
Fig. 4.
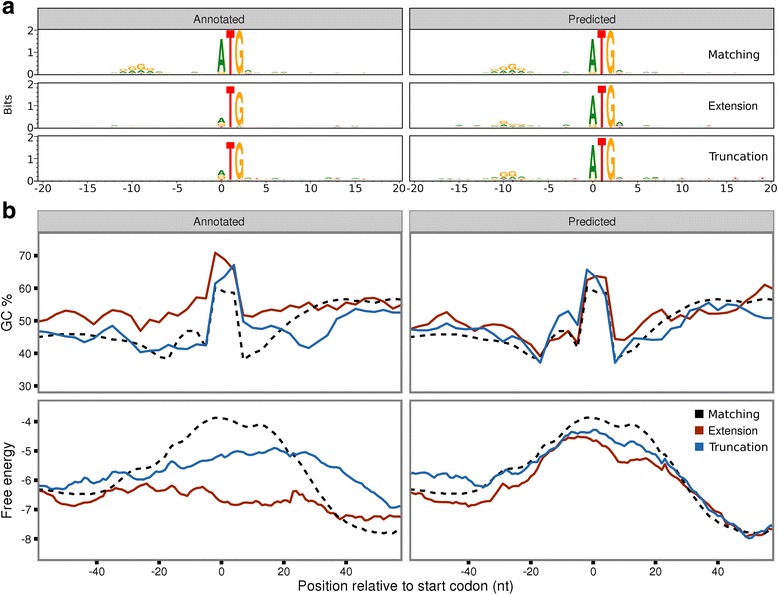
Sequence and structure features support re-annotation of translation initiation sites (TISs). a Sequence motifs relative to annotated or predicted TISs in the same genes. ‘Matching’ (n = 3853) are identical, while predicted extensions (n = 214) and truncations (n = 205) have stronger Shine-Dalgarno sequences than their annotated counterparts. b Meta-profiles relative to annotated or predicted TISs, with lines representing open reading frames matching annotated genes (dashed black), predicted extensions (red) and predicted truncations (blue). (upper) Mean guanine-cytosine (GC) content at third codon positions, averaged over 9 nt sliding windows. Predicted TISs match the expected profile more closely than annotated positions (Wilcoxon rank sum test W = 463,640, P = 0.001665 for extensions, W = 453,510, P = 0.0001546 for truncations), showing an increase in GC content immediately after the start codon, whereas annotated extensions and truncations are less similar to the expected profile (Wilcoxon rank sum test W = 493,250, P = 1.226 × 10–9 for extensions, Wilcoxon rank sum test W = 460,810, P = 5.395 × 10–5 for truncations), showing shifts down or upstream in annotated TIS. Peaks over the zero position correspond to nucleotide biases in start codon selection. (lower) Meta-profiles of mean free energy averaged in 39 nt sliding windows. Peaks of low secondary structure potential, expected to occur over start codons, are centred over predicted TIS, but are clearly shifted down or upstream of annotated TIS, in predicted extensions and truncations