to be downregulated in JL 24 at 3DAI. The ethylene-responsive transcription factor (ERF) proteins were upregulated at 1DAI and repressed at 2DAI and 3DAI in the case of JL 24. Microsomal omega-3 fatty acid desaturase was found to be upregulated in the case of infected samples of JL 24 at 3DAI as compared to J 11. Other genes like Kunitz-type trypsin inhibitor, desiccation protectant late embryogenesis abundant (LEA) 14 proteins, pectate lyase, subtilisin-like proteases, expansin, and serine-threonine protein kinases were found to be upregulated at 7DAI in J 11 as compared to JL 24. Also, the allergen genes (Ara h 2) were found to be repressed in J 11.
An important observation was made in case of genes involved in oxylipin production that are known to be involved in signal transduction i.e., LOX, peroxygenase 1 like proteins, 4-hydroxy-3-methylbut-2-en-1-yl diphosphate, phytyltransferases. All these genes were found to be marginally induced initially in JL 24 and repressed at the later stages. In J 11, the expression of LOX and peroxygenase was marginally decreased at 1DAI but increased at 7DAI. The expression of other two genes got induced at 3DAI in J 11.
Genome-wide distribution of differentially expressed genes
A total of 19,398 and 20,227 genes were mapped to the pseudomolecules of A and B subgenomes, respectively. A total of 2,811 genes were mapped on A03, followed by 2,772 on B03. Among all, A07 (1,504) and B02 (1,660) had minimum number of genes mapped. The relative gene abundance, calculated from all the genes expressed in J 11 and JL 24 under control and infected conditions were plotted on A and B subgenomes. Interestingly, relative abundance of expression of genes was significantly higher in the case of pseudomolecule A10 (2.6 million reads) and B07 (2.3 million reads) of the A and B subgenomes, respectively. The frequency of mapped-reads was higher in infected samples compared to their control counterparts. The results clearly showed an increase in the level of expression in the case of J 11 during infection as compared to JL 24 (Fig. 2b). When the relative abundance of DEGs was checked, the DEGs were distributed across the groundnut genome and was higher in case of A10 pseudomolecule and distributed across A01, A10, B04 and B07 pseudomolecules (Supplementary Fig. 1).
Out of 4,445 DEGs identified, 2,007 DEGs (45.15%) were mapped on A subgenome and 2,438 (54.84%) DEGs on the B subgenome. These DEGs were distributed across pseudomolecules and the highest number of DEGs (376) were present on pseudomolecule B03 followed by A03 (313) and lowest number of DEGs were located on pseudomolecule A07 (143) with an average of 220 DEGs per pseudomolecule (Fig. 2c).
Gene ontology (GO) and pathway analysis of differentially expressed genes
GO analysis annotated a total of 1,412 DEGs (70.35% of 2,007 DEGs in A subgenome) and 1,647 DEGs (67.55% of 2,438 DEGs in B subgenome) (Supplementary Fig. 2). Most of the DEGs were assigned to “Biological process” (3,321; 55%) followed by “Molecular function” (1,511; 25.33%) and “Cellular component” (1,132; 18.98%) on the A subgenome. Similarly functional categorization of the DEGs on B subgenome were assigned to “Biological process” (3,666; 55.27%) followed by, “Molecular function” (1,675; 25.25%) and “Cellular component” (1,291; 19.46%). Majority of the DEGs were related to response to stress/stimulus, signal transduction mechanisms, secondary metabolite mechanisms and lipid metabolism processes. These DEGs were more often related to molecular functions including binding, catalytic, hydrolytic and transporter activities and localized in cellular, membrane and intracellular part of the cells.
The pathway based analysis was done to further understand the molecular mechanisms of the differentially expressed genes. The DEGs were found to represent 119 and 171 pathways when mapped to A and B subgenomes, respectively and 89 non-redundant pathways were affected or influenced by their expression (Supplementary Table 4).
Majority of the pathways could be assigned to three general categories: (1) Primary metabolism including carbohydrate, lipid, amino acid and vitamin metabolisms. Carbohydrate metabolism pathways that are influenced in the present study include: citrate cycle, starch and sucrose metabolism, glycolysis, gluconeogenesis, galactose metabolism, etc. Similarly, lipid metabolism pathways like fatty acid biosynthesis and degradation, glycerolipid metabolism, sphingolipid metabolism, etc. were affected. Furthermore, amino acid metabolism pathways such as biosynthesis of phenylalanine, tryptophan, tyrosine, glutathione, etc. and several pathways related to the metabolism of vitamins were affected. (2) Secondary metabolic pathways dependent upon biosynthesis of terpenoids including biosynthesis of zeatin, ubiquinone, terpenoid-quinone, steroid, carotenoid, flavonoid biosynthesis, cutin, suberin, wax, etc. (3) Shikimate derivative dependent pathways including biosynthesis phenylpropanoid, stilbenoid, diarylheptanoid, gingerol, flavonoid, isoflavonoid, cyanoamino acid metabolism, etc.
Differentially co-expressed gene modules
DiffCoEx outlined differentially co-expressed modules between J 11 and JL 24 genotypes by grouping genes according to their shared, yet sophisticated, varied differential correlation patterns. Altogether, the analysis suggested significant differences between both the genotypes leading to the identification of 23 and 18 modules with different built-in color codes comprising of 19,251 and 19,923 genes in case of A and B subgenomes, respectively (Fig. 3). Three of these modules (darkgreen- 4707 genes; darkgoldenrod4-4755 genes; aquamarine4-1700 genes) were significantly highly correlated in the case of J 11 on A subgenome. In the case of B subgenome, three modules (forestgreen- 1808 genes; brown4-1960 genes; lightseagreeen-967 genes) were significantly more highly correlated in J 11 genotype and one module (red3-1500 genes) was found to be highly expressed and correlated in JL 24. Two interesting modules in each of the genomes viz., darkgoldenrod4 from A subgenome and red3 from B subgenomes were discussed here. The darkgoldenrod4 module from A subgenome comprised of the genes encoding plant defense such as PR proteins, chalcone synthase, chalcone-flavanone isomerase, resveratrol synthase, GST, senescence-associated proteins, WRKY, bHLH, leucine-rich repeat (LRR), chitinases, LEA proteins, calmodulin binding and transporting proteins and so on. The red3 module from B subgenome has genes encoding the binding and transporter activities like ABC (ATP binding cassette) transporters, plasma-membrane associated cation-binding protein, sugar transporters (SWEET), SNARE interacting proteins, autophagy related proteins, patatin like proteins and so on.
Figure 3.
