

Abstract
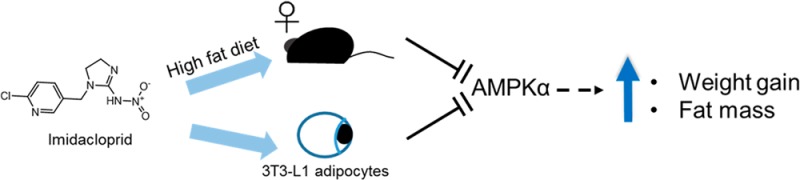
Imidacloprid, a neonicotinoid insecticide, was previously reported to enhance adipogenesis and resulted in insulin resistance in cell culture models. It was also reported to promote high fat diet-induced obesity and insulin resistance in male C57BL/6J mice. Thus, the goal of the present study was to determine the effects of imidacloprid and dietary fat interaction on the development of adiposity and insulin resistance in female C57BL/6J mice. Mice were fed with a low (4% w/w) or high fat (20% w/w) diet containing imidacloprid (0.06, 0.6, or 6 mg/kg bw/day) for 12 weeks. Mice fed with imidacloprid (0.6 mg/kg bw/day) significantly enhanced high fat diet-induced weight gain and adiposity. Treatment with imidacloprid significantly increased serum insulin levels with high fat diet without effects on other markers of glucose homeostasis. AMPKα activation was significantly inhibited by 0.6 and 6 mg imidacloprid/kg bw/day in white adipose tissue. Moreover, AMPKα activation with 5-aminoimidazole-4-carboxamide ribonucleotide abolished the effects of imidacloprid (10 μM) on enhanced adipogenesis in 3T3-L1 adipocytes. N-Acetyl cysteine also partially reversed the effects of imidacloprid on reduced phosphorylation of protein kinase B (AKT) in C2C12 myotubes. These results indicate that imidacloprid may potentiate high fat diet-induced adiposity in female C57BL/6J mice and enhance adipogenesis in 3T3-L1 adipocytes via the AMPKα-mediated pathway. Imidacloprid might also influence glucose homeostasis partially by inducing cellular oxidative stress in C2C12 myotubes.
Keywords: imidacloprid, neonicotinoids, adiposity, insulin resistance, AMPKα, AKT
Introduction
Neonicotinoids are the largest single class of insecticides currently on the market, which occupies approximately 27% of global insecticide use in 2013.1 Since its commercial introduction in 1991, imidacloprid has become the most successful neonicotinoid insecticide, representing 41.5% of neonicotinoid use.2,3 Imidacloprid is used on various vegetables, grains, and turf to control insect pests as well as on pets to control ectoparasitic arthropods.2,4,5 Imidacloprid is also known to persist in soil with a photolysis half-life of 39 days and an aerobic half-life of ∼3 years.6 These findings suggest high potential for human exposure to imidacloprid. In addition, the use of imidacloprid and two other neonicotinoids were restricted by the European Commission in 2013 due to their potential role in the collapse of bee populations.7
Epidemiological studies have suggested a link between persistent organic pollutants, including insecticides, and the risk of obesity and its associated pathology.8−12 Our previous studies have demonstrated that several pesticides, including imidacloprid, promotes adipogenesis in 3T3-L1 adipocytes and induces insulin resistance in C2C12 myotubes.13−17 Our recent publication also reported that oral imidacloprid exposure enhanced high fat diet-induced adiposity and insulin resistance in male C57BL/6J mice.18 It is not known, however, whether imidacloprid exposure alone or together with high fat diet will exacerbate obesity and insulin resistance symptoms in female mice. Thus, the present study aimed to determine if oral exposure to imidacloprid would aggravate high fat diet-induced adiposity and insulin resistance in female C57BL/6J mice. The role of AMPKα in imidacloprid-induced adipogenesis in 3T3-L1 adipocytes was also determined.
Materials and Methods
Materials
3T3-L1 preadipocytes and C2C12 myoblasts were from the American Type Culture Collection (Manassas, VA). Horse serum (HS), fetal bovine serum (FBS), methylisobutylxanthin, insulin, dexamethasone, phosphatase and protease inhibitor cocktail, and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Imidacloprid (>98%, CAS: 138261-41-3) was from Chem Service Inc. (West Chester, PA, USA). Five-week-old female C57BL/6J mice were from the Jackson Laboratory (Bar Harbor, ME, USA). Semipurified powdered diets (TD 94048, 4% fat w/w, low fat diet and TD 07518, 20% fat w/w, high fat diet) were prepared based on Harlan Laboratories (Madison, WI, USA) diets. Food ingredients were purchased from Bio-Serv (Flemington, NJ, USA). Serum insulin level was determined with mouse insulin ELISA kit from ALPCO (Salem, NH, USA). Nonesterified fatty acid assay kit was from Wako Life Sciences, Inc. (Mountain View, CA, USA). Total cholesterol and glucose assay kits were from Genzyme Diagnostics (Charlottetown, PE, Canada). Mouse leptin assay kit was obtained from R&D Systems (Minneapolis, MN, USA). Mem-PER Plus Membrane Protein Extraction Kit, BCA protein assay kit, Infinity triglycerides reagent, dichlorodihydrofluorescein diacetate (H2DCFDA), N-acetyl cysteine (NAC), and protein A-sepharose 4B conjugate were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Radioimmunoprecipitation assay (RIPA) buffer supplemented with 1% protease inhibitor was obtained from Boston Bioproducts Inc. (Ashland, MA, USA). Rabbit antibodies for mouse acetyl-CoA carboxylase (ACC), phosphorylated ACC (pACC), AMP-activated protein kinase α (AMPKα), phosphorylated AMPKα (pAMPKα), and horseradish peroxidase-conjugated goat antirabbit IgG were obtained from Cell Signaling Technology (Beverly, MA, USA). Rabbit antibodies for β-actin were from Santa Cruz Biotechnology (Dallas, Texas, USA). Rabbit antibody for Ca2+/calmodulin-dependent protein kinase β (CAMKKβ) was purchased from Abcam Inc. (Cambridge, MA, USA). Human recombinant insulin (Novolin, 100 U/ml) was bought from Novo Nordisk Pharmaceuticals Industries Inc. (Seattle, WA, USA). Dextrose (50%) and bacteriostatic 0.9% sodium chloride solution were from Hospira, Inc. (Lake Forest, IL, USA). Dulbecco’s modified Eagle medium was from Mediatech, Inc. (Manassas, VA). (DMEM), dimethyl sulfoxide (DMSO), and other chemicals were purchased from Fisher Scientific (Pittsburgh, PA, USA).
