

Abstract
The generalized gradient approximation (GGA) function based on density functional theory is adopted to investigate the optimized geometrical structure, electron structure and hydrogen storage performance of Sc modified porous graphene (PG). It is found that the carbon ring center is the most stable adsorbed position for a single Sc atom on PG, and the maximum number of adsorbed H2 molecules is four with the average adsorption energy of −0.429 eV/H2. By adding a second Sc atom on the other side of the system, the hydrogen storage capacity of the system can be improved effectively. Two Sc atoms located on opposite sides of the PG carbon ring center hole is the most suitable hydrogen storage structure, and the hydrogen storage capacity reach a maximum 9.09 wt % at the average adsorption energy of −0.296 eV/H2. The adsorption of H2 molecules in the PG system is mainly attributed to orbital hybridization among H, Sc, and C atoms, and Coulomb attraction between negatively charged H2 molecules and positively charged Sc atoms.
Keywords: first principles, Sc modification, porous graphene, hydrogen storage
1. Introduction
Considerable research has been conducted to search for clean, renewable energy to meete growing energy demand [1]. Hydrogen has many advantages such as being recyclable, highly abundant, pollution-free, of high gravimetric energy density, and high combustion heat. Therefore hydrogen is considered to be an ideal carrier for energy storage [2]. However, the primary obstacle to hydrogen application is safe and efficient storage under ambient conditions [3,4]. To meet the criteria of the U.S. Department of Energy (DOE) and the International Energy Agency (IEA), the desirable hydrogen storage capacity of a promising material should be greater than 5.5 wt %. The adsorption energy between H2 molecules and materials should be from 0.2 to 0.7 eV [5]. Therefore, it is appropriate that hydrogen stores in solid materials in a molecular form [6].
Carbon nanostructures, such as graphene [7,8], fullerene [9,10], and nanotubes [11] have been investigated for solid-state hydrogen storage because of their high specific surface area, fast kinetics, and reversible hydrogen storage etc. However, H2 molecules bind weakly with pristine graphene, which is chemically too inert to act as a promising hydrogen storage material. Many studies have been performed to improve the chemical activity of graphene, such as doping or decoration by alkali metals [12,13], alkaline-earth metals [14,15,16], and transition metals [17,18,19]. The adsorption energy of hydrogen molecules on graphene can significantly be improved by the modification of transition metal (TM) atoms [20]. However, the large cohesive energy of TM atoms can easily lead to the formation of clusters on graphene. Research shows that the introduction of impurity atoms or vacancies can prevent the TM atoms from aggregating, thus increasing the hydrogen storage capacity of graphene [21]. Therefore, graphene which has a foraminiferous defect and doped atoms, decorated with TM atoms, has attracted attention.
Luo et al. [22] noted that the Sc-decorated single-vacancy defect graphene with three N atoms doped was the best medium for hydrogen storage, using Vienna Ab-initio Simulation Package (VASP) code within the GGA functional calculation, because each Sc atom could adsorb five H2 molecules with the adsorption energies from 0.2 to 0.4 eV. Using DMol3 code within the local density approximation (LDA) functional calculation, Omar Faye et al. [23] found that up to eight H2 molecules could be adsorbed by double-sided Pd-functionalized graphene. In addition, Omar Faye et al. [24] also studied hydrogen storage in Cu-functionalized boron-doped graphene by the same method. A gravimetric hydrogen density of 4.231 wt % was reached when H2 adsorbed on double-sided Cu-functionalized graphene. Using the DMol3 package within the LDA functional calculation, Y decorated B-doped graphene was studied as a potential carrier for hydrogen storage, in which the hydrogen storage capacity was 5.78 wt %. The corresponding average adsorption energy was −0.568 eV per H2 molecule [25]. Although the average adsorption energy of H2 molecules is suitable for reversible hydrogen storage in these systems, the hydrogen storage capacity is close to the minimum 5.5 wt % of United States DOE.
Because of the good performance of graphene, other graphene-like materials have also attracted attention. Examples are silicene [26,27,28], monolayer black phosphorus [29], arsenene [30] and porous graphene [31]. Porous graphene (PG) was successfully fabricated by Bieri et al [32]. The inherent nanopores in porous graphene are well-developed and uniformly distributed, which portend highly effective hydrogen storage by a high specific surface area [33]. Du et al. [12] investigated Li-decorated PG by VASP code within the LDA, which found that the hydrogen storage capacity was up to 12 wt %. However, the average binding energy of 0.243 eV per H2 was overestimated due to the LDA function. Reunchan et al. [34] investigated the Ca-decorated PG and found that an isolated Ca atom could adsorb five hydrogen molecules with the average adsorption energy of 0.230 eV. Recently, Yuan et al. [35] studied Y-decorated porous graphene and they found that the maximum number of adsorbed hydrogen molecules around the Y atom was six and the average adsorption energy was −0.297 eV.