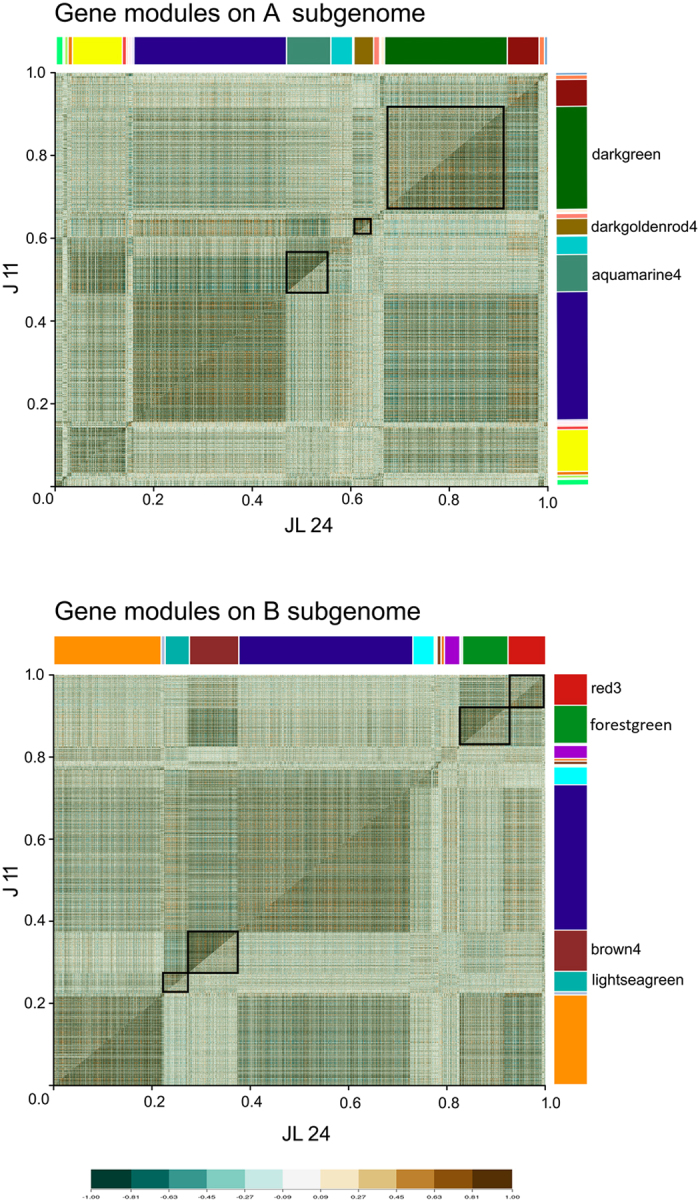
Differentially co-expressed modules between J 11 and JL 24 in A and B subgenomes of groundnut. The comparative correlation heat maps with brown color corresponding to positive correlation and blue corresponding to negative correlations are depicted in the figure. The upper diagonal of the main matrix shows a correlation between pairs of genes among J 11 (resistant) transcripts while the lower diagonal of the heat map shows a correlation between the same gene pairs in the JL 24 (susceptible). Modules are identified in the heat map by different built-in color bars on the right side of the heat map. Distinguished gene modules are demarcated by black squares.
Marker development and validation
A total of 39,742 and 40,946 sequence variants were identified between J 11 and JL 24 when aligned on A and B subgenomes, respectively. The variants on ‘A’ subgenome included 994 homozygous SNPs, 85 Indels and 38,663 heterozygous SNPs and Indels, whereas on ‘B’ genome included 1,651 homozygous SNPs, 72 Indels, and 39,223 heterozygous SNPs/Indels. Of the total SNPs, 50 and 118 SNPs/Indels were present in the 98 unique DEGs collectively from both A (30) and B (68) subgenomes, respectively. A maximum number of sequence variants were located on ‘B’ subgenome- B10 (22), followed by B02 (18), B05 (16) and B03 (15). The number of sequence variants was comparatively lesser in the case of ‘A’ subgenome, the highest being on pseudomolecule A05 (10). All 168 sequence variants were checked for their effect on the function of the gene (Supplementary Table 5). Maximum number of SNPs were non-synonymous coding (62) followed by synonymous coding (51), intronic (23), UTR_3_prime (12), UTR_5_prime (11), stop_gained (2), stop_lost (1) and others (5) when in case of both ‘A’ and ‘B’ subgenomes together (Table 2). About 5,025 selected SNPs from the available transcriptome were considered while designing an SNP array (58 K) chip in groundnut for further use in molecular breeding34. The identified sequence variants (SNPs and Indels) on DEGs across pseudomolecules, similarity relation of DEGs in both subgenomes have been shown in the Fig. 4. A total of 73 allele-specific primer pairs were designed to validate the sequence variants between J 11 and JL 24 (Supplementary Table 6). Nineteen polymorphic markers between J 11 and JL 24 were identified in genes like resveratrol synthase, calmodulin, GST, flavonoid glucosyltransferase, disease resistance proteins rpp13 and TIR-NBS-LRR (Supplementary Fig. 3). After thorough screening and validation, these functional/expression markers can further be used in genomics-enabled aflatoxin resistance breeding.
Table 2.
Distribution of SNPs in differentially expressed genes across A and B subgenomes of groundnut.
| SNP categories | A subgenome | B subgenome | Total |
|---|---|---|---|
| Non-synonymous coding | 20 | 42 | 62 |
| Synonymous coding | 18 | 33 | 51 |
| Intron | 4 | 19 | 23 |
| UTR_3 prime | 2 | 10 | 12 |
| UTR_5 prime | 2 | 9 | 11 |
| Others | 3 | 2 | 5 |
| Stop_gained | 0 | 2 | 2 |
| Frame shift | 1 | 0 | 1 |
| Stop_lost | 0 | 1 | 1 |
| Total | 50 | 118 | 168 |
Figure 4.

Distribution of single nucleotide polymorphisms (SNPs), Indels and differentially expressed genes (DEGs) across the pseudomolecules of progenitor reference genomes of groundnut. (a) Groundnut pseudomolecules from A subgenome are depicted as A01 to A10 and that of B subgenome are depicted as B01 to B10 (b) SNP density depicted in blue (c) InDel density depicted in green (d) both upregulated (with upward arrow) and downregulated (with downward arrow) differentially expressed genes depicted in brown. Synteny of the differentially expressed genes among the pseudomolecules are depicted with the different colored lines with respect to representative colors of the pseudomolecules.
Validation of 15 DEGs including PR proteins, resveratrol synthase, cationic peroxidases, heat shock proteins-83 like, microsomal omega-3 fatty acid desaturase, ERF060, lipoxygenases, Kunitz type trypsin inhibitor, desiccation protectant LEA protein, seed linoleate 9s- lipoxygenases, chalcone reductase, isoflavone reductase, allergen Ara h2, chitinases and subtilisin like protease was carried out. qRT-PCR plots were drawn that indicated relative expression of these genes in case of J 11 and JL 24 (Fig. 5). The majority of the genes showed the similar pattern of expression as indicated by RNA seq results. There was slight variation in the expression of resveratrol synthase and ERF060 at the different time period of the infection though the overall expression was unvaried.