Animals and Diet
All animal work was performed using the guidelines approved by the Institutional Animal Care and Use Committee at the University of Massachusetts under protocol number 2016-0011. Mice were housed in a temperature and humidity-controlled room with a 12 h light/dark cycle. The compositions of diets were as previously described.18 Water and diet were given to mice ad libitum, and each was changed twice a week. Body weight and food intake were monitored weekly. After adaptation, the mice were fed a control diet (4% fat w/w) and subjected to baseline insulin tolerance and glucose tolerance tests. Mice were then divided into low fat and high fat dietary groups with four imidacloprid treatment doses (0, 0.06, 0.6, and 6 mg/kg bw/day). The treatment lasted for 12 weeks. Doses of imidacloprid were based on a previous publication;18 the highest imidacloprid dose was no observed adverse effect level (NOAEL) of imidacloprid at 5.7 mg/kg bw/day, and the lowest dose of imidacloprid was estimated average daily intake of imidacloprid of 60 μg/kg body weight/day.19−21 Imidacloprid contents in low fat diets were 0, 0.47, 4.7, and 47 mg of imidacloprid/kg diet. The contents of imidacloprid in high fat diets were 0, 0.636, 6.36, and 63.6 mg of imidacloprid/kg diet. In the end, the average imidacloprid consumptions were 0, 0.07, 0.69, and 6.69 mg/kg bw/day in low fat dietary groups and 0, 0.08, 0.74, and 6.66 mg/kg bw/day in high fat dietary groups. There were no differences in imidacloprid intakes between low vs high fat diet groups at the same doses.
At the end of the experiment, mice were fasted for 4 h before being sacrificed by CO2 asphyxiation. Blood was collected by cardiac puncture and then centrifuged at 2000g for 20 min to collect serum. Part of the omental adipose tissue was first fixed in 10% phosphate-buffered formalin and then used for paraffin sectioning.22 The adipocyte size was measured as previously described.18 The other parts of the omental adipose tissue and all other organs were snap frozen with liquid nitrogen and kept at −80 °C. The levels of total nonesterified fatty acids (NEFA), leptin, cholesterol, triglyceride (TG), and insulin in serum were determined with commercial kits following the manufacturers’ protocols. The homeostasis model assessment-insulin resistance (HOMA-IR) score, insulin tolerance test (ITT), and glucose tolerance test (GTT) were performed as previously described.18
mRNA Expression Analysis
Total RNA was extracted as previously described.18 Tumor necrosis factor α (TNF-α) and 18S rRNA (18S rRNA) were analyzed. Real-time polymerase chain reaction (PCR) was carried out as previously described.18 Mm00443258_m1 (TNFα) and Mm03928990_g1 (18S rRNA) taqman probe-based gene expression assay kits were from Thermo Fisher Scientific (Waltham, MA, USA).
Immunoprecipitation and Immunoblotting
Proteins from mouse tissues were extracted, quantified, and immunoblotted as previously described.17,18 For the detection of the phosphorylation of serine 307 on insulin receptor substrate (IRS1) (p-IRS1), cell lysates (500 μg of protein) were immunoprecipitated with IRS1 and protein G beads overnight at 4 °C before immunoblotting.
Cell Culture
3T3-L1 preadipocytes were maintained and differentiated as previously described.17 3T3-L1 cells were cultured in DMEM with 10% FBS at 37 °C. Two days after confluence (day 0), cells were differentiated into adipocytes with a mixture of dexamethasone (1 μM), methylisobutylxanthin (0.5 mM), and insulin (1 μg/mL) in DMEM supplemented with 10% FBS. On day 2, the medium was replaced with DMEM with 10% FBS and insulin (1 μg/mL). Starting from day 4, medium was changed with DMEM containing 10% FBS every 2 days until day 8. 3T3-L1 cells were treated with imidacloprid (10 μM) and/or AICAR (40 μM) for 8 days by adding a stock solution of imidacloprid or AICAR in dimethyl sulfoxide (DMSO) with all treatments having a final concentration of 0.02% DMSO. The concentration of imidacloprid and AICAR were based on our previous studies.13,14,17
C2C12 cells were maintained in DMEM with 10% FBS at 37 °C. When the cells reach 80% confluence, they were differentiated into myotubes in DMEM with 2% horse serum for 6 days. The myotubes were pretreated with or without NAC (10 mM) for 30 min,23 and then 10 μM imidacloprid was added for an additional 2 days.
Determination of Cellular Oxidative Status
We measured the cellular oxidative status based on a previous method,24 where the nonfluorescent dye H2DCFDA is converted to fluorescent 2′,7′-dichlorofluorescein (DCF) after being oxidized by a variety of oxidants.24 After imidacloprid treatments, cells were incubated with 10 μM H2DCFDA for 30 min. Cells were washed with PBS three times, immediately harvested, and lysed with RIPA buffer. The fluorescence of the cell lysate supernatant was determined at excitation/emission 485/520 nm with a SpectraMax spectrophotometer (Molecular Devices, Sunnyvale, CA). The fluorescence was then normalized to the protein concentration of the cell lysate supernatant as determined by the BCA protein kit.
Statistical Analyses
The PROC MIXED procedure was utilized to analyze data with the SAS software (version 9.3, SAS Institute Inc., Cary, NC, USA). For the result of body weight (Figure 1A), two-way repeated measure Analysis of Variance (ANOVA) and the slice option in the Least Square (LS) means statement were used to determine the differences at each time point. For all other results in mice, two-way ANOVA along with LS means statement was used. The Tukey–Kramer’s method was applied for the multiple comparisons among the groups. If there were significant interactions between diet and imidacloprid, letters were used in the figures to present the differences between each group. When there were no interactions between diet and imidacloprid, brackets were used in the figures to represent differences between imidacloprid treatments and control groups. Cell culture data (Figure 4 and 5) were analyzed by one-way ANOVA followed by PROC mixed with LS means statement. Tukey’s test was utilized for the multiple comparisons among the groups. P < 0.05 was considered as statistically significant.
Figure 1.

Effects of imidacloprid on body weight, body weight gain, adipocyte size, and food intake. Mice were fed with low or high fat diet supplemented with imidacloprid (0, 0.06, 0.6, and 6 mg/kg bw/day) for 12 weeks. (A) Body weight was monitored weekly. (B) Body weight gain over 12 weeks. (C) Total calorie intake. (D) Representative pictures of epididymal adipose tissues after H&E staining (100× magnifications). (E) Adipocyte size. The mean area of 50 cells from each sample was measured with Image J. Numbers are means ± SE (n = 5–7 for A, B, and D and n = 3–4 for E). Means with different letters are significantly different at P < 0.05.