Sc is the lightest TM atom, indicating that Sc-decorated PG could produce higher hydrogen storage capacity. In this paper, we calculate the most stable adsorption structure of Sc atoms on PG and the corresponding adsorption energy. In addition, the adsorption properties and adsorption mechanism of H2 molecules on the Sc decorated PG system are analyzed to better understand the effect of Sc atoms modified PG on the hydrogen storage properties.
2. Calculation Details
Our studies are performed using the plane wave CASTEP package [36] with the Perdew-Burke-Ernzerhof (PBE) of the generalized gradient approximation (GGA) exchange-correlation function [37]. Since the GGA function may underestimate the relatively weak adsorption energies, a pragmatic method to resolve this issue has been given by the dispersion-corrected density functional theory (DFT-D) approach. Calculations are performed using the DFT-D method to consider the van der Waals forces. All the atoms are relaxed in our calculations such that the force on each atom is less than 0.01 eV/Å. The convergence tolerance energy is 5.0 × eV/atom to realize the energy minimization. The self-consistent field (SCF) convergence threshold is 1.0 × eV/atom. The calculation of the porous graphene unit cell satisfies the periodic boundary conditions. The vacuum thickness of 20 Å is adopted to minimize the interlayer interaction. In order to ensure convergence and improve the accuracy of our calculations, the cutoff energy and the k-point are tested. Considering the test results and the computational cost, the cutoff energy is set to 500 eV and the k-point is set to 7 × 7 × 1.
The average adsorption energy of Sc atoms on PG layer is defined as:
| (1) |
where , and represent the total energy of the PG system with Sc atoms, the total energy of PG and the total energy of a free Sc atom, respectively. Moreover, n represents the number of adsorbed Sc atoms.
The adsorption energy and average adsorption energy () of adsorbed H2 molecules are respectively defined as:
| (2) |
| (3) |
where and are the total energy of the Sc-decorated PG system with and () H2 molecules adsorbed, respectively. represents the energy of an isolated H2 molecule.
The fully optimized structure of a porous graphene unit cell is shown in Figure 1. The calculated lattice constant is 7.49 Å, which agrees well with the experimental value of 7.40 Å [32]. There are two kinds of C atoms on PG. C1 is connected with three C atoms, while C2 is connected with two C atoms and one H atom. The bond length of C1–C1 is 1.489 Å, C1–C2 is 1.399 Å, C2–H is 1.085 Å. Our results are very close to the values of Brunetto [38], who gets the results using the DMol3 package with the GGA-BLYP function. In addition, Li [39] calculates the bond length of C2–H at 1.080 Å using the DMol3 package with the GGA-PW91 function. The result is consistent with our value of 1.085 Å. The direct band gap is calculated to be 2.399 eV using the GGA with the PBE function, which coincides well with the results of Rao [37] who obtains the value of band gap at 2.400 eV based on VASP code with the PBE of the GGA function. The above test results show that the calculation method and parameters used in this paper are reliable.
Figure 1.
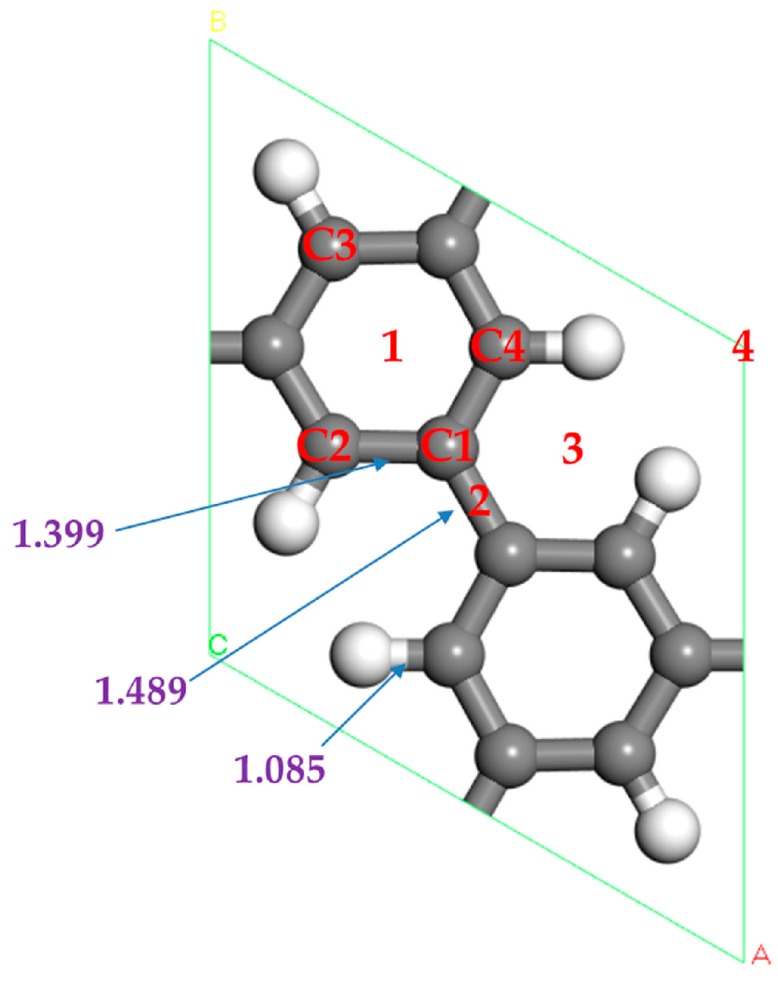
The optimized atomic structure of porous graphene. The gray and white balls represent C and H respectively.