Figure 5.

Validation of selected genes through qRT-PCR across different time periods of Aspergillus infection in J 11 (resistant) and JL 24 (susceptible) genotypes. Relative gene expression for 15 selected candidate genes like pathogenesis-related protein, resveratrol synthase, cationic peroxidase, lipoxygenase, heat shock protein 83-like, microsomal omega-3 fatty acid desaturase, ethylene-responsive transcription factor erf060-like, Kunitz-type trypsin inhibitor-like 2 protein, desiccation protectant protein Lea14, seed linoleate 9s-lipoxygenase-2, chalcone reductase, isoflavone reductase, allergen Ara h 2, class ii chitinase and subtilisin-like protease was calculated at four different time points of infection in case of J 11 and JL 24.
Differentially expressed genes (DEGs) in fungus during infection
Expression analysis of unmapped groundnut transcripts on Aspergillus genome indicated differential expression of 578 genes consisting of 390 induced and 188 repressed genes. Interestingly, number of induced genes were more in infected JL 24 sample as compared to J 11 at 3DAI and that of repressed were more at 2DAI. The list included several genes like cytochrome p450 monooxygenases, conidial hydrophobin, cytochrome b5, O-methyl transferase (O-MTA), oxidoreductases, Ver 1, aflE norA aad adh-2 NOR reductase dehydrogenase, aflD nor 1 reductase and several transporter proteins.
The highest number of DEGs (113) were found to be induced in the case of 3DAI followed by 1DAI in case of JL 24 infected samples (compatible host-pathogen interaction). However, in the case of J 11 infected samples (incompatible interaction), highest number of DEGs (125) were induced at 2DAI (Supplementary Table 7). In compatible interaction, at the initiation of pathogenesis (1DAI), the genes involved in fatty acid biosynthesis (stearic acid desaturase, acyl-binding family, fatty acid elongase Gns1), cell-wall related (hydrophobin, chitin binding, pectate lyase), growth related (early growth response, conidiation-specific, conidial hydrophobin, developmental regulator), transporters [ABC transporters, major facilitator superfamily (MFS) transporters], and kinases (calcium calmodulin-dependent kinases, phosphofructokinases) were found to be induced. There was no or meager expression of these genes in incompatible interaction. At 2DAI, the majority of DEGs belonged to metabolic processes such as transcription with metal/ion binding functions were expressed. The genes that are involved in aflatoxin biosynthesis viz., Ver-1, aflE norA aad adh-2 NOR reductase dehydrogenase, aflD nor-1 reductase, Noranthrone monooxygenase and that of peroxidase activity (thioredoxin reductase, cytochrome p450, GST, glutathione peroxidase, glutaredoxin) were found to be highly upregulated in compatible interaction at 3DAI. Besides, the proteins that are involved in endocytosis and signaling viz., synaptobrevin (member of SNARE complex involved in vesicular trafficking and endocytosis), and dynamin-like proteins were induced at 3DAI in compatible interaction. Interestingly majority of these enzymes were integral part of the membranes and have metal/ion binding and transferase functions as per the gene ontology studies. During incompatible interaction, the genes involved in transport (phthalate transporter, uridine permease), hydrolysis (glucanases, ureidoglycolate hydrolase, epoxide hydrolase), proteolysis (serine peptidase, metalloendopeptidase, cupin domain) and oxidation-reduction reactions (xanthine dehydrogenase, lignostilbene dioxygenase, etc.) were upregulated as compared to the compatible reactions especially at 2DAI. The genes involved in aflatoxin biosynthesis were not observed in incompatible interaction.
Discussion
Groundnut is a crop of global importance and aflatoxin contamination is the major bottleneck for international trade and industry, in addition to posing a health risk. Although sources of resistance to aflatoxin contamination in groundnut have been identified but there is no proper understanding of the molecular mechanisms associated with aflatoxin resistance. In this context, the present study provides a better understanding of the molecular mechanisms involved with resistance to IVSC through transcriptomic profiling of the crucial stages of fungal growth and infection. The mycelial growth and amount of toxin estimated on the 3rd day (3DAI) were exceptionally high in JL 24 as compared to J 11. This period is considered to be the invasive growth for Aspergillus during infection and also in few other fungi like rice blast fungus35.
The plant defense response to IVSC resistance seems to be a multifaceted endeavor that includes the recognition of a pathogen, the activation of a number of genes that lead to changes in the plant cell wall, changes in ion flux across the plasma membrane, formation of reactive oxygen species (ROS), production of phytoalexins, PR related proteins that induce resistance against pathogen attack (Fig. 6). The defense mechanism involves the expression of several TFs, systemic acquired resistance (SAR), mediated by SA, JA and ethylene signaling pathways36–38.
Figure 6.

Illustration of cross-talk between groundnut (Arachis hypogaea) and fungus (Aspergillus flavus) during in-vitro seed colonization. The genes/components illustrated represent the defense responsive molecules identified from groundnut-A. flavus interaction during in-vitro seed colonization studies. The components marked within the green rectangular box (with green arrows) are those induced in groundnut and those which are in light blue rectangular box (blue arrows) are the components induced in pathogen in compatible reaction through mixed transcriptome analysis. The components represented in dark blue boxes have important role in host-pathogen interaction in this study. Briefly, the pathogenesis initiation occurs at plant cell wall where NBS-LRR, elicitors and oxylipins have important role in host-pathogen interactions. JA and SA signaling pathways mediated by transcription factors like WRKY, NAC, MYB and ERFs play important role in plant defense. During the process of defense the phytoalexins like resveratrol synthase/stillbene synthase, PR proteins, LOX, chalcone synthase and PAL were expressed. Basal resistance works with the expression of senescence associated proteins that give hypersensitive response to combat the pathogen entry further. The chitinases, glucanases, PGIPs, PAL, PR proteins induce cell wall degradation of fungi during pathogen entry. Due to pathogen attack, there is oxidative burst in the plant cell that regulates the oxidation of fatty acids that in turn involved in signal transduction. Transcriptome analysis of fungus indicated that the RLKs, SNARE complex, elicitors and oxylipins play important role in plant recognition and infection. Most of these proteins are integral part of cell wall and membrane. Genes involved in fungal growth, aflatoxin synthesis and transport were highly expressed in Aspergillus. Abbreviations: ROS- reactive oxygen species; SOD-superoxide dismutase; PR-pathogenesisrelated proteins; HR-hypersensitive response; JA-Jasmonic acid; SA- Salicylic acid; SAR-Systemic acquired resistance; LOX-lipoxygenase; PAL- Phenylalanine ammonia-lyase; Transcription factors-NAC (NAM, ATAF1/2 and CUC2 domain proteins), ERF (Ethylene responsive factors); MAPK- Mitogen-activated protein kinases; SNARE- soluble N-ethylmaleimide sensitive factor attachment receptor; RLK- receptor-like kinase; cAMP- Cyclic adenosine monophosphate, OmtA- O-methyl transferase; AFB1-aflatoxin B1.