Figure 4.

Influence of AMPKα activation on adipogenesis induced by imidacloprid in 3T3-L1 adipocytes. (A) Triglyceride; (B) representative pictures; (C) C/EBPα, CAATT element binding protein-α; (D) pAMPKα/AMPKα, phosphorylated AMP-activated protein kinase-α/AMP-activated protein kinase-α. Cells were treated with imidacloprid (10 μM) or AICAR (40 μM) for 8 days. Numbers represent mean ± SE (n = 3). Means with different letters are significantly different at P < 0.05.
Figure 5.

Effects of imidacloprid on insulin signaling and oxidative stress in C2C12 myotubes. (A) pAKT/AKT, phosphorylated AKT/AKT; (B) pIRS1/IRS1, phosphorylated insulin receptor substrate 1 (Serine 307)/insulin receptor substrate 1; (C) effects of imidacloprid on oxidative stress; (D) N-acetylcysteine (NAC, 10 mM) partially restored the imidacloprid-induced decrease of pAKT. Immunoprecipitation was first performed with the IRS1 antibody and then immunoblotted with pIRS1 or IRS1 antibodies. Cells were differentiated into myotubes for 6 days in DMEM with 2% horse serum, and then the myotubes were treated with imidacloprid (10 μM) for 2 days. One day before insulin stimulation, media were changed to F-12K with 2% horse serum. Protein expression was determined after treatment with or without insulin (100 nM) for 15 min. Numbers represent mean ± SE (n = 3). Means with different letters are significantly different at P < 0.05.
Results
Imidacloprid Enhanced Body Weight Gain and Adipocyte Size in High Fat Diet-Fed Mice
Overall, there were significant effects of dietary fat, imidacloprid, time, and their interactions (imidacloprid × diet × time) on body weight (Figure 1A). Both dietary fat and imidacloprid had significantly increased body weight gain with a significant interaction between dietary fat and imidacloprid (Figure 1B, 133% increase in high fat compared to low fat diet and 10% increase in imidacloprid treatments compared to the controls). No differences in body weight were observed among all groups in low fat diet-fed mice (Figure 1A). In high fat diet-fed mice, imidacloprid (0.6 mg/kg bw/day) led to a significant increase in body weight compared to the control group starting from week 5 and maintained this trend until the end of the experiment (∼15–22% increase compared to the controls, P < 0.05 for all weeks from 5 to 12) (Figure 1A). Among high fat diet-fed groups, imidacloprid at 0.6 mg/kg bw/day treatment group had greater weight gain, 71% increase compared to that of the high fat diet-fed control (P = 0.0167) (Figure 1B).
The calorie consumption of mice is shown in Figure 1C. Both dietary fat and imidacloprid significantly affected the calorie intake without interaction. High fat diet-fed mice consumed 12% more calories than mice fed a low fat diet (Figure 1C), and imidacloprid fed mice have a 6% decrease in calorie intake compared to that of mice in the control groups. However, mice fed 6 mg/kg bw/day of imidacloprid consumed 12% fewer calories compared to that of the control groups (P = 0.0223) (Figure 1C).
Effects of Imidacloprid on Organs and Tissue Weights
Organs and tissues weights are shown in Table 1. There was a significant dietary fat effect, but neither an imidacloprid effect nor its interactions were evident on liver, heart, spleen, and kidney weights. Neither dietary fat nor imidacloprid treatments resulted in any effects on pancreas. There were significant effects of dietary fat and imidacloprid treatments, and with their interactions on omental and total adipose tissue weights (92–98% increase in high fat compared to low fat diet and 11–17% increase in imidacloprid treatments compared to the controls). In mice fed the high fat diet, animals with imidacloprid (0.6 mg/kg bw/day) had greater omental (45% increase, P = 0.0416) and total adipose tissue weights (40% increase, P = 0.0391) when compared with the control group.
Table 1. Organ Weightsa.
| low
fat diet |
high
fat diet |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| imidacloprid
(mg/kg bw/day) |
imidacloprid
(mg/kg bw/day) |
P-value |
|||||||||
| control | 0.06 | 0.6 | 6 | control | 0.06 | 0.6 | 6 | dietary fat | imidacloprid | interaction | |
| % of Body Weight | |||||||||||
| liver | 4.04 ± 0.27 | 4.45 ± 0.35 | 4.56 ± 0.17 | 4.87 ± 0.25 | 3.45 ± 0.26 | 3.63 ± 0.16 | 3.79 ± 0.13 | 3.36 ± 0.15 | <0.0001 | n.s. | n.s. |
| heart | 0.52 ± 0.02 | 0.54 ± 0.03 | 0.50 ± 0.01 | 0.48 ± 0.01 | 0.47 ± 0.03 | 0.46 ± 0.02 | 0.40 ± 0.02 | 0.42 ± 0.03 | <0.0001 | n.s. | n.s. |
| spleen | 0.37 ± 0.03 | 0.43 ± 0.07 | 0.35 ± 0.01 | 0.36 ± 0.02 | 0.37 ± 0.04 | 0.31 ± 0.01 | 0.26 ± 0.01 | 0.29 ± 0.01 | 0.017 | n.s. | n.s. |
| kidney | 1.30 ± 0.06 | 1.31 ± 0.08 | 1.27 ± 0.02 | 1.19 ± 0.02 | 1.17 ± 0.05 | 1.15 ± 0.05 | 1.05 ± 0.03 | 1.09 ± 0.04 | 0.0002 | n.s. | n.s. |
| pancreas | 1.04 ± 0.09 | 1.06 ± 0.08 | 0.96 ± 0.04 | 0.93 ± 0.03 | 1.06 ± 0.16 | 0.93 ± 0.10 | 0.82 ± 0.05 | 0.85 ± 0.04 | n.s. | n.s. | n.s. |
| Adipose Tissue | |||||||||||
| omental | 3.00 ± 0.28c | 2.82 ± 0.23c | 2.60 ± 0.27c | 3.34 ± 0.51bc | 4.62 ± 0.38b | 4.71 ± 0.69ab | 6.69 ± 0.39a | 6.63 ± 1.19a | <0.0001 | 0.010 | 0.0375 |
| subcutaneous | 2.45 ± 0.19abc | 2.32 ± 0.17c | 2.22 ± 0.24c | 2.24 ± 0.39c | 4.26 ± 0.41a | 4.22 ± 0.68ab | 5.89 ± 0.31a | 5.69 ± 0.38a | <0.0001 | n.s. | 0.029 |
| retroperitoneal | 1.01 ± 0.13 | 0.91 ± 0.17 | 0.90 ± 0.13 | 1.34 ± 0.18 | 2.16 ± 0.21 | 2.10 ± 0.39 | 2.93 ± 0.16 | 2.82 ± 0.12 | <0.0001 | n.s. | n.s. |
| mesenteric | 0.88 ± 0.15 | 0.80 ± 0.09 | 0.81 ± 0.12 | 1.07 ± 0.16 | 1.47 ± 0.16 | 1.50 ± 0.35 | 2.03 ± 0.19 | 1.80 ± 0.15 | <0.0001 | n.s. | n.s. |
| total | 7.34 ± 0.85cd | 6.85 ± 0.35d | 6.53 ± 0.81d | 7.99 ± 0.97bcd | 12.5 ± 1.25b | 10.0 ± 3.09bc | 17.5 ± 1.06a | 16.9 ± 1.08a | <0.0001 | 0.026 | 0.028 |
Mice were treated with three doses of imidacloprid (0.06, 0.6, and 6 mg/kg bw/day). Values represent means ± SE (n = 5–7). Means with different superscripts within the same row are significantly different at P < 0.05. Abbreviations: n.s., not significant.
