

Abstract
There is no good science in bad models. Cell culture is especially prone to artifacts. A number of novel cell culture technologies have become more broadly available in the 21st century, which allow overcoming limitations of traditional culture and are more physiologically relevant. These include the use of stem-cell derived human cells, cocultures of different cell types, scaffolds and extracellular matrices, perfusion platforms (such as microfluidics), 3D culture, organ-on-chip technologies, tissue architecture, and organ functionality. The physiological relevance of such models is further enhanced by the measurement of biomarkers (e.g., key events of pathways), organ specific functionality, and more comprehensive assessment cell responses by high-content methods. These approaches are still rarely combined to create microphysiological systems. The complexity of the combination of these technologies can generate results closer to the in vivo situation but increases the number of parameters to control, bringing some new challenges. In fact, we do not argue that all cell culture needs to be that sophisticated. The efforts taken are determined by the purpose of our experiments and tests. If only a very specific molecular target to cell response is of interest, a very simple model, which reflects this, might be much more suited to allow standardization and high-throughput. However, the less defined the end point of interest and cellular response are, the better we should approximate organ- or tissue-like culture conditions to make physiological responses more probable. Besides these technologic advances, important progress in the quality assurance and reporting on cell cultures as well as the validation of cellular test systems brings the utility of cell cultures to a new level. The advancement and broader implementation of Good Cell Culture Practice (GCCP) is key here. In toxicology, this is a major prerequisite for meaningful and reliable results, ultimately supporting risk assessment and product development decisions.
Graphical Abstract
INTRODUCTION
The statement that “there is no good science in bad models” might sound very apodictic. However, the best available model is the one which gives us most likely the best result, i.e., the most reliable (reproducible) and the most relevant one. Simple and complex models are both valuable depending simply on the task at hand. Simple models are usually more reproducible, and if we are looking for something simple, they can be very relevant. It is about being fit for purpose. However, many questions in toxicology are very complex, involving networked responses of the biological model. Thus, often the closer our model comes to, in doubt human, physiology, the more likely the results are relevant. In an area of science, where little is reproduced, many results for regulatory toxicology are not published, but important decisions for the safety of consumers and patients as well as for the fate of a product are taken, and we need to strive for such relevance. We cannot leave it to time that wrong results are simply forgotten as we do in most preclinical research. Formal validation is the key tool to ensure reliability and relevance for a given purpose. Good models are the starting point for successful validation and before validation for obtaining meaningful results.
Human cell cultures (Figure 1) are becoming more and more the prime tool of research and preclinical drug development. While the number of researchers is continuously increasing, the number of animals used dropped strongly, by about 50% from the mid-seventies to 2000 according to U.K. statistics,1 though this development has plateaued, or there is even an increase again because genetically modified animals are increasingly used in academic research nowadays.2
Figure 1.
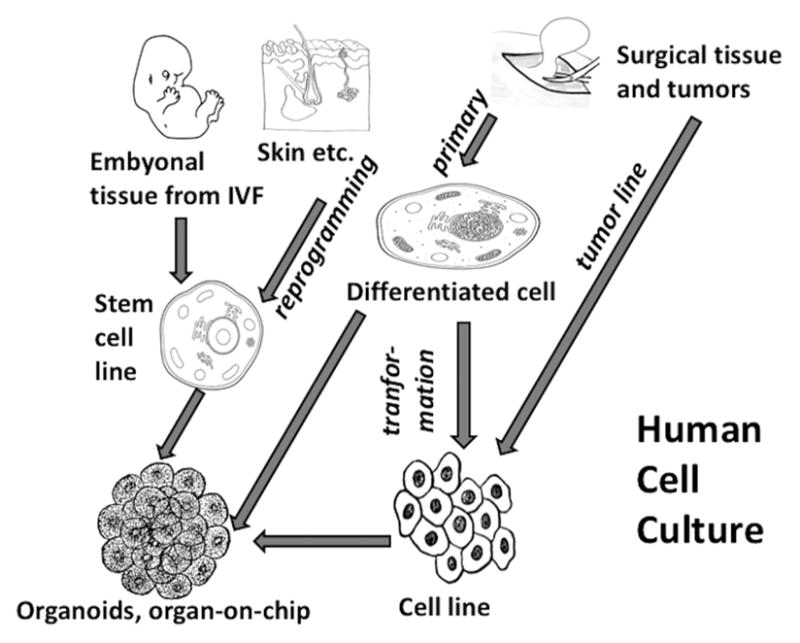
Derivation of different types of human cell cultures. IVF = in vitro fertilization.