3. Results and Discussion
3.1. Single Sc Atom Decorated PG
3.1.1. The Adsorption Structure of Single Sc Atom Decorated PG
Now we study the adsorption of one Sc atom on PG. There are four different symmetric positions for a Sc atom adsorption, which are shown in Figure 1 and named 1, 2, 3, 4, indicating the hollow center of C ring, the bridge of C–C bond, the hollow center of a half C ring, and a large hexagonal pore, respectively. It is found that the 1 position is the favorite adsorption site for a Sc atom on PG, and this position is the same as the adsorption of a Sc atom on graphene [22]. The favorite configuration after full relaxation is shown in Figure 2a. We find that the Sc atom is a little deviation from the hollow center of the C-hexagon, and the adsorption energy is −2.143 eV for a Sc atom on PG.
Figure 2.

The optimized atomic structure of Sc atom decorated PG. (a) single Sc atom decorated PG; (b) two Sc atoms decorated single-sided PG; (c) two Sc atoms decorated double-sided PG at the same hole; (d) two Sc atoms decorated double-sided PG at the adjacent hole. The gray, white and pink balls in this and following figures denote C, H and Sc atoms, respectively.
We also calculate the adsorption of Sc on perfect graphene to further investigate the effect of graphene defects on Sc adsorption. The adsorption energy of the Sc atom on the intact graphene is −1.499 eV, which is calculated using the CASTEP package within the PBE of the GGA function. This is very close to the value of −1.550 eV, which is calculated based on VASP code with the PBE of the GGA function [22]. The adsorption energy of Sc atoms on perfect graphene is far less than that on PG. After adsorption, the positive charges of the Sc atom on the perfect graphene and PG are 1.24 e, 1.33 e, respectively. In other words, more charge transfers in the Sc-decorated PG system, and this stems from the fact that the interaction between Sc atoms and PG is stronger than the interaction between Sc atoms and perfect graphene. From the partial densities of states (PDOS) of Sc and C in Figure 3, we can learn the peak of the C 2p orbital overlap with the peak of the Sc 3d orbital at −2.50 eV, and this suggests a strong hybridization between C and Sc atoms.
Figure 3.

PDOS of Sc-decorated PG system.
3.1.2. The Adsorption of H2 Molecules on Single Sc-Decorated PG
The optimized geometries for H2 molecules adsorbed on the Sc-decorated porous graphene are shown in Figure 4, and Table 1 lists the adsorption energy and the average adsorption energy of H2 molecules. There are several positions for single H2 molecule adsorption, including the bridge of the C–C bond, the bridge of the C–H bond, the top of the C atom and the top of the Sc atom. It is found that the favorite position for the first H2 molecule is above the C–H bond. The binding energy is −0.401 eV, and the bond length is elongated to 0.818 Å. In order to study the hydrogen storage capacity of the system, we add other H2 molecules to the system one by one. From Table 1, we can see that the adsorption energy of H2 molecules first increases and then decreases for the consecutive adsorption of the H2 molecule. However, (the nearest distance between the Sc atom and the C atom of porous graphene) is the opposite, which means that the adsorption of H2 molecules influences the binding between the Sc atom and porous graphene. It is very interesting to note that almost all H2 molecules are symmetrically distributed around the Sc atom because of the symmetry of the bonding configuration of H2 molecules. In addition, the first four H2 molecules are parallel to the PG sheet and stay on the same plane. When the fifth H2 molecule is added to the system, it moves to an upper layer after relaxation, as shown in Figure 4e. This effect may be due to the limited space around the Sc atom and the repulsive interaction between the adsorbed H2 molecules [40]. Therefore, the H–H bond length of the fifth H2 molecule is 0.756 Å and this H2 molecule has the smallest adsorption energy of −0.093 eV, which is weak for hydrogen storage application [41]. The system of the single Sc atom decorated porous graphene can adsorb a maximum of 4 H2 molecules because of the two-layer arrangement. Therefore, the hydrogen storage capacity is 3.94 wt % and the average adsorption energy is −0.429 eV/H2, which is higher than that of Sc modified defect graphene with three N atoms doped [22].
Figure 4.

The optimized atomic structures of the Sc atom decorated PG with (a) one H2 molecule; (b) two H2 molecules; (c) three H2 molecules; (d) four H2 molecules; (e) five H2 molecules adsorbed.
Table 1.