Histological analysis showed significantly increased effects of both dietary fat and imidacloprid treatments as well as their interaction on omental adipocyte size (52% increase in high fat compared to low fat diet and 17% increase in imidacloprid treatments compared to the controls, Figure 1D and E). In high fat diet-fed mice, imidacloprid (0.6 mg/kg bw/day) treatment markedly increased adipocyte size (45% increase, P = 0.0063) compared to the control group (Figure 1D and E).
Effects of Imidacloprid on Serum Markers
Analyses of serum markers are shown in Table 2. There were significant effects of both dietary fat and imidacloprid treatments (without interaction) on serum insulin, leptin, and TG levels. There was a 43% increase for insulin, 167% increase for leptin, and 9% decrease in the high fat over low fat diet-fed mice. There was a 20% increase for insulin, 46% increase for leptin, and 19% increase in imidacloprid treatments over the controls. Mice treated with 6 mg of imidacloprid/kg bw/day had higher blood insulin and leptin levels compared with the respective control groups (50 and 87% increase with P = 0.0363 and 0.0271, respectively). Mice fed imidacloprid (0.06 mg/kg bw/day) had higher levels of TG (23% increase, P = 0.0458) than the control groups. Dietary fat, but not imidacloprid, had significant effects on cholesterol levels (16% increase in high fat compared to low fat diet groups). Neither dietary fat nor imidacloprid had any effect on serum glucose or NEFA levels in the current study.
Table 2. Serum Parametersa.
| low
fat diet |
high
fat diet |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| imidacloprid
(mg/kg BW/day) |
imidacloprid
(mg/kg BW/day) |
P-value |
|||||||||
| control | 0.06 | 0.6 | 6 | control | 0.06 | 0.6 | 6 | dietary fat | imidacloprid | interaction | |
| glucose (mg/dL) | 154.3 ± 27.1 | 139.9 ± 23.7 | 145.1 ± 10.1 | 144.3 ± 5.9 | 152.5 ± 15.1 | 150.9 ± 13.2 | 170.6 ± 9.3 | 141.9 ± 9.4 | n.s. | n.s. | n.s. |
| insulin (ng/mL) | 0.70 ± 0.09 | 0.79 ± 0.13 | 0.62 ± 0.08 | 1.14 ± 0.18 | 1.02 ± 0.10 | 1.00 ± 0.21 | 1.19 ± 0.25 | 1.45 ± 0.14 | 0.0025 | 0.0269 | n.s. |
| leptin (ng/mL) | 16.1 ± 5.0 | 19.8 ± 4.2 | 12.3 ± 3.8 | 29.1 ± 6.8 | 36.8 ± 5.0 | 47.7 ± 21.7 | 52.5 ± 4.0 | 69.7 ± 11.0 | <0.0001 | 0.0387 | n.s. |
| TG (mg/dL) | 63.1 ± 1.9 | 83.7 ± 7.2 | 75.3 ± 7.2 | 73.0 ± 3.8 | 59.9 ± 5.1 | 67.9 ± 3.7 | 66.3 ± 1.5 | 73.1 ± 2.1 | 0.0378 | 0.0343 | n.s. |
| cholesterol (mg/dL) | 65.3 ± 4.9 | 73.0 ± 6.5 | 71.3 ± 3.6 | 62.4 ± 5.2 | 73.1 ± 2.4 | 68.6 ± 2.8 | 89.3 ± 8.3 | 84.8 ± 8.5 | 0.0098 | n.s. | n.s. |
| NEFA (mEq/L) | 1.3 ± 0.3 | 1.5 ± 0.3 | 1.3 ± 0.3 | 1.0 ± 0.1 | 1.2 ± 0.2 | 1.3 ± 0.1 | 1.4 ± 0.2 | 1.8 ± 0.4 | n.s. | n.s. | n.s. |
Mice were treated with three doses of imidacloprid (0.06, 0.6, and 6 mg/kg bw/day). Values represent means ± SE (n = 5–7). Means with different superscripts within the same row are significantly different at P < 0.05. Abbreviations: n.s., not significant; TG, triglyceride; NEFA, nonesterified fatty acid.
Influence of Imidacloprid on Glucose Homeostasis
The effects of treatment with imidacloprid or dietary fat on glucose homeostasis were determined by ITT, GTT, and HOMA-IR (Figure 2 and Supplementary Figure 1). There were no differences of the ITT between any of the groups over time as shown in Figure 2A–C. There was a significant effect of dietary fat on GTT values in weeks 6 and 11 (7 and 19% increase in high fat groups compared to those in low fat groups, respectively) without imidacloprid or interaction effects (Figure 2E and F). There was only dietary fat effect on HOMA-IR without any effects of imidacloprid or interaction (P = 0.0047, Figure 2).
Figure 2.