Noteworthy, in Europe, over the past decade drug development despite increased investments showed at the same time 30% decline in animal use. This suggests that this very strategic, progressive, and less resource-limited area of the life sciences is transitioning to new approaches.3
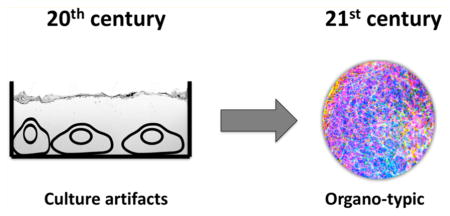
Technologies to maintain cells in culture to enable their study and their use for the testing of substances have continuously improved over the 20th century. Especially, the access to human cells of high quality was very limited, until human stem cells and the respective differentiation protocols became most recently available. Stem cell technologies are strongly impacting on all life sciences including toxicology;4 they are extensively reviewed here as this perspective discusses more how we culture our cells than what cells to culture. It requires an understanding of the shortcomings of traditional culture to systematically improve and approximate the physiological situation. The advances in cell technologies and bioengineering are accelerating and in combination creating synergistic effects, promising to become a disruptive technology, i.e., one that displaces an established technology and shakes up the industry; organs-on-chip or even multiorgans-on-chip, often referred to as human-on-chip, come into reach.5
Efforts to modernize the field of toxicology, often termed Toxicology for the 21st Century,6 rely very strongly on in vitro approaches.7–9 Here the prospects and challenges of current cell culture developments shall be addressed. We first discuss the shortcomings of traditional cell cultures, especially the limitations of cell materials used, their culture conditions, and lack of architecture and organ functionality. Second, the technological possibilities to overcome these deficits are summarized. Third, we will argue for a fit-for-purpose compromise of sophistication of our cell cultures. Finally, the need for quality assurance and the assessment of whether the purpose is met, i.e., validation, will be discussed.
1. SHORTCOMINGS OF TRADITIONAL CELL CULTURES
The art of cell culture too often leads to the artifacts of cell culture: The high number of conditions to control and the many parameters to measure lead to artifacts as a result of cell culture procedures. A few limitations, not really comprehensive, are shown in Figure 2.
Figure 2.
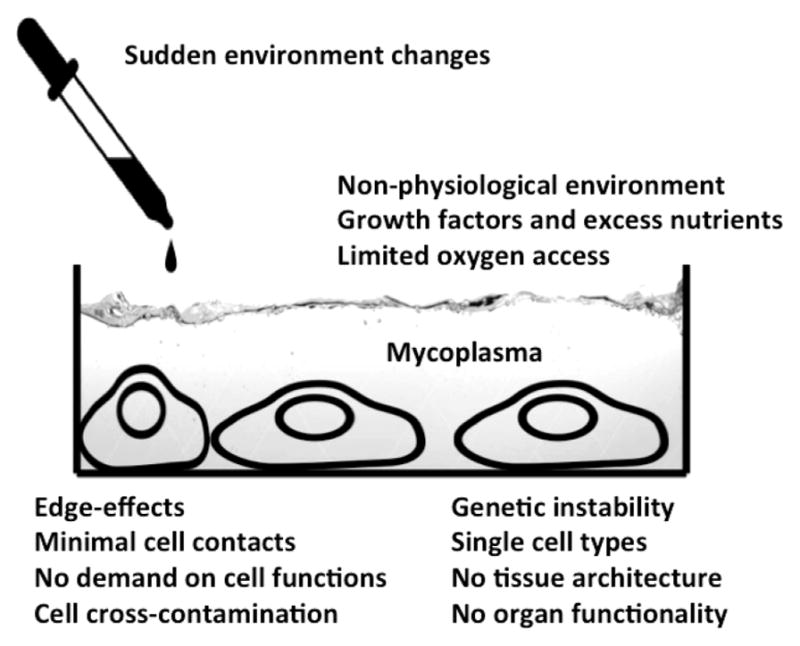
Some pertinent problems of traditional cell culture.
1.1. Cell Sources
Human cells are difficult to obtain in high quality and quantity; with very few exceptions, such as blood, bone marrow, placenta, fat, and skin, it is difficult to obtain healthy human tissue. However, these tissue donations are the prerequisite for human tissue models and are nowadays regulated.10,11 The infrastructures to make surgical tissues and organ donations available commercially and by biobanking are improving,12 but limitations in quantity, costs, and interindividual differences persist. Some human primary cells can be passaged and thereby expanded, but the total number of cell divisions is typically limited. Technologies to immortalize cells represent a prime opportunity to increase the available quantities, with many positive examples such as LUHMES cells,13 i.e., human mesencephalic cells conditionally immortalized with a v-myc retroviral vector allowing to generate dopaminergic neurons, or the telomerase-immortalized human renal epithelial cell line RPTEC/TERT114 but unavoidably mean maintenance culture (passaging) with the risks of dedifferentiation and genetic instability (see below).