The adsorption energy, average adsorption energy and the nearest distance between Sc and C of porous graphene for H2 adsorbed on single Sc decorated PG.
| Number of H2 | (eV) | (eV) | (Å) |
|---|---|---|---|
| 1H2 | −0.401 | −0.401 | 2.348 |
| 2H2 | −0.718 | −0.559 | 2.217 |
| 3H2 | −0.422 | −0.514 | 2.291 |
| 4H2 | −0.174 | −0.429 | 2.451 |
| 5H2 | −0.093 | −0.361 | 2.477 |
In order to investigate the interaction between the Sc atom and adsorbed H2 molecules, the partial densities of states for the H2 molecules and the Sc atom are plotted in Figure 5. It can be observed that there is a band broadening around −9 eV, which indicates an H2–H2 interaction. In addition, there is a strong hybridization between H2 1s orbitals and Sc 3d orbitals around −10.5 to −8.0 eV, since the orbital overlap is between 1s of H2 and 3d of Sc. In the range −1.5 to 0 eV, the adsorption of 1~4 H2 molecules can be seen to have overlapping peaks between H2 1s orbital and Sc 3d orbital, suggesting that in this interval H2 molecules and the Sc atom also exist in hybridization, which may be one of the reasons why the first four H2 molecules have a strong interaction with the Sc atom. From the PDOS of H2 molecules, we can see that the 1s orbital mainly distributes in σ bonding state, namely a bonding state dominates the 1s orbital and there is no H2 molecular dissociation.
Figure 5.

PDOS of Sc-decorated PG with one to five H2 molecules adsorbed.
Given the Mulliken charge population before and after H2 molecules adsorption, the bond strength and charge transfer between atoms could be analyzed. The Mulliken population analysis of Sc-PG system with and without one H2 molecule adsorbed are shown in Table 2. H1 and H2 represent the two hydrogen atoms of the adsorbed hydrogen molecule, and C2, C3 and C4 are the three C atoms that charge transfer most in the PG layer (as shown in Figure 1). After the first H2 adsorption, the charge of each atom of the H2 molecule is −0.13 e, while the Sc atom carries the positive charge of 1.56 e. According to Figure 5, Sc 3d orbital and the H2 molecule of bonding and antibonding state are hybridizations. Therefore, H2 molecule transfers charge to the Sc atom, and then Sc atom back donates charge to the antibonding orbital of the H2 molecule. As a result, the H2 molecule carries a more negative charge. There is Coulomb attraction between the negatively charged H2 molecule and positively charged Sc atom. This is consistent with the case of Ca-decorated graphene for hydrogen storage [42]. The Coulomb interaction enhances the adsorption of the H2 molecule. Although the bond length of the H2 molecule is stretched, the H2 molecule remains its molecular bond. After one H2 molecule adsorption, the negative charge of C2 and C4 atoms increase. However, the negative charge of C3 nearest the H2 molecule is reduced by about 0.15 e, which indicates that these C atoms also play a role in H2 molecular adsorption.
Table 2.
Mulliken population analysis of the Sc-PG system before and after one H2 molecule adsorption.
| Atom | Before Adsorption (e) | After Adsorption (e) | ||||||
|---|---|---|---|---|---|---|---|---|
| s | p | d | Charge | s | p | d | Charge | |
| H1 | 1.00 | - | - | - | 1.13 | - | - | –0.13 |
| H2 | 1.00 | - | - | - | 1.13 | - | - | –0.13 |
| C2 | 1.19 | 3.19 | - | –0.38 | 1.19 | 3.23 | - | –0.42 |
| C3 | 1.20 | 3.28 | - | –0.48 | 1.18 | 3.15 | - | –0.33 |
| C4 | 1.19 | 3.20 | - | –0.39 | 1.19 | 3.23 | - | –0.42 |
| Sc | 0.18 | 5.84 | 1.65 | 1.33 | 0.04 | 5.63 | 1.77 | 1.56 |
The charge transfer of the Sc-PG system can be intuitively observed through charge density difference. Therefore, the charge density difference is defined as:
| (4) |
where is the electron charge density of the total system, is the charge density of i adsorbed H2 molecules, and is the charge density of the Sc decorated PG system. The charge density difference for the system Sc-PG with H2 molecules adsorbed is shown in Figure 6. The blue and yellow isosurface represents space charge accumulation and depletion, and the value for the isosurface is 0.007 e/. It is easy to see that the charge transfer is mainly distributed in regions between H2 molecules and the Sc atom, indicating a strong interaction between them. The charge transfer of C atoms in PG is also observed, which indicates that these C atoms also play a role in H2 molecular adsorption. It demonstrates the H2 molecules are polarized such that charge depletion and accumulation at both ends of H2 molecules can be observed in Figure 6b. Combining the Mulliken charge population, each H atom of H2 molecules carries a negative charge of −0.16 e and −0.14 e, and it has a Coulomb attraction with a positively charged Sc atom after polarization. Consequently, the adsorption of hydrogen molecules in the Sc-decorated PG system is due to the orbital hybridization among H, Sc, C atoms, and Coulomb attraction between negatively charged H2 molecules and positively charged Sc atoms.
Figure 6.