Effects of imidacloprid on insulin tolerance test (ITT) (A–C) and glucose tolerance test (GTT) (D–F) as determined as the area under the curve (AUC) and HOMA-IR (G). Mice were fed a low or high fat diet supplemented with imidacloprid (0, 0.06, 0.6, and 6 mg/kg bw/day) for 12 weeks. ITT was measured during the adaptation period and weeks 5 and 9. GTT was measured during the adaptation period and weeks 6 and 11. Blood was collected from the tail vein, and glucose levels were determined at 0 min. Insulin (ITT) or glucose solutions (GTT) were subsequently administered by intraperitoneal injection, and the glucose level was further measured at 15, 30, 60, and 120 min post-injection. HOMA-IR score was calculated at week 12 with a HOMA-IR calculator. Numbers represent means ± SE (n = 5–8).
Effects of Imidacloprid on the AMPKα Pathway and TNFα in White Adipose Tissue
The activation of AMPK, a master regulator of energy metabolism, results in the inhibition of lipogenesis.25 On the basis of previous publications,13,18 we have determined the effects of imidacloprid and high fat diet on AMPK, one of its downstream markers (acetyl Co-A carboxylase, ACC), and one of its upstream regulators (Ca2+/calmodulin-dependent protein kinase β, CaMKKβ)26,27 from omental adipose tissue (Figure 3A–D). There were both dietary fat and imidacloprid effects on the expression of CaMKKβ without interaction (20% decrease in high fat compared to low fat diet and 18% decrease in imidacloprid treatments compared to the controls, Figure 3A). Imidacloprid (0.6 and 6 mg/kg bw/day) reduced the expression of CaMKKβ compared to controls (21 and 24% reduction with P = 0.0099 and 0.0023, respectively) (Figure 3A). There were significant effects of imidacloprid and imidacloprid and dietary fat interactions on the ratio of pAMPKα/AMPKα (7% decrease in high fat compared to low fat diet and 22% decrease in imidacloprid treatments compared to the controls, Figure 3B). High fat diet and imidacloprid (0.6 and 6 mg/kg bw/day) fed mice had a decreased ratio of pAMPKα/AMPKα compared to the high fat diet-fed control (45 and 58% reduction with P = 0.0064 and 0.0002, respectively) (Figure 3B). There were effects of dietary fat and imidacloprid as well as their interactions on the ratio of phosphorylated ACC/ACC (pACC/ACC) (14% decrease in high fat compared to low fat diet and 16% decrease in imidacloprid treatments compared to the controls, Figure 3C). High fat diet-fed mice treated with imidacloprid at 0.06 and 6 mg/kg bw/day had significantly lower pACC/ACC ratios compared to the high fat diet-fed control (48 and 43% reduction with P = 0.0061 and 0.0172, respectively) (Figure 3C). The current results indicate that oral administration of imidacloprid enhances lipogenesis in part via an AMPKα-dependent pathway in mice white adipose tissue.
Figure 3.

Effects of imidacloprid on AMPKα pathway and TNFα expression in white adipose tissue. Mice were fed with low or high fat diet supplemented with imidacloprid (0, 0.06, 0.6, and 6 mg/kg bw/day) for 12 weeks. (A) CaMKKβ, Ca2+/calmodulin-dependent protein kinase β; (B) pAMPKα/AMPKα, phosphorylated AMP-activated protein kinase-α/AMP-activated protein kinase-α; (C) pACC/ACC, phosphorylated acetyl-CoA carboxylase/acetyl-CoA carboxylase; (D) representative pictures; (E) TNFα, tumor necrosis factor α. Numbers represent mean ± SE (n = 4–6). Means with different letters are significantly different at P < 0.05.
There were effects of dietary fat and imidacloprid as well as their interactions on TNF-α mRNA level (127% increase in high fat compared to low fat diet and 70% increase in imidacloprid treatments compared to the controls, Figure 3E). High fat diet-fed mice treated with imidacloprid at 0.6 and 6 mg/kg bw/day had higher levels of TNFα compared to high fat diet-fed controls (165 and 182% increase, respectively, with P < 0.0001 for both) (Figure 3E).
Effects of AMPKα Activation on Imidacloprid-Induced Adipogenesis
To further determine the role of AMPKα activation on imidacloprid-induced adipogenesis, 3T3-L1 adipocytes were cotreated with imidacloprid and AICAR, an AMPK activator.17 Consistent with results in Figure 3 and a previous report,13 imidacloprid treatment alone increased fat accumulation compared to that of the control group (P = 0.0012) (Figure 4A). Treatment with AICAR alone reduced fat accumulation (P < 0.0001) as expected.17 However, cotreatment of AICAR and imidacloprid resulted in significantly decreased fat accumulation to a level similar to that of AICAR alone (P = 0.0012, compared to control). Figure 4B–D further shows that AICAR and imidacloprid cotreatment significantly decreased C/EBPα expression (P < 0.0001) while increasing the ratio of pAMPKα/AMPKα to a level similar to that of AICAR alone. These findings are consistent with previously reported findings where AMPK activation was reported to decrease the expression of C/EBPα.28 These results suggest that imidacloprid promotes adipogenesis via the AMPKα-mediated pathway in 3T3-L1 adipocytes.
Effects of Imidacloprid on the Insulin Signaling Pathway in C2C12 Myotubes
Phosphorylation of IRS1 serine 307 (pIRS1 S307) is associated with the inhibition of insulin signaling.29 Panels A and B in Figure 5 show that imidacloprid significantly decreases insulin-stimulated phosphorylation of protein kinase B (AKT) while increasing the phosphorylation of IRS1 S307. Imidacloprid (10 μM) was further observed to elevate oxidative stress (Figure 5C), and NAC, an oxygen-free radical chelator,30 partially reversed the decreased phosphorylation of AKT induced by imidacloprid (Figure 5D). These results indicate that the elevated cellular oxidative stress is partially responsible for insulin resistance induced by imidacloprid.