Human tumors have for long been the source of naturally immortalized cells. Since 1951, when the HeLa cell was brought into culture as first human tumor cells successfully maintained, many lines have been established, shared between laboratories and cell banks, and led to hundreds of thousands of scientific articles. HeLa alone were used in more than 70,000 articles and 11,000 patents. However, tumor cells differ dramatically in their genetic repertoire and thus physiology: In 2015, Frattini and co-workers15 reported the tremendous genetic differences in HeLa cells from different laboratories. The four samples showed gain and loss of genomic material varying widely between laboratories, drastically affecting basal gene expression. We recently showed16 similar findings using MCF-7 cells. This cell line has been used since 1970 in more than 23,000 articles. We found by comparative genome hybridization in cells obtained from a leading cell bank, that about 10% of genome was lost, 50% of genes were present with less than the normal two copies, and that 30% were multiplied with often more than 16 copies. A worrying finding was that substantial differences in genetic repertoire were found even in different frozen samples from the same lot obtained from the cell bank, which corresponded with different morphology, growth characteristics, estrogen responses, and transcriptomic and metabolomic measurements.16 Tumor cell lines like HeLa and MCF-7 are so strongly genetically distorted that rarely a physiological response can be expected. Furthermore, this genetic instability continues, and we must expect that after some more passages we face a culture with again a different genotype and often phenotype. These changes obviously do not occur in all cells of our cell culture flasks at the same time. What we often forget is that, in many cases, we handle a mix of different variants of the cell in culture, even if at some point they were monoclonal. This becomes very evident in comparative genome hybridization experiments:16 we would have discrete values of 0, 1, 2, 3, 4, etc. genes if all cells were the same; however, we observe a continuum of values, and not even neighboring genes show similar values. This can only be explained by many subpopulations. Consequently, any response measured in tumor cells will be the mixed response of the subpopulations. Sometimes we see indeed, that, for example, not all cells can be killed by a given substance and that other responses show a plateau below 100% of cells reacting. There must be a real evolutionary process of competition and survival of the fittest in our culture flasks. With the selection pressure of our nonhomeostatic culture conditions and driving cell growth (both to be discussed later), we maximize the evolutionary process in the cell culture flask. Perhaps this is an interesting model to study evolution at large. As a consequence, the proportions of these subpopulations can change from passage to passage and affect our results more quickly than genetic drift would make us expect.
As to be discussed below, as a basic quality measure, cell identification is required in order to work according to Good Cell Culture Practice (GCCP). A technical solution has been introduced by the leading cell banks (ATCC, CellBank Australia, ECACC, JCRB, sDSMZ, and RIKEN), i.e., short tandem repeat (STR) microsatellite sequences.17 It is considered the reference technique for identity control of human cell lines and should be done regularly (i.e., for each cell line) to exclude cross-contamination by cells of other cell types or form other species. ISCBI has published guidance related with the best practices on identification of PSCs.18
1.2. Cell Differentiation and Organ Functionality
The cellular phenotype is very much determined by gene expression. For most applications, terminally differentiated (mature) cells are ideal. However, a key element of cell culture is the expansion, i.e., production of more cells, for future experiments. To be able to grow the cells we need, we have to control differentiation and proliferation. For example, in the case of stem cell culture, it is key to maintain pluripotency of the cells, which in many cases is a tricky and tedious procedure. Growth is the opposite of differentiation, and too often, we sacrifice differentiation for cell expansion, not necessarily being able or even only seeking to reestablish a tissue-like phenotype. This process requires often paracrine growth factors from other cell types, extracellular matrix, functional demands, and time. All of this is not present in simple culture systems. Instead we use nonphysiological or embryonal factors, such as fetal calf serum, recombinant growth factors, superphysiological insulin, etc. to drive growth but hindering the expression of the desired phenotype.
Many cell functions are expressed because there is demand. A good example is the metabolism of chemicals. We know well that exposure to chemicals induces the expression of the enzymes, which do metabolize them. However, during maintenance culture, we do not expose the cells to any chemicals and are then surprised that these capabilities are not found in the cells, when testing substances.19
The lack of differentiation obviously limits organ functionality. Continuous growth together with usually only one or two cell types present and two-dimensional cultures do not allow replicating tissue architecture in many tissues necessary for organ functionality.