Electronic charge density difference for Sc-decorated PG system in the presence of (a) one H2 molecule; (b) two H2 molecules; (c) four H2 molecules.
3.2. Two Sc Atoms Decorated PG
3.2.1. The Adsorption Structure of Two Sc Atoms Decorated PG
The cohesive energy of Sc atoms is 3.90 eV [43], which is much bigger than the binding energy of Sc atoms on PG. In order to test the possibility of Sc aggregation, we add the second Sc atom in the system on the same side. Figure 2b presents the most stable optimized geometry for two Sc atoms adsorbed on PG. We find that the second Sc atom is more likely to adsorb on the center of another C hexagon rather than aggregate with the first Sc atom, and this is mainly because the Coulomb repulsion between two Sc atoms (both Sc atoms carry 0.87 eV positive charge) and the strong interaction between Sc atoms and PG. The average binding energy of two Sc atoms on PG is −3.195 eV, which implies that the interaction between the second Sc atom and PG is stronger than the first. In addition, the large distance of 4.322 Å between two Sc atoms may be another reason for resistance clusters.
To examine further the space for hydrogen storage, this paper also studies two Sc atoms decorated double-sided PG. Each transition metal atom is an active adsorption point, and transition metal atoms modified double-sided PG can increase the hydrogen storage area. Based on the Figure 2a system, there are four adsorption positions for the second Sc atom on the opposite side of PG. The stable structures after relaxation are shown in Figure 2c,d and the average adsorption energies of the Sc atom are −2.813 eV and −2.537 eV. The hydrogen adsorption energy and storage properties of these two models are investigated in our work since the binding energy of Sc atoms has little difference.
3.2.2. The Hydrogen Storage Capacity of Two Sc Atoms Modified PG System
As shown in Figure 7a, about ten H2 molecules can be adsorbed in the two Sc single-sided modified PG system without aggregation. The average adsorption energy is about −0.268 eV/H2, and the hydrogen storage capacity is 7.69 wt %. The cohesive energy of the Sc atom is higher than its binding energy on PG, and after H2 adsorption the two Sc atoms are slightly closer to each other. This means that Sc aggregation may occur on the porous graphene, but from the optimized structure we not find Sc atom bonding with each other although Sc atoms can move freely. The average binding energy of two Sc atoms on PG is −3.195 eV, which means that the adsorption structure of the two Sc atoms on the PG is more stable than that of a single Sc on the PG. This indicates adsorption structures are stable. After testing, we found that low concentrations of Sc atoms do not aggregate on PG. This phenomenon may be due to the Coulomb repulsion between two Sc atoms and the strong interaction between Sc atoms and PG, as well as the pore structure of the porous graphene. Given that the hydrogen storage space of PG on a single side is limited, to further improve the hydrogen storage capacity, two Sc atoms modified double-sided PG are also considered, as shown in Figure 2c,d. After hydrogen storage, the optimized structures are shown in Figure 7b,c. And the distance between the Sc atoms adsorbed on both sides of PG is large enough to prevent the aggregation. The Figure 7b system can adsorb ten H2 molecules, the average adsorption energy of H2 molecules is −0.292 eV, and the hydrogen storage capacity is 7.69 wt %. The system of Figure 7c contains twelve H2 molecules adsorbed on Sc-decorated both sides of PG. The average adsorption energy of hydrogen in this system is −0.296 eV/H2 higher than that of −0.23 eV/H2 in the Y-PG system, and the hydrogen storage capacity is up to 9.09 wt %, higher than that of 7.87 wt % in the Y-PG system [35].
Figure 7.

The optimized geometries for H2 molecules adsorbed on Sc-decorated PG. (a) two Sc atoms decorated single-sided PG; (b) two Sc atoms decorated double-sided PG at the same hole; (c) two Sc atoms decorated double-sided PG at the adjacent hole.
The ideal adsorption energy between H2 molecules and materials should remain in the range of 0.2~0.7 eV/H2, which is between the physical and chemical adsorption state. It will be beneficial to hydrogen storage and release under the environmental temperature and pressure. In our work, the average adsorption energies of H2 molecules adsorbed on graphene are from −0.060 to −0.075 eV/H2. The adsorption energy is in the range of −0.061~−0.104 eV/H2 for H2 molecules adsorbed on PG. The adsorption energy of H2 molecules at the 4 sites in Figure 1 (in the same plane with PG) is positive and is the most unstable adsorption site. Therefore, the adsorption energy of H2 molecules adsorb on intrinsic graphene and PG is very small, which is not conducive to hydrogen storage.
The three systems of two Sc atoms modified PG for hydrogen storage are shown in Figure 7a–c. Their average adsorption energies are −0.268 eV/H2, −0.292 eV/H2, −0.296 eV/H2 respectively, which is small in the ideal range. The systems of Figure 7a,b have the same hydrogen storage capacity, but the average adsorption energy of Figure 7b is bigger than that of Figure 7a. The hydrogen storage capacity of Figure 7c is the largest of the three systems, and the average adsorption energy of H2 molecules is the largest. As a result, the system of Figure 7c is the best structure for reversible hydrogen storage.