Discussion
In the current in vivo study, oral exposure to imidacloprid at or lower than the NOAEL aggravated high fat diet-induced weight gain and elevated serum insulin levels in C57BL/6J female mice. The current in vitro results indicate that imidacloprid may elicit these effects by post-translational regulation of the AMPK pathway in white adipose tissue. Although there was no significant effect of imidacloprid on insulin resistance in the current female study, imidacloprid might induce insulin resistance via TNFα and/or an oxidative stress-mediated mechanism as seen in a previous male study.18
Previously, oral administration of imidacloprid to mice or rats at 15 or 20 mg/kg bw/day significantly reduced body weight, whereas mice exposed to 5 or 10 mg/kg bw/day revealed no change in body weight when fed chow diet (low fat diet).31,32 Our previous publication first reported the interaction between oral imidacloprid administration (at lower than 6 mg/kd bw/day) and high fat diet in male mice, resulting in significantly induced weight gain in male mice.18 We now show that imidacloprid and high fat diet also significantly increased weight gain in female mice. There is only one other study reporting the role of high fat diet potentiating the effect of environmental contaminants on weight gain where persistent organic pollutants (POP) in crude fish oil were reported to potentiate high fat diet (20 wt %/wt corn oil)-induced visceral adipose tissue and weight gain compared with refined salmon oil (without POP) after 4 weeks in male Sprague–Dawley rats.33 This study failed, however, to compare the role of POP in a low fat diet. Therefore, it was not clear if high fat diet contributed to the effects of POP in this model. Thus, the significance of the current study and our previous male study is the interaction between low dose imidacloprid (at or lower than an NOAEL of 5.7 mg of imidacloprid/kg bw/day) and high fat diet on the development of obesity. The current results further support that effects of imidacloprid and high fat diet on weight gain were independent of the sex of the mice. Although the mechanism of how a high fat diet induces increased weight gain following imidacloprid treatment is still unknown, we speculate that the high fat diet and imidacloprid interaction is independent of type of fat used because both the current study (using soybean oil) and a study from Ruzzin et al. (2010) (using corn oil) observed similar weight gain by treatment and high fat diet. It is possible that the high fat diet induces metabolic stress and imidacloprid further exacerbates this process, resulting in excess weight gain in the high fat diet-fed mice. Alternatively, we cannot exclude the possibility that low and high fat diets might have different effects on the bioavailability of insecticides, thus leading to the differences observed in the current study.
In addition to high fat diet-induced weight gain, Ruzzin et al.33 also reported that persistent organic pollutants (POP) in crude fish oil promoted high fat diet (20 wt %/wt corn oil)-induced insulin resistance compared to that of refined salmon oil (without POP) in male rats. Others have reported that oral administration of 20 mg of imidacloprid/kg bw/day for 90 days with chow diet (low fat diet) increased the blood glucose level, whereas 10 mg of imidacloprid/kg bw/day did not affect blood glucose in female rats.31 Our previous study reported that imidacloprid treatment (0.6 and 6 mg/kg bw/day for 12 weeks) exacerbated high fat diet (soybean oil)-induced insulin resistance in male mice.18 In the current study, however, the 12 week imidacloprid exposure significantly increased serum insulin levels without having an influence on insulin and glucose tolerance in the high fat diet-fed female mice. These findings suggest a potential sex difference in the susceptibility to high fat diet-induced insulin resistance. It is possible that, compared with male mice, female mice may require a longer time and/or higher treatment dose to develop metabolic disorders under the experimental conditions used. In support of this contention, others have reported that female mice were resistant to high fat diet-induced glucose intolerance and insulin resistance,34 which may be due to estrogens that are protective against the development of metabolic syndrome and insulin resistance in female rodents.35
In the previous male study,18 there were interactions between imidacloprid and dietary fat on CaMKKβ expression and AMPK phosphorylation in adipocytes. In the current study, however, there is only an interaction between imidacloprid and dietary fat on AMPK phosphorylation but not on CaMKKβ expression. Thus, whether imidacloprid affects AMPKα activation via CaMKKβ or other upstream regulators in high fat diet-fed female mice needs to be further determined. In addition, imidacloprid is an agonist on nicotinic acetylcholine receptors at low concentrations but blocks them at high concentrations.36 It is not clear from the results of the current study whether imidacloprid induces adipogenesis and/or insulin resistance by way of or independent of its action on nicotinic acetylcholine receptors in muscle or adipose tissue.
Imidacloprid (10 μM) exposure was previously reported to induce insulin resistance by decreasing both insulin-stimulated glucose uptake and phosphorylation of AKT in myotubes.14 Imidacloprid was also reported to increase TNF-α, interleukin-1β, and interleukin-6 in mouse liver and brain.37 TNFα is a proinflammatory cytokine released mainly from macrophages in response to other physiological processes38 and is known to inhibit insulin signaling by activating protein tyrosine phosphatase by removing tyrosine phosphate groups from IRS-1.38 In addition, binding of TNFα to its receptors triggers a broad pattern of signaling cascades, including c-Jun amino-terminal kinase (JNK), which cause insulin resistance by promoting serine phosphorylation of IRS1 at 307, a negative regulator of insulin signaling.38 In this study, imidacloprid increased TNFα expression in adipose tissue, which might contribute to the development of insulin resistance by imidacloprid.
Chronic oxidative stress is thought to be one of contributing factors to the development of chronic diseases, including insulin resistance.39 Oxidative stress was reported to stimulate the serine kinase of p38 mitogen-activated protein kinase (p38 MAPK), which is correlated with reduced insulin signaling and glucose transport.40 Imidacloprid was previously reported to cause oxidative stress,37,41 and our current results from C2C12 muscle cells suggest that imidacloprid reduces insulin-stimulated phosphorylation of AKT partially mediated by oxidative stress, further supporting the potential role of oxidative stress on imidacloprid’s effects on insulin responsiveness. However, the in vivo effect of imidacloprid on insulin responsiveness, especially in females, needs to be further determined.
Imidacloprid is readily absorbed from the gastrointestinal tract and distributed in almost all tissues.19 Once absorbed, imidacloprid is degraded into several metabolites including 6-chloronicotinic acid, which is also an indicator of imidacloprid exposure;19,42 thus, determining the blood concentrations of imidacloprid and its metabolites would also be helpful for identifying the compound responsible for adiposity in future studies. Imidacloprid is regarded as moderately toxic with an oral LD50 of 420 mg/kg bw in rats, and the typical symptoms of poisoning include cramps, fatigue, and muscle weakness, which were not observed in the current study due to the low doses used.20
In conclusion, the current study reports that imidacloprid exposure (0.6 and 6 mg/kg bw/day) promotes high fat diet-induced obesity in female C57BL/6J mice, which is equivalent to 0.049 and 0.49 mg/kg bw/day for humans based on the body surface area normalization method.43 Although the detailed mechanisms on how imidacloprid interacted with high fat diet still needs to be determined, the current results are significant in demonstrating a potential correlation between low level imidacloprid exposure and high fat diet in augmenting obesity as well as insulin resistance.
Acknowledgments
The China Scholarship Council supported Q.S. This project is supported by NIH R21ES023128.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jafc.7b02584.