The present study identified about 89 non-redundant pathways that were influenced by the DEGs. The primary metabolic pathways that were affected mainly include carbohydrate, fatty acid, and amino acid metabolisms. Glucose is the preferred carbon source of A. flavus hence, up-regulation of sucrose hydrolyzing enzymes that promote disease development by providing a steady supply of nutrients to the pathogen. Earlier studies also indicated that pathogens can manipulate plant’s metabolism to create a congenial environment for their growth39, 40. Starch degradation and hexose mobilization were reported in A. flavus infected maize41.
Oxygenase enzymes act on fatty acids and produce oxylipins that play important role in signaling pathways and conidial growth in fungi and also they exhibit a common structural motif in plant and pathogen42, 43. The role of fatty acids was also evident in aflatoxin production, as the first acting enzymes in the aflatoxin biosynthesis pathway are fatty acid synthases (fsa1, fsa2)44. These fatty acids also act as precursor molecules for the production of cuticular components and phytohormone JA that are involved in plant defense. The direct roles for fatty acids in plant defense by modulating the basal, effector-triggered and systemic immunity responses have also been demonstrated45. Secondary metabolic pathways that include terpenoids, flavonoids, and steroid biosynthesis pathways have also shown to be affected during the Aspergillus infection that play an important role in plant defense by anti-oxidative properies.
The flavonoids are known to quench ROS generated both by the pathogens and the plant as a result of the infection46. Biosynthetic pathway of flavonoids includes the synthesis of chalcone catalyzed by CHS and chalcone isomerase. In the present study, we observed an increased expression of CHS and chalcone-flavanone isomerase proteins under stress condition in J 11 as compared to JL 24. The role of CHS in SA driven signaling during plant defense is reported47. Phytoalexins especially resveratrol synthase/ stilbene synthase were the most abundant enzymes that were produced in response to Aspergillus infection in case of J 11 and JL 24 as compared to their control counterparts during early stages of infection. The level of expression was higher in the case of J 11 as compared to JL 24. Interestingly the control samples showed meager expression indicated the expression of these genes upon the trigger from pathogen attack. Accumulation of phytoalexins at post-pathogen or elicitor induction is shown in many plants including Brassica 48, chickpea49 and soybean50. In the case of groundnut, resveratrol, arachidin and arahypin phytoalexins were found to be induced when infected by Aspergillus caelatus 51. A recent report about the effect of resveratrol on A. flavus indicated decreased growth and aflatoxin production in fungus52. SA is derived from phenylpropanoid pathway, and their glucoside conjugates play important role in infection initiation. It was interesting to note in the current study that the salicylate o-methyltransferase, enzyme involved in SA pathway was found to be induced during 3DAI in J 11 and JL 24 cultivars in infected samples and level of expression was more in JL 24 as compared to J 11, indicating the hypersensitive responses. The enzymes like PAL and GST were found to be highly up-regulated in J 11. GSTs under stress have peroxidase activity that protects the cells from oxidative injuries53. Senescence-associated proteins, geraniol 8-hydroxylase, serine-threonine protein phosphatase, seed linoleate 9s-lipoxygenase (LOX) and PR proteins were the other important enzymes that were expressed at higher levels in J 11 compared to JL 24 during infection. The senescence-associated proteins were found to be expressed in J 11 and JL 24 at different time points, i.e., initial induction was seen in J 11 and later induction was seen in JL 24. This indicated the dual function of senescence-associated proteins that initially expressed in J 11 as a defense response to trigger basal immunity of plant by SAR and then the increased expression in JL 24 at later stages as seeds got invaded by fungus and initiates senescence. LOX enzymes catalyze the oxygenation of fatty acids to produce oxylipins and increased LOX activity may contribute to resistance by generating signal molecules such as JA, methyl-JA or lipid peroxidases54 that was illustrated in maize during A. flavus infection55, 56. There are controversial studies regarding the activity of LOX genes in resistance mechanism pertaining to Aspergillus infection in soybean57. Hence there is a need to study the effect of the LOX genes thoroughly in groundnut. The geraniol 8-hydroxylase involved in production of iridoid glycosides reduce the fungal growth was reported58. PR proteins are produced in plants upon pathogen attack and induce SAR that play important role in cell rescue and defense. These proteins include endoglucanases, chitinases, peroxidases, defensin proteins, heat shock proteins that either act on fungal cell wall or act as signaling molecules to trigger the basal resistance and pathogen associated molecular pattern (PAMP). These molecules also act on cross-linking of molecules of cell wall to deposit lignin, callose molecules that prevent the pathogen entry inside the plant/ seed59. In the present study, some PR-like proteins were also found to be accumulated in the both infected and uninfected samples. PR-like proteins were found to have role in development of plant tissues in a developmentally controlled manner besides their role in defense response to stresses60. LEA proteins were also over-expressed in infected samples, of Arabidopsis 61 and maize62, but limited information is available about their role in plant defense. The relation between aflatoxin and allergens is not very well established, but some reports showed abundance of allergens storage proteins during A. flavus infection in groundnut63, 64. However the relation between aflatoxins and allergens in groundnut need to be thoroughly understood.
The differential co-expression studies also indicated a correlation pattern between expression of genes in J 11 and JL 24 genotypes. In fact, when two differentially correlated gene modules that were highly expressed and correlated in resistant (darkgoldenrod4) and susceptible (red3) genotypes were studied, they tend to contain a bunch of genes that have interesting roles in plant-pathogen interactions. The gene modules that were highly expressed in resistant genotype contained defense related genes like disease resistance response proteins/ PR proteins, chalcone synthase, resveratrol synthase, GST, senescence- associated proteins etc. The genes that were highly expressed and correlated in susceptible genotype present in red3 modules were involved in pathogen recognition, binding, transporter activity. The high expression of SNARE interacting proteins in JL 24 and also in fungus suggests the presence of common motif/complex that is evolutionarily selected to ensure the survival of pathogens within the eukaryotic environment65. Genetic markers for screening aflatoxin resistance in groundnut are very limited and this study provided the much needed genetic markers (SNPs and Indels) which upon validation can be used in GAB for enhancing resistance to aflatoxin contamination.