In addition, in comparing these systems (Figure 4e and Figure 7a,c) we find an interesting phenomenon: The H2 molecules in Figure 7a are distributed in three layers, so the average adsorption energy is small (−0.268 eV/H2). In Figure 4e and Figure 7c, there are two layers of H2 molecules, and the average adsorption energies of −0.429, −0.296 eV/H2 are larger. To sum up, two Sc atoms located above the adjacent C ring in double-sided PG is the most suitable structure for hydrogen storage. Therefore, Sc-decorated PG is a promising material for hydrogen storage.
4. Conclusions
In summary, based on the density functional theory, we studied the hydrogen storage properties of the Sc-PG system. For a single Sc atom, the most stable adsorption position on PG is the carbon hexagon center with an adsorption energy of −2.143 eV. Four H2 molecules can be adsorbed around an Sc atom with an average adsorption energy of −0.429 eV/H2. Analyzing the electronic structure of the system, the adsorption of H2 molecules in the PG system is mainly attributed to orbital hybridization among H, Sc, C atoms. The largest hydrogen storage capacity structure is two Sc atoms located above the adjacent C ring in a double side of PG. The system can absorb twelve H2 molecules with an average adsorption energy of −0.296 eV/H2, and the hydrogen storage capacity is 9.09 wt %. Therefore, Sc-decorated porous graphene may become one of the most promising hydrogen storage materials.
Acknowledgments
This work is supported by the National Natural Science Foundation of China (No. 51562022) and the Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase).
Author Contributions
Yuhong Chen designed the project, Jing Wang and Lihua Yuan performed the calculations, Yuhong Chen and Jing Wang prepared the manuscript, Cairong Zhang revised the paper, Lihua Yuan and Meiling Zhang analyzed the data, and all authors discussed the results and commented on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Masika E., Mokaya R. Exceptional gravimetric and volumetric hydrogen storage for densified zeolite templated carbons with high mechanical stability. Energy Environ. Sci. 2014;7:427–434. doi: 10.1039/C3EE42239A. [DOI] [Google Scholar]
- 2.Wang Y., Ji Y., Li M., Yuan P., Sun Q., Jia Y. Li and Ca Co-decorated carbon nitride nanostructures as high-capacity hydrogen storage media. J. Appl. Phys. 2011;110:94311. doi: 10.1063/1.3656454. [DOI] [Google Scholar]
- 3.Dillon A.C., Jones K.M., Bekkedahl T.A., Kiang C.H., Bethune D.S., Heben M.J. Storage of hydrogen in single-walled carbon nanotubes. Nature. 1997;386:377–379. doi: 10.1038/386377a0. [DOI] [Google Scholar]
- 4.Steele B.C., Heinzel A. Materials for fuel-cell technologies. Nature. 2001;414:345–352. doi: 10.1038/35104620. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z.W., Li J.C., Jiang Q. Hydrogen Adsorption on Eu/SWCNT Systems: A DFT Study. J. Phys. Chem. C. 2010;114:7733–7737. doi: 10.1021/jp100017y. [DOI] [Google Scholar]
- 6.Sahaym U., Norton M.G. Advances in the application of nanotechnology in enabling a ‘hydrogen economy’. J. Mater. Sci. 2008;43:5395–5429. doi: 10.1007/s10853-008-2749-0. [DOI] [Google Scholar]
- 7.Ataca C., Aktürk E., Ciraci S., Ustunel H. High-capacity hydrogen storage by metallized graphene. Appl. Phys. Lett. 2008;93:43123. doi: 10.1063/1.2963976. [DOI] [Google Scholar]
- 8.Kim G., Jhi S.-H., Lim S., Park N. Crossover between multipole Coulomb and Kubas interactions in hydrogen adsorption on metal-graphene complexes. Phys. Rev. B. 2009;79 doi: 10.1103/PhysRevB.79.155437. [DOI] [Google Scholar]
- 9.Chandrakumar K.R.S., Ghosh S.K. Alkali-Metal-Induced Enhancement of Hydrogen Adsorption in C60 Fullerene: An ab Initio Study. Nano Lett. 2008;8:13–19. doi: 10.1021/nl071456i. [DOI] [PubMed] [Google Scholar]
- 10.Sun Q., Wang Q., Jena P. Functionalized heterofullerenes for hydrogen storage. Appl. Phys. Lett. 2009;94:13111. doi: 10.1063/1.3058678. [DOI] [Google Scholar]