Effects of imidacloprid on insulin tolerance test (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Sparks T. C.; Nauen R. Irac: Mode of action classification and insecticide resistance management. Pestic. Biochem. Physiol. 2015, 121, 122–128. 10.1016/j.pestbp.2014.11.014. [DOI] [PubMed] [Google Scholar]
- Jeschke P.; Nauen R.; Schindler M.; Elbert A. Overview of the status and global strategy for neonicotinoids. J. Agric. Food Chem. 2011, 59, 2897–2908. 10.1021/jf101303g. [DOI] [PubMed] [Google Scholar]
- Jeschke P.; Nauen R. Neonicotinoids—from zero to hero in insecticide chemistry. Pest Manage. Sci. 2008, 64, 1084–1098. 10.1002/ps.1631. [DOI] [PubMed] [Google Scholar]
- Herms D.; McCullough D.; Smitley D.; Sadof C.; Cranshaw W.. Insecticide options for protecting ash trees from emerald ash borer. http://www.emeraldashborer.info/documents/Multistate_EAB_Insecticide_Fact_Sheet.pdf (accessed 06/2017).
- Badgujar P. C.; Jain S. K.; Singh A.; Punia J. S.; Gupta R. P.; Chandratre G. A. Immunotoxic effects of imidacloprid following 28 days of oral exposure in BALB/c mice. Environ. Toxicol. Pharmacol. 2013, 35, 408–418. 10.1016/j.etap.2013.01.012. [DOI] [PubMed] [Google Scholar]
- Fossen M.Environmental Fate of Imidacloprid. California Department of Pesticide Regulation, 2006; pp 1–16. http://www.cdpr.ca.gov/docs/emon/pubs/fatememo/Imidclprdfate2.pdf
- The European Commission. Decision by the European Commission to restrict the use of imidacloprid, thiamethoxam and clothianidin (Regulation EU No 485/2013). http://eurlex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2013:139:0012:0026:EN:PDFS (accessed 06/2017).
- Lee D.-H.; Steffes M. W.; Sjödin A.; Jones R. S.; Needham L. L.; Jacobs D. R. Jr Low dose organochlorine pesticides and polychlorinated biphenyls predict obesity, dyslipidemia, and insulin resistance among people free of diabetes. PLoS One 2011, 6, e15977. 10.1371/journal.pone.0015977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezg R.; Mornagui B.; El-Fazaa S.; Gharbi N. Organophosphorus pesticides as food chain contaminants and type 2 diabetes: a review. Trends Food Sci. Technol. 2010, 21, 345–357. 10.1016/j.tifs.2010.04.006. [DOI] [Google Scholar]
- Son H.-K.; Kim S.-A.; Kang J.-H.; Chang Y.-S.; Park S.-K.; Lee S.-K.; Jacobs D.; Lee D.-H. Strong associations between low-dose organochlorine pesticides and type 2 diabetes in Korea. Environ. Int. 2010, 36, 410–414. 10.1016/j.envint.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Montgomery M.; Kamel F.; Saldana T.; Alavanja M.; Sandler D. Incident diabetes and pesticide exposure among licensed pesticide applicators: Agricultural Health Study, 1993–2003. Am. J. Epidemiol. 2008, 167, 1235–1246. 10.1093/aje/kwn028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boada L. D.; Lara P. C.; Álvarez-León E. E.; Losada A.; Zumbado M. L.; Limiñana-Cañal J. M.; Apolinario R.; Serra-Majem L.; Luzardo O. P. Serum levels of insulin-like growth factor-I in relation to organochlorine pesticides exposure. Growth Horm. IGF Res. 2007, 17, 506–511. 10.1016/j.ghir.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Park Y.; Kim Y.; Kim J.; Yoon K. S.; Clark J.; Lee J.; Park Y. Imidacloprid, a Neonicotinoid Insecticide, Potentiates Adipogenesis in 3T3-L1 Adipocytes. J. Agric. Food Chem. 2013, 61, 255–259. 10.1021/jf3039814. [DOI] [PubMed] [Google Scholar]
- Kim J.; Park Y.; Yoon K. S.; Clark J. M.; Park Y. Imidacloprid, a neonicotinoid insecticide, induces insulin resistance. J. Toxicol. Sci. 2013, 38, 655–660. 10.2131/jts.38.655. [DOI] [PubMed] [Google Scholar]
- Kim J.; Park Y.; Yoon K. S.; Clark J. M.; Park Y. Permethrin alters adipogenesis in 3T3-L1 adipocytes and causes insulin resistance in C2C12 myotubes. J. Biochem. Mol. Toxicol. 2014, 28, 418–424. 10.1002/jbt.21580. [DOI] [PubMed] [Google Scholar]
- Kim J.; Sun Q.; Yue Y.; Yoon K. S.; Whang K. Y.; Marshall Clark J.; Park Y. 4,4′-Dichlorodiphenyltrichloroethane (DDT) and 4,4′-dichlorodiphenyldichloroethylene (DDE) promote adipogenesis in 3T3-L1 adipocyte cell culture. Pestic. Biochem. Physiol. 2016, 131, 40–45. 10.1016/j.pestbp.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Qi W.; Yang J. J.; Yoon K. S.; Clark J. M.; Park Y. Fipronil promotes adipogenesis via AMPKα-mediated pathway in 3T3-L1 adipocytes. Food Chem. Toxicol. 2016, 92, 217–223. 10.1016/j.fct.2016.04.011. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Xiao X.; Kim Y.; Kim D.; Yoon K. S.; Clark J. M.; Park Y. Imidacloprid Promotes High Fat Diet-Induced Adiposity and Insulin Resistance in Male C57BL/6J Mice. J. Agric. Food Chem. 2016, 64, 9293–9306. 10.1021/acs.jafc.6b04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agency, C. E. P. Imidacloprid risk characterization document dietary and drinking water exposure. http://www.cdpr.ca.gov/docs/risk/rcd/imidacloprid.pdf (accessed 06/2017).
- Marshall I.; Rhtherford S.. Health Investigation Level for Endosulfan in Soil. Proceedings of the Fifth National Workshop on the Assessment of Site Contamination Environmental Protection and Heritage Council. http://www.nepc.gov.au/system/files/resources/27b740ab-880f-d7f4-d10e-bab0fa964e48/files/asc-wkshoppaper-15-pest-marshall-endosulfan-200301.pdf (accessed 06/2017).