The simultaneous analysis of mixed transcriptomes of groundnut- Aspergillus provided the insights into the host-pathogen interactions. Similar approach was successfully used to study rice-blast fungus66 and sorghum-leaf spot fungus67 interactions. In present study, the pathogen transcriptome analysis indicated expression of genes related to fungal growth, aflatoxin production and transport. The genes related to initiation of pathogenesis, fatty acid biosynthesis, conidiation, transportation were regulated at initial period (1DAI) and that of aflatoxin production, peroxidation process were upregulated at 3DAI in compatible interaction. This observation can be correlated to the higher level of aflatoxin estimated at 3DAI. The presence of transporters implies an important role in fungal nutrient uptake as well as in signal transduction. Most of transporter proteins detected in the present study were integral part of the membrane proteins that might play very important role in inter-kingdom cross-talks.
The initiation of pathogenesis starts with the recognition of receptor like kinases (RLK) by the plant cell for pathogen detection. In the present study, the expression of large number of RLKs viz., serine-threonine protein kinase (br1-like, nak types), wall-associated receptor kinase-like 9, LRR family protein, etc. were observed in both compatible and incompatible interactions. NBS-LRR genes are the TF binding genes involved in plant defense68. Mitogen activated protein kinase (MAPK) cascade activated by RLKs participate in innate or induced plant defense response by their interaction with WRKY TFs69. During pathogenesis there is out-burst of ROS that is utilized in lipid peroxidation. Hyper-oxidant status of cell is pre-requisite for the onset of aflatoxin biosynthesis and there seems a close link between peroxisome metabolism and aflatoxin synthesis in A. flavus 70.
In summary, the comparative analysis of transcriptome of resistant and susceptible cultivars during Aspergillus infection has provided insights into various genes involved in several pathways that coherently induce plant defense mechanisms through basal and induced resistance. Further the key candidate genes viz., resveratrol synthase, chalcone synthase, chitinases, 9s-LOX, PR proteins, GSTs can be used in groundnut improvement by either genomics-enabled breeding or modern transgenic approaches. Most importantly, in addition to greater insights on the genetic mechanism and discovery of candidate genes, this study also provided genetic markers for IVSC resistance in groundnut.
Methods
Materials and methods
Plant and fungal materials
The resistant (J 11) and susceptible (JL 24) genotypes of groundnut and highly toxigenic A. flavus strain AF 11-4, characterized at Groundnut Pathology Unit of ICRISAT, were used for conducting transcriptomic study for IVSC resistance. The strain in pure culture form was grown on Potato Dextrose Agar for 7 days and conidial suspension was prepared (1 × 106 spores/ml).
Screening of in-vitro seed colonization
A total of ~200 healthy seeds for J 11 and JL 24 were surface sterilized with 0.1% HgCl2 for 3 min followed by three washes with sterile distilled water. Two different sets (control and infected) were made for each genotype. One set of sterilized seeds i.e., ~100 seeds for each genotypes were transferred to sterile filter papers on petri dishes and were used as control. The second set with remaining ~100 sterilized seeds were infected with the spore suspension of A. flavus toxigenic strain ‘AF 11-4’ with optimum concentration of 106 colony forming units/ml. Both the sets were incubated in a moist chamber with 100% relative humidity at 28 °C in dark. The samples were taken at 1 day after inoculation (1DAI), 2DAI, 3DAI and 7DAI from the control and infected samples of J 11 and JL 24 genotypes. About 10-12 seeds at each time intervals were frozen in liquid N2 until further use and the remaining seeds were used for aflatoxin estimation and microscopic observation of seed coat. This experiment was carried out twice and each set was considered as independent biological replicates. In total, 16 samples (2 genotypes × 4 stages × 2 treatments) were analyzed for aflatoxin estimation and RNA isolation.
Aflatoxin quantification and microscopic observation of seed coat
Indirect competitive enzyme-linked immunosorbent assay (ELISA) using polyclonal antibodies produced against Aflatoxin B1 (AFB1) was used for quantitative estimation of total aflatoxins accumulated under control and infected treatments following the protocol explained by Waliyar et al.71. The seed coats of the control and infected genotypes stained with coomassie brilliant blue and were observed through Zeiss Axio Scope.A1 Florescence microscope (Carl Zeiss, Germany).
RNA isolation and sequencing
Total RNA was isolated from the seeds using “NucleoSpin® RNA Plant” kit (Macherey-Nagel, Germany) following manufacturer’s protocol. RNA quality and quantity was determined using Nanodrop 1000 spectrophotometer (Thermo Fisher Scientific Inc, USA) and Bioanalyzer RNA Nano chip (Agilent Technologies, USA). Approximately 5 µg of total RNA pooled in equal quantity from two biological replicates were used for the construction of cDNA library using mRNA-Seq Sample Prep kit (Illumina Inc., USA) following manufacturer’s instructions. The RNA samples with 260/280 ratio of 1.8 to 2.1, 260/230 ratio of 2.0 to 2.3 and RIN (RNA integrity number) more than 7.0, were used for sequencing on Illumina HiSeq 2500 platform. The samples were sequenced as 2 × 100 bp paired-end reads. Filtered reads were obtained after running the quality control (QC) using NGS-QC box72.
Read alignment and gene expression estimation
The reads were mapped to pseudomolecule level genome assemblies30 using TopHat273 using a reference GTF file using the −G option and the inner distance between the mating pairs was set to 180 bp with a mate standard deviation of 75. The reads that were not aligned to the reference genomes were used to align on the Aspergillus genome (A. flavus NRRL3357 GCF_000006275.2) available at NCBI74.
Read counts were normalized by calculating the fragments per kilobase of exon per million fragments mapped (FPKM) value for each genes. Reads were assembled into transfrags using Cufflinks v2.1.175. Genes with FPKM ≥ 1 were further considered for analysis. Differentially expressed genes were identified using Cuffdiff76. The log2 fold change values of ≥ + 2 and ≤ − 2 (up- and down-regulated) with significance level ‘yes’ were considered for DEGs.
Functional categorization of the DEGs was studied using GoSlim (http://www.agbase.msstate.edu/cgi-bin/tools/goslimviewer_select.pl) while gene ontology (GO) terms were assigned to the expressed genes and assignment of KEGG pathways was carried out using Blast2GO77. DiffCoEx analysis method was used to identify differentially coexpressed gene modules based on Weighted Gene Coexpression Network Analysis (WGCNA) framework78. The clustering was done using standard hierarchical clustering with average linkage, followed by coexpressed gene modules detection from the resulting dendrogram, using a fixed cut height of 0.3, minimum module size of 30, and the soft thresholding power of 10. Each module was depicted with different built-in colors.
The alignment (bam) files were used for variant discovery with Genome Analysis Tool Kit (GATK)79. A position was reported as a variant for a genotype if phred quality score > 30 supported by a minimum read depth of 5. Distribution of DNA polymorphisms was assessed by calculating their frequency in a window size of 100 Kb along each pseudomolecule. For identification of effects of synonymous and non-synonymous SNPs and Indels, SnpEff program80 was used. In-house Perl scripts were used to analyze the distribution of the variations (SNPs and Indels) across the genome. BatchPrimer3 tool was used to design allele-specific primers.