- 11.Seenithurai S., Pandyan R.K., Kumar S.V., Saranya C., Mahendran M. Al-decorated carbon nanotube as the molecular hydrogen storage medium. Int. J. Hydrogen Energy. 2014;39:11990–11998. doi: 10.1016/j.ijhydene.2014.05.184. [DOI] [Google Scholar]
- 12.Du A., Zhu Z., Smith S.C. Multifunctional Porous Graphene for Nanoelectronics and Hydrogen Storage: New Properties Revealed by First Principle Calculations. J. Am. Chem. Soc. 2010;132:2876–2877. doi: 10.1021/ja100156d. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y., Meng Z., Liu Y., You D., Wu K., Lv J., Wang X., Deng K., Rao D., Lu R. Lithium decoration of three dimensional boron-doped graphene frameworks for high-capacity hydrogen storage. Appl. Phys. Lett. 2015;106:63901. doi: 10.1063/1.4907975. [DOI] [Google Scholar]
- 14.Hussain T., Pathak B., Ramzan M., Maark T.A., Ahuja R. Calcium doped graphane as a hydrogen storage material. Appl. Phys. Lett. 2012;100:183902. doi: 10.1063/1.4710526. [DOI] [Google Scholar]
- 15.Song N., Wang Y., Zheng Y., Zhang J., Xu B., Sun Q., Jia Y. New template for Li and Ca decoration and hydrogen adsorption on graphene-like SiC: A first-principles study. Comput. Mater. Sci. 2015;99:150–155. doi: 10.1016/j.commatsci.2014.12.016. [DOI] [Google Scholar]
- 16.Gao Y., Zhao N., Li J., Liu E., He C., Shi C. Hydrogen spillover storage on Ca-decorated graphene. Int. J. Hydrogen Energy. 2012;37:11835–11841. doi: 10.1016/j.ijhydene.2012.05.029. [DOI] [Google Scholar]
- 17.Lebon A., Carrete J., Gallego L.J., Vega A. Ti-decorated zigzag graphene nanoribbons for hydrogen storage. A van der Waals-corrected density-functional study. Int. J. Hydrogen Energy. 2015;40:4960–4968. doi: 10.1016/j.ijhydene.2014.12.134. [DOI] [Google Scholar]
- 18.Chu S., Hu L., Hu X., Yang M., Deng J. Titanium-embedded graphene as high-capacity hydrogen-storage media. Int. J. Hydrogen Energy. 2011;36:12324–12328. doi: 10.1016/j.ijhydene.2011.07.015. [DOI] [Google Scholar]
- 19.Liu Y., Ren L., He Y., Cheng H.-P. Titanium-decorated graphene for high-capacity hydrogen storage studied by density functional simulations. J. Phys. Condens. Matter. 2010;22:445301. doi: 10.1088/0953-8984/22/44/445301. [DOI] [PubMed] [Google Scholar]
- 20.Rojas M.I., Leiva E.P.M. Density functional theory study of a graphene sheet modified with titanium in contact with different adsorbates. Phys. Rev. B. 2007;76 doi: 10.1103/PhysRevB.76.155415. [DOI] [Google Scholar]
- 21.Kim G., Jhi S.-H., Park N., Louie S.G., Cohen M.L. Optimization of metal dispersion in doped graphitic materials for hydrogen storage. Phys. Rev. B. 2008;78 doi: 10.1103/PhysRevB.78.085408. [DOI] [Google Scholar]
- 22.Luo Z., Fan X., Pan R., An Y. A first-principles study of Sc-decorated graphene with pyridinic-N defects for hydrogen storage. Int. J. Hydrogen Energy. 2017;42:3106–3113. doi: 10.1016/j.ijhydene.2016.11.039. [DOI] [Google Scholar]
- 23.Faye O., Szpunar J.A., Szpunar B., Beye A.C. Hydrogen adsorption and storage on Palladium—Functionalized graphene with NH-dopant: A first principles calculation. Appl. Surf. Sci. 2017;392:362–374. doi: 10.1016/j.apsusc.2016.09.032. [DOI] [Google Scholar]
- 24.Faye O., Eduok U., Szpunar J., Szpunar B., Samoura A., Beye A. Hydrogen storage on bare Cu atom and Cu-functionalized boron-doped graphene: A first principles study. Int. J. Hydrogen Energy. 2017;42:4233–4243. doi: 10.1016/j.ijhydene.2016.10.031. [DOI] [Google Scholar]
- 25.Liu W., Liu Y., Wang R. Prediction of hydrogen storage on Y-decorated graphene: A density functional theory study. Appl. Surf. Sci. 2014;296:204–208. doi: 10.1016/j.apsusc.2014.01.087. [DOI] [Google Scholar]
- 26.Sivek J., Sahin H., Partoens B., Peeters F.M. Adsorption and absorption of boron, nitrogen, aluminum, and phosphorus on silicene: Stability and electronic and phonon properties. Phys. Rev. B. 2013;87 doi: 10.1103/PhysRevB.87.085444. [DOI] [Google Scholar]