- Craig M. S.; Gupta R. C.; Candery T. D.; Britton D. A. Human exposure to imidacloprid from dogs treated with advantage(r). Toxicol. Mech. Methods 2005, 15, 287–291. 10.1080/15376520590968842. [DOI] [PubMed] [Google Scholar]
- Berry R.; Church C.; Gericke M. T.; Jeffery E.; Colman L.; Rodeheffer M. S. Methods in Enzymology (MIE): Methods of Adipose Tissue Biology-: Chapter 7: Imaging of Adipose Tissue. Methods Enzymol. 2014, 537, 47–73. 10.1016/B978-0-12-411619-1.00004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiaschi T.; Cirelli D.; Comito G.; Gelmini S.; Ramponi G.; Serio M.; Chiarugi P. Globular adiponectin induces differentiation and fusion of skeletal muscle cells. Cell Res. 2009, 19, 584–597. 10.1038/cr.2009.39. [DOI] [PubMed] [Google Scholar]
- Wu W.; Li Y.; Wu Y.; Zhang Y.; Wang Z.; Liu X. Lutein suppresses inflammatory responses through Nrf2 activation and NF-kappaB inactivation in lipopolysaccharide-stimulated BV-2 microglia. Mol. Nutr. Food Res. 2015, 59, 1663–1673. 10.1002/mnfr.201500109. [DOI] [PubMed] [Google Scholar]
- Viollet B.; Foretz M.; Guigas B.; Horman S.; Dentin R.; Bertrand L.; Hue L.; Andreelli F. Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. J. Physiol. 2006, 574, 41–53. 10.1113/jphysiol.2006.108506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A.; Dickerson K.; Heath R.; Hong S. P.; Momcilovic M.; Johnstone S. R.; Carlson M.; Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Zhang B. B.; Zhou G.; Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009, 9, 407–416. 10.1016/j.cmet.2009.03.012. [DOI] [PubMed] [Google Scholar]
- Habinowski S. A.; Witters L. A. The effects of AICAR on adipocyte differentiation of 3T3-L1 cells. Biochem. Biophys. Res. Commun. 2001, 286, 852–856. 10.1006/bbrc.2001.5484. [DOI] [PubMed] [Google Scholar]
- Aguirre V.; Uchida T.; Yenush L.; Davis R.; White M. F. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J. Biol. Chem. 2000, 275, 9047–9054. 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Lau S. S.; Monks T. J. The Cytoprotective Effect of N-acetyl-L-cysteine against ROS-Induced Cytotoxicity Is Independent of Its Ability to Enhance Glutathione Synthesis. Toxicol. Sci. 2011, 120, 87–97. 10.1093/toxsci/kfq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj S.; Srivastava M. K.; Kapoor U.; Srivastava L. P. A 90 days oral toxicity of imidacloprid in female rats: morphological, biochemical and histopathological evaluations. Food Chem. Toxicol. 2010, 48, 1185–1190. 10.1016/j.fct.2010.02.009. [DOI] [PubMed] [Google Scholar]
- Arfat Y.; Mahmood N.; Tahir M. U.; Rashid M.; Anjum S.; Zhao F.; Li D.-J.; Sun Y.-L.; Hu L.; Zhihao C.; Yin C.; Shang P.; Qian A.-R. Effect of imidacloprid on hepatotoxicity and nephrotoxicity in male albino mice. Toxicol. Rep. 2014, 1, 554–561. 10.1016/j.toxrep.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzin J.; Petersen R.; Meugnier E.; Madsen L.; Lock E.-J.; Lillefosse H.; Ma T.; Pesenti S.; Sonne S. B.; Marstrand T. T.; Malde M. K.; Du Z.-Y.; Chavey C.; Fajas L.; Lundebye A.-K.; Brand C. L.; Vidal H.; Kristiansen K.; Frøyland L. Persistent Organic Pollutant Exposure Leads to Insulin Resistance Syndrome. Environ. Health Perspect. 2010, 118, 465–471. 10.1289/ehp.0901321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson U. S.; Waldén T. B.; Carlsson P.-O.; Jansson L.; Phillipson M. Female Mice are Protected against High-Fat Diet Induced Metabolic Syndrome and Increase the Regulatory T Cell Population in Adipose Tissue. PLoS One 2012, 7, e46057. 10.1371/journal.pone.0046057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louet J.-F.; LeMay C.; Mauvais-Jarvis F. Antidiabetic actions of estrogen: Insight from human and genetic mouse models. Curr. Atheroscler. Rep. 2004, 6, 180–185. 10.1007/s11883-004-0030-9. [DOI] [PubMed] [Google Scholar]
- Gervais J. A.; Luukinen B.; Buhl K.; Stone D.. Imidacloprid technical fact sheet. National Pesticide Information Center, Oregon State University Extension Services. http://npic.orst.edu/factsheets/imidacloprid (accessed 06/2017).
- Duzguner V.; Erdogan S. Chronic exposure to imidacloprid induces inflammation and oxidative stress in the liver & central nervous system of rats. Pestic. Biochem. Physiol. 2012, 104, 58–64. 10.1016/j.pestbp.2012.06.011. [DOI] [Google Scholar]
- Borst S. E. The role of TNF-alpha in insulin resistance. Endocr. J. 2004, 23, 177–182. 10.1385/ENDO:23:2-3:177. [DOI] [PubMed] [Google Scholar]
- Park K.; Gross M.; Lee D.-H.; Holvoet P.; Himes J. H.; Shikany J. M.; Jacobs D. R. Oxidative Stress and Insulin Resistance: The Coronary Artery Risk Development in Young Adults study. Diabetes Care 2009, 32, 1302–1307. 10.2337/dc09-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen E. J.; Diamond-Stanic M. K.; Marchionne E. M. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radical Biol. Med. 2011, 51, 993–999. 10.1016/j.freeradbiomed.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor U.; Srivastava M. K.; Bhardwaj S.; Srivastava L. P. Effect of imidacloprid on antioxidant enzymes and lipid peroxidation in female rats to derive its No Observed Effect Level (NOEL). J. Toxicol. Sci. 2010, 35, 577–581. 10.2131/jts.35.577. [DOI] [PubMed] [Google Scholar]
- Uroz F. J.; Arrebola F. J.; Egea-Gonzalez F. J.; Martinez-Vidal J. L. Monitoring of 6-chloronicotinic acid in human urine by gas chromatography-tandem mass spectrometry as indicator of exposure to the pesticide imidacloprid. Analyst 2001, 126, 1355–1358. 10.1039/b101167g. [DOI] [PubMed] [Google Scholar]
- Reagan-Shaw S.; Nihal M.; Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.