Quantitative real time PCR (qRT-PCR) analysis
To validate the expression analysis of key candidate genes, primers were designed using Primer 3 plus tool (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi). cDNA was prepared using superscript first strand synthesis followed by second strand synthesis according to the instructions of manufactures (Invitrogen, USA). The primer pairs used for qRT-PCR are presented in Supplementary Table 8. The primer efficiency was checked using 10-fold dilution of cDNA and primers with efficiency ranging from 90–110% were used further for qRT-PCR. The qRT-PCR was performed on the Applied Biosystems 7500 Real-Time PCR systems using SYBRGreen chemistry following the manufacturer’s instructions (Invitrogen, USA). At least two independent biological replicates and three technical replicates at four different stages (1DAI, 2DAI, 3DAI and 7DAI) were used for qRT-PCR analysis. Alcohol dehydrogenase (adh3) was used as a reference gene and relative expression of the candidate genes was calculated using the delta delta Ct method81.
Electronic supplementary material
Acknowledgements
We acknowledge Dr. Manish Vishwakarma for designing allele specific primers and Ms. Vanika Garg for preparing circos diagram for the manuscript. This research was partially funded by the USAID-US University Collaboration Grant, Peanut & Mycotoxin Innovation Lab (PMIL), National Natural Science Foundation of China (No. 31461143022) funded China-CGIAR Collaborative Research Project and MARS Chocolate North America, USA. This work has been undertaken as part of the CGIAR Research Program on Grain Legumes. ICRISAT is a member of CGIAR Consortium.
Author Contributions
S.N.N. performed the experiments, assisted in data analysis, interpretation and writing of manuscript. G.A. and S.P. performed the data analysis. M.K.P. contributed towards designing of experiment, interpretation of results and manuscript writing, H.K.S. contributed towards the designing of experiment, toxin estimation and manuscript revision. A.S.J. and A.D. assisted in RNA preparations and validation experiments, L.W., B.L. and B.G. contributed towards interpretation and manuscript revision. R.K.V. conceived and supervised the project, revised and submitted the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-09260-8
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Williams JH, et al. HIV and hepatocellular and esophageal carcinomas related to consumption of mycotoxin-prone foods in sub-Saharan Africa. The American Journal of Clinical Nutrition. 2010;92:154–160. doi: 10.3945/ajcn.2009.28761. [DOI] [PubMed] [Google Scholar]
- 2.Andrade P, Caldas E. Aflatoxins in cereals: worldwide occurrence and dietary risk assessment. World Mycotoxin Journal. 2015;8:415–431. doi: 10.3920/WMJ2014.1847. [DOI] [Google Scholar]
- 3.Gong Y, et al. Post weaning exposure to aflatoxin results in impaired child growth: a longitudinal study in Benin, West Africa. Environmental Health Perspectives. 2004;112:1334–1338. doi: 10.1289/ehp.6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang Y, et al. Aflatoxin-related immune dysfunction in health and in human immunodeficiency virus disease. Clinical and Developmental Immunology. 2008;2008 doi: 10.1155/2008/790309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jolly PE. Aflatoxin: does it contribute to an increase in HIV viral load? Future Microbiology. 2014;9:121–124. doi: 10.2217/fmb.13.166. [DOI] [PubMed] [Google Scholar]
- 6.Wild CP, Montesano R. A model of interaction: aflatoxins and hepatitis viruses in liver cancer aetiology and prevention. Cancer Letters. 2009;286:22–28. doi: 10.1016/j.canlet.2009.02.053. [DOI] [PubMed] [Google Scholar]
- 7.Upadhyaya, H.D., Nigam, S.N. & Thakur R.P. Genetic enhancement for resistance to aflatoxin contamination in groundnut, In F. Waliyar, Adomou M, eds Summary proceedings of the 7th ICRISAT regional groundnut meeting for Western and Central Africa, Patancheru, India, 29–36 (2002).
- 8.Waliyar F, et al. Prevalence and distribution of aflatoxin contamination in groundnut (Arachis hypogaea L.) in Mali, West Africa. Crop Protection. 2015;70:1–7. doi: 10.1016/j.cropro.2014.12.007. [DOI] [Google Scholar]
- 9.Liang XA, Luo M, Guo BZ. Resistance Mechanisms to Aspergillus falvus infection and aflatoxin contamination in peanut (Arachis hypogaea) Plant Pathology Journal. 2006;5:115–124. doi: 10.3923/ppj.2006.115.124. [DOI] [Google Scholar]
- 10.Mehan, V. K. et al. Screening groundnuts for resistance to seed invasion by Aspergillus flavus and to aflatoxin production. In International Workshop on Aflatoxin Contamination of Groundnut, Patancheru, India, 6–9 Oct. ICRISAT. 323–334 (1989).