- 27.Vogt P., De Padova P., Quaresima C., Avila J., Frantzeskakis E., Asensio M.C., Resta A., Ealet B., Le Lay G. Silicene: Compelling Experimental Evidence for Graphenelike Two-Dimensional Silicon. Phys. Rev. Lett. 2012;108 doi: 10.1103/PhysRevLett.108.155501. [DOI] [PubMed] [Google Scholar]
- 28.Zhao H. Strain and chirality effects on the mechanical and electronic properties of silicene and silicane under uniaxial tension. Phys. Lett. A. 2012;376:3546–3550. doi: 10.1016/j.physleta.2012.10.024. [DOI] [Google Scholar]
- 29.Liu H., Neal A.T., Zhu Z., Luo Z., Xu X., Tománek D., Ye P.D. Phosphorene: An Unexplored 2D Semiconductor with a High Hole Mobility. ACS Nano. 2014;8:4033–4041. doi: 10.1021/nn501226z. [DOI] [PubMed] [Google Scholar]
- 30.Zhang S., Yan Z., Li Y., Chen Z., Zeng H. Atomically Thin Arsenene and Antimonene: Semimetal-Semiconductor and Indirect-Direct Band-Gap Transitions. Angew. Chem. Int. Ed. 2015;54:3112–3115. doi: 10.1002/anie.201411246. [DOI] [PubMed] [Google Scholar]
- 31.Xie X., Zhou Y., Bi H., Yin K., Wan S., Sun L. Large-range Control of the Microstructures and Properties of Three-dimensional Porous Graphene. Sci. Rep. 2013;3 doi: 10.1038/srep02117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bieri M., Treier M., Cai J., Aït-Mansour K., Ruffieux P., Gröning O., Gröning P., Kastler M., Rieger R., Feng X., et al. Porous graphenes: Two-dimensional polymer synthesis with atomic precision. Chem. Commun. 2009 doi: 10.1039/b915190g. [DOI] [PubMed] [Google Scholar]
- 33.Huang C., Wu H., Deng K., Tang W., Kan E. Improved permeability and selectivity in porous graphene for hydrogen purification. Phys. Chem. Chem. Phys. 2014;16:25755–25759. doi: 10.1039/C4CP04385E. [DOI] [PubMed] [Google Scholar]
- 34.Reunchan P., Jhi S.-H. Metal-dispersed porous graphene for hydrogen storage. Appl. Phys. Lett. 2011;98:93103. doi: 10.1063/1.3560468. [DOI] [Google Scholar]
- 35.Yuan L., Chen Y., Kang L., Zhang C., Wang D., Wang C., Zhang M., Wu X. First-principles investigation of hydrogen storage capacity of Y-decorated porous graphene. Appl. Surf. Sci. 2017;399:463–468. doi: 10.1016/j.apsusc.2016.12.054. [DOI] [Google Scholar]
- 36.Sun J., Wang H.-T., He J., Tian Y. Ab initio investigations of optical properties of the high-pressure phases of ZnO. Phys. Rev. B. 2005;71 doi: 10.1103/PhysRevB.71.125132. [DOI] [Google Scholar]
- 37.Rao D., Lu R., Meng Z., Wang Y., Lu Z., Liu Y., Chen X., Kan E., Xiao C., Deng K., et al. Electronic properties and hydrogen storage application of designed porous nanotubes from a polyphenylene network. Int. J. Hydrogen Energy. 2014;39:18966–18975. doi: 10.1016/j.ijhydene.2014.09.112. [DOI] [Google Scholar]
- 38.Brunetto G., Autreto P.A.S., Machado L.D., Santos B.I., Dos Santos R.P.B., Galvão D.S. Nonzero Gap Two-Dimensional Carbon Allotrope from Porous Graphene. J. Phys. Chem. C. 2012;116:12810–12813. doi: 10.1021/jp211300n. [DOI] [Google Scholar]
- 39.Li Y., Zhou Z., Shen P., Chen Z. Two-dimensional polyphenylene: Experimentally available porous graphene as a hydrogen purification membrane. Chem. Commun. 2010;46:3672. doi: 10.1039/b926313f. [DOI] [PubMed] [Google Scholar]
- 40.Ao Z.M., Peeters F.M. High-capacity hydrogen storage in Al-adsorbed graphene. Phys. Rev. B. 2010;81 doi: 10.1103/PhysRevB.81.205406. [DOI] [Google Scholar]
- 41.Bhattacharya A., Bhattacharya S., Majumder C., Das G.P. Transition-Metal Decoration Enhanced Room-Temperature Hydrogen Storage in a Defect-Modulated Graphene Sheet. J. Phys. Chem. C. 2010;114:10297–10301. doi: 10.1021/jp100230c. [DOI] [Google Scholar]
- 42.Ataca C., Aktürk E., Ciraci S. Hydrogen storage of calcium atoms adsorbed on graphene: First-principles plane wave calculations. Phys. Rev. B. 2009;79 doi: 10.1103/PhysRevB.79.041406. [DOI] [Google Scholar]
- 43.Chen M., Yang X.-B., Cui J., Tang J.-J., Gan L.-Y., Zhu M., Zhao Y.-J. Stability of transition metals on Mg(0001) surfaces and their effects on hydrogen adsorption. Int. J. Hydrogen Energy. 2012;37:309–317. doi: 10.1016/j.ijhydene.2011.09.065. [DOI] [Google Scholar]