- 11.Waliyar F, et al. Resistance to pre-harvest aflatoxin contamination in ICRISAT’s groundnut mini core collection. European Journal of Plant Pathology. 2016;145:901–913. doi: 10.1007/s10658-016-0879-9. [DOI] [Google Scholar]
- 12.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin L, Fei Z, Giovannoni J, Rose JKC. Catalyzing plant science research with RNA-seq. Frontiers in Plant Science. 2013;4 doi: 10.3389/fpls.2013.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Payton P, et al. Gene expression profiling in peanut using high density oligonucleotide microarrays. BMC Genomics. 2009;10 doi: 10.1186/1471-2164-10-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen X, et al. Comparison of gene expression profiles in cultivated peanut (Arachis hypogaea) under strong artificial selection. Plant Breeding. 2012;131:620–630. doi: 10.1111/j.1439-0523.2012.01997.x. [DOI] [Google Scholar]
- 16.Li HF, et al. Transcriptome profiling of peanut (Arachis hypogaea) gynophores in gravitropic response. Functional Plant Biology. 2013;40:1249–1260. doi: 10.1071/FP13075. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, et al. De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in Peanut (Arachis hypogaea L.) BMC Genomics. 2012;13 doi: 10.1186/1471-2164-13-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia H, et al. Transcriptome profiling of peanut gynophores revealed global reprogramming of gene expression during early pod development in darkness. BMC Genomics. 2013;14 doi: 10.1186/1471-2164-14-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X, et al. Deep sequencing analysis of the transcriptomes of peanut aerial and subterranean young pods identifies candidate genes related to early embryo abortion. Plant Biotechnology Journal. 2013;11:115–127. doi: 10.1111/pbi.12018. [DOI] [PubMed] [Google Scholar]
- 20.Yin D, et al. De novo assembly of the peanut (Arachis hypogaea L.) seed transcriptome revealed candidate unigenes for oil accumulation pathways. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0073767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao C, et al. Small RNA and degradome deep sequencing reveals peanut microRNA roles in response to pathogen infection. Plant Molecular Biology Reporter. 2015;33:1013–1029. doi: 10.1007/s11105-014-0806-1. [DOI] [Google Scholar]
- 22.Clevenger J, Chu Y, Scheffler B, Ozias-Akins P. A developmental transcriptome map for allotetraploid Arachis hypogaea. Frontiers in Plant Science. 2016;7 doi: 10.3389/fpls.2016.01446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo DP, et al. Identification of transcripts involved in resistance responses to leaf spot disease caused by Cercosporidium personatum in peanut (Arachis hypogaea L. Phytopathology. 2005;95:381–387. doi: 10.1094/PHYTO-95-0381. [DOI] [PubMed] [Google Scholar]
- 24.Guimarães PM, et al. Global transcriptome analysis of two wild relatives of peanut under drought and fungi infection. BMC Genomics. 2012;13 doi: 10.1186/1471-2164-13-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, et al. Dynamics in the resistant and susceptible peanut (Arachis hypogaea L.) root transcriptome on infection with the Ralstonia solanacearum. BMC Genomics. 2014;15 doi: 10.1186/1471-2164-15-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo B, et al. Peanut gene expression profiling in developing seeds at different reproduction stages during Aspergillus parasiticus infection. BMC Developmental Biology. 2008;8 doi: 10.1186/1471-213X-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo B, et al. Gene expression profiling and identification of resistance genes to Aspergillus flavus infection in peanut through EST and microarray strategies. Toxins. 2011;3:737–753. doi: 10.3390/toxins3070737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo M, et al. Microarray-based screening of differentially expressed genes in peanut in response to Aspergillus parasiticus infection and drought stress. Plant Science. 2005;169:695–703. doi: 10.1016/j.plantsci.2005.05.020. [DOI] [Google Scholar]
- 29.Wang H, et al. Comparative transcript profiling of resistant and susceptible peanut post-harvest seeds in response to aflatoxin production by Aspergillus flavus. BMC Plant Biology. 2016;16 doi: 10.1186/s12870-015-0700-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bertioli DJ, et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nature Genetics. 2016;48:438–446. doi: 10.1038/ng.3517. [DOI] [PubMed] [Google Scholar]
- 31.Chen X, et al. Draft genome of the peanut A-genome progenitor (Arachis duranensis) provides insights into geocarpy, oil biosynthesis, and allergens. Proceedings of the National Academy of Sciences. 2016;113:6785–6790. doi: 10.1073/pnas.1600899113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varshney RK, Nayak SN, May GD, Jackson SA. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends in Biotechnology. 2009;27:522–530. doi: 10.1016/j.tibtech.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 33.Varshney, R. K., Terauchi, R. & McCouch, S. R. Harvesting the promising fruits of genomics: applying genome sequencing technologies to crop breeding. PLoS Biology12, e1001883 (2014). [DOI] [PMC free article] [PubMed]
- 34.Pandey, M. K. et al. Development and evaluation of a high density genotyping ‘Axiom_Arachis’ array with 58K SNPs for accelerating genetics and breeding in groundnut. Scientific Reports7, 40577 (2017). [DOI] [PMC free article] [PubMed]
- 35.Mathioni SM, Beló A, Rizzo CJ, Dean RA, Donofrio NM. Transcriptome profiling of the rice blast fungus during invasive plant infection and in vitro stresses. BMC Genomics. 2011;12 doi: 10.1186/1471-2164-12-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh KB, Foley RC, Oñate-Sánchez L. Transcription factors in plant defense and stress responses. Current Opinion in Plant Biology. 2002;5:430–436. doi: 10.1016/S1369-5266(02)00289-3. [DOI] [PubMed] [Google Scholar]
- 37.Durrant W, Dong X. Systemic Acquired Resistance. Annual Review of Phytopathology. 2004;42:185–209. doi: 10.1146/annurev.phyto.42.040803.140421. [DOI] [PubMed] [Google Scholar]
- 38.Kunkel BN, Brooks DM. Cross talk between signaling pathways in pathogen defense. Current Opinion in Plant Biology. 2002;5:325–331. doi: 10.1016/S1369-5266(02)00275-3. [DOI] [PubMed] [Google Scholar]
- 39.Govrin EM, Levine A. The hypersensitive response facilitates plant infection by the necrotrophic pathogen Botrytis cinerea. Current Biology. 2000;10:751–757. doi: 10.1016/S0960-9822(00)00560-1. [DOI] [PubMed] [Google Scholar]
- 40.Doehlemann G, et al. Reprogramming a maize plant: transcriptional and metabolic changes induced by the fungal biotroph Ustilago maydis. The Plant Journal. 2008;56:181–195. doi: 10.1111/j.1365-313X.2008.03590.x. [DOI] [PubMed] [Google Scholar]
- 41.Dolezal AL, et al. Aspergillus flavus infection induces transcriptional and physical changes in developing maize kernels. Frontiers in Microbiology. 2014;5 doi: 10.3389/fmicb.2014.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brodhagen M, et al. Reciprocal oxylipin-mediated cross-talk in the Aspergillus-seed pathosystem. Molecular Microbiology. 2008;67:378–391. doi: 10.1111/j.1365-2958.2007.06045.x. [DOI] [PubMed] [Google Scholar]
- 43.Amaike S, Keller NP. Aspergillus flavus. Annual Review of Phytopathology. 2011;49:107–133. doi: 10.1146/annurev-phyto-072910-095221. [DOI] [PubMed] [Google Scholar]
- 44.Yu J, et al. Clustered pathway genes in aflatoxin biosynthesis. Applied and Environmental Microbiology. 2004;70:1253–1262. doi: 10.1128/AEM.70.3.1253-1262.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kachroo A, Kachroo P. Fatty acid-derived signals in plant defense. Annual Review of Phytopathology. 2009;47:153–176. doi: 10.1146/annurev-phyto-080508-081820. [DOI] [PubMed] [Google Scholar]
- 46.Mierziak J, Kostyn K, Kulma A. Flavonoids as Important Molecules of Plant Interactions with the Environment. Molecules. 2014;19:16240–16265. doi: 10.3390/molecules191016240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dao TTH, Linthorst HJM, Verpoorte R. Chalcone synthase and its functions in plant resistance. Phytochemistry Reviews. 2011;10:397–412. doi: 10.1007/s11101-011-9211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pedras MSC, Zheng Q-A, Gadagi RS, Rimmer SR. Phytoalexins and polar metabolites from the oilseeds canola and rapeseed: Differential metabolic responses to the biotroph Albugo candida and to abiotic stress. Phytochemistry. 2008;69:894–910. doi: 10.1016/j.phytochem.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 49.Arman M. LC-ESI-MS characterisation of phytoalexins induced in chickpea and pea tissues in response to a biotic elicitor of Hypnea musciformis (red algae) Natural Product Research. 2011;25:1352–1360. doi: 10.1080/14786419.2011.553952. [DOI] [PubMed] [Google Scholar]
- 50.Lee MR, Chun JY, Kim HJ, Jeong JI. Induction of glyceollins by fungal infection in varieties of Korean soybean. Journal of Microbiology and Biotechnology. 2010;20:1226–1229. doi: 10.4014/jmb.1005.03047. [DOI] [PubMed] [Google Scholar]
- 51.Sobolev VS, Neff SA, Gloer JB. New stilbenoids from peanut (Arachis hypogaea) seeds challenged by an Aspergillus caelatus Strain. Journal of Agricultural and Food Chemistry. 2009;57:62–68. doi: 10.1021/jf802891v. [DOI] [PubMed] [Google Scholar]
- 52.Wang, H. et al. Deep sequencing analysis of transcriptomes in Aspergillus flavus in response to resveratrol. BMC Microbiology15, 182 (2015). [DOI] [PMC free article] [PubMed]
- 53.Dixon DP, Lapthorn A, Edwards R. Plant glutathione transferases. Genome Biology. 2002;3:3004.1–3004.10. doi: 10.1186/gb-2002-3-3-reviews3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hammond-Kosack KE, Jones JD. Resistance gene-dependent plant defense responses. The Plant Cell. 1996;8 doi: 10.1105/tpc.8.10.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gao X, Kolomiets MV. Host-derived lipids and oxylipins are crucial signals in modulating mycotoxin production by fungi. Toxin Reviews. 2009;28:79–88. doi: 10.1080/15569540802420584. [DOI] [Google Scholar]
- 56.Gao X, et al. Inactivation of the lipoxygenase ZmLOX3 increases susceptibility of maize to Aspergillus spp. Molecular Plant-Microbe Interactions. 2009;22:222–231. doi: 10.1094/MPMI-22-2-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mellon JE, Cotty PJ. No Effect of Soybean Lipoxygenase on Aflatoxin Production in Aspergillus flavus–Inoculated Seeds. Journal of Food Protection. 2002;65:1984–1987. doi: 10.4315/0362-028X-65.12.1984. [DOI] [PubMed] [Google Scholar]
- 58.Whitehead SR, Tiramani J, Bowers MD. Iridoid glycosides from fruits reduce the growth of fungi associated with fruit rot. Journal of Plant Ecology. 2015;9:357–366. doi: 10.1093/jpe/rtv063. [DOI] [Google Scholar]
- 59.Ebrahim S, Usha K, Singh B. Pathogenesis related (PR) proteins in plant defense mechanism. Science against Microbial Pathogens. 2011;2:1043–1054. [Google Scholar]
- 60.Golshani F, Fakheri BA, Behshad E, Vashvaei RM. PRs proteins and their mechanism in plants. Biological Forum – An International Journal. 2015;7:477–495. [Google Scholar]
- 61.Hanin M, et al. Plant dehydrins and stress tolerance: versatile proteins for complex mechanisms. Plant Signaling & Behavior. 2011;6:1503–1509. doi: 10.4161/psb.6.10.17088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu Y, et al. ZmLEA3, a multifunctional group 3 LEA protein from maize (Zea mays L.), is involved in biotic and abiotic stresses. Plant and Cell Physiology. 2013;54:944–959. doi: 10.1093/pcp/pct047. [DOI] [PubMed] [Google Scholar]
- 63.Liang XQ, Holbrook CC, Lynch RE, Guo BZ. β-1,3-Glucanase activity in peanut seed (Arachis hypogaea) is induced by inoculation with Aspergillus flavus and copurifies with a conglutin-like protein. Phytopathology. 2005;95:506–511. doi: 10.1094/PHYTO-95-0506. [DOI] [PubMed] [Google Scholar]
- 64.Wang Z, Yan S, Liu C, Chen F, Wang T. Proteomic analysis reveals an aflatoxin-triggered immune response in cotyledons of Arachis hypogaea infected with Aspergillus flavus. Journal of Proteome Research. 2012;11:2739–2753. doi: 10.1021/pr201105d. [DOI] [PubMed] [Google Scholar]
- 65.Wesolowski J, Paumet F. SNARE motif: a common motif used by pathogens to manipulate membrane fusion. Virulence. 2010;1:319–324. doi: 10.4161/viru.1.4.12195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kawahara Y, et al. Simultaneous RNA-seq analysis of a mixed transcriptome of rice and blast fungus interaction. PLoS One. 2012;7 doi: 10.1371/journal.pone.0049423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yazawa, T., Kawahigashi, H., Matsumoto, T. & Mizuno, H. Simultaneous transcriptome analysis of Sorghum and Bipolaris sorghicola by using RNA-seq in combination with de novo transcriptome assembly. PLoS ONE8, e62460 (2013). [DOI] [PMC free article] [PubMed]
- 68.Belkhadir Y, Subramaniam R, Dangl JL. Plant disease resistance protein signaling: NBS–LRR proteins and their partners. Current Opinion in Plant Biology. 2004;7:391–399. doi: 10.1016/j.pbi.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 69.Bent AF, Mackey D. Elicitors, Effectors, and R Genes: The new paradigm and a lifetime supply of questions. Annual Review of Phytopathology. 2007;45:399–436. doi: 10.1146/annurev.phyto.45.062806.094427. [DOI] [PubMed] [Google Scholar]
- 70.Fountain, J. C. et al. Environmental influences on maize-Aspergillus flavus interactions and aflatoxin production. Frontiers in Microbiology5, 40 (2014). [DOI] [PMC free article] [PubMed]
- 71.Waliyar, F., Reddy, S. V. & Kumar P. L. Estimation of Aspergillus flavus infection and aflatoxin contamination in seeds. Laboratory Manual. International Crops Research Institute for the Semi-Arid Tropics, Patancheru, India, 26–27 (2005).
- 72.Katta, M. A., Khan, A. W., Doddamani, D., Thudi, M. & Varshney, R. K. NGS-QC box and Raspberry for parallel, automated and rapid quality control analysis of large-scale next generation sequencing (Illumina) data. PLoS ONE10, e0139868 (2015). [DOI] [PMC free article] [PubMed]
- 73.Trapnell C, Pachter L, Salzberg SL. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nierman WC, et al. Genome Sequence of Aspergillus flavus NRRL 3357, a Strain That Causes Aflatoxin Contamination of Food and Feed. Genome Announcements. 2015;3:e00168–15. doi: 10.1128/genomeA.00168-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Conesa A, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 78.Tesson BM, Breitling R, Jansen RC. DiffCoEx: a simple and sensitive method to find differentially coexpressed gene modules. BMC Bioinformatics. 2010;11 doi: 10.1186/1471-2105-11-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McKenna A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.