1.3. Nonhomeostatic and Nonphysiological Culture Conditions
The artificial cell and tissue culture environments differ in many respects from those in vivo.20,21 Traditional cell culture changes media ever 2–3 days. In this time, the medium, i.e., the environment of the cells, has changed dramatically: nutrients were extracted, other substances degraded, oxidized, metabolized, or attached to culture devices; waste and metabolic end products as well as secreted factors have accumulated; the pH often changed as indicated by color changes in phenol red typically included in our media; oxygen has been extracted and replenishment depending on the liquid/air interface; and access to cells is diffusion-limited. This all changes within seconds, when media are first removed and then with some delay with all of the disturbing impact on the culture, in an instant replaced with fresh media, most likely not only different in composition from what was removed but also temperature, pH, osmotic strength, etc. All this differs dramatically from the homeostasis of the environment, which most cells in the body are exposed to, where thanks to the liver most other organs see no or slow changes in exposures taking place. The necessary consequence of this regular stress to the cells is the need to adapt quickly, something undifferentiated cells are more apt to.
Many other parameters of our cultures are set to values not necessarily reflecting the physiological values of the species in culture: body temperatures, pH, and osmotic strength of our media mimic humans not necessarily other species. There is very little insight on how this affects cell physiology.
1.4. Cell Culture Contaminations
There are various types of contaminations, other mammalian cells, microbial ones (viruses and bacteria, especially mycoplasma), and chemical ones. Contaminations can have a serious impact on the results, producing genetic instability, transformation, changes in normal physiological function, and changes in virus susceptibility (Table 1).
Table 1.
Types of Microbial and Other Contaminations in Cell Culture
| “can be known”, where we have tests | mycoplasma, cell authentication, Gram-negative pyrogens, i.e., endotoxins |
| “known unknowns “, where we do not have routine tests | human pathogenic viruses, nonendotoxin pyrogens |
| “unknown unknowns”, where we do not think of as today | prions, chemicals in culture materials |
The astonishing experience is that all these are much more frequent than most researchers think. Data available are typically coming from the more conscious laboratories and those who can afford controls (both financially and practically), thus likely even underestimating the problem. What we learn from these laboratories actually shows that we should say we cannot afford not to control for this. When even cell banks some 10 years ago reported 15% wrongly identified cells in 529 cell lines analyzed,22,23 often not even from the species claimed, this is indeed alarming. We have to assume that people depositing cells to cell banks truly know what they are submitting. Cell banks have reacted, and authentication has improved dramatically, but how many old lines are still around, and how many new cross-contaminations take place all the time? A recent study from China24 showed 25% misidentification in close to 400 samples; most strikingly 86% of cell lines established in China were misidentified. There is even a Web site,25 which lists cell lines now known to be nothing but, e.g., HeLa cells. However, too few researchers and reviewers consult them, and articles continue to be published assuming to work with very different organs and not a cervix carcinoma as in the case of Hela cells. Bad news travels slow in science.
Mycoplasma are another contamination, for which we can control but do not do it everywhere and not always often enough.26 The reason is that they are less visible and less acute than other bacterial infections. However, their impact can be very profound, perturbing completely the cell physiology. Some common sources of mycoplasma are (primary) tissue isolates, culture reagents (predominantly fetal bovine serum), laboratory personnel, and cross-contaminations from infected cultures and were found in a cell bank at 28% of 426 submitted cell lines.22 Trypsin produced from slaughterhouse materials, a major mycoplasma source in the past, is nowadays better controlled by the producers, but half of the species commonly found are actually human pathogens, suggesting that laboratory personnel are the source. Still, the percentage of infected cell cultures is probably somewhere in the 15–30% range.23 A more recent study from Brazil found 34% of 88 cell culture samples from eight laboratories contaminated27 and one in India with 23% of 77 cultures.28 In the biopharmaceutical industry, the problem is more closely controlled, but still up to 8% contaminated cultures were found.29 Other contaminations are less on our radar, such as porcine circovirus 1 (PCV1), bovine leukemia virus (BLV), or bovine viral diarrhea virus (BVDV), but not necessarily less frequent.27 This means that we have in an academic setting about 50% chances that work on cell lines is either not the right cell or one infected with mycoplasma. Who is still surprised about reproducibility issues?30
2. NOVEL CELL CULTURE TECHNOLOGIES TO IMPROVE CELL CULTURE
The many problems of traditional culture make it poor luck when they reflect physiology in a given experimental setup. This explains the need for validation of cellular responses, something we have called mechanistic validation.31 From a different point of view, it is not the reflection of physiology but just the fitness for purpose, as we will argue in the third section. However, when looking for more complex phenomena and studying unknown biology, we will better come as close as possible to physiological conditions. There are many technical solutions to each and every problem, but we do so rarely and arguably can address them all at the same time, though there is synergy in producing an “organo-typic culture” (Figure 3) to achieve what is often referred to as microphysiological systems.32
Figure 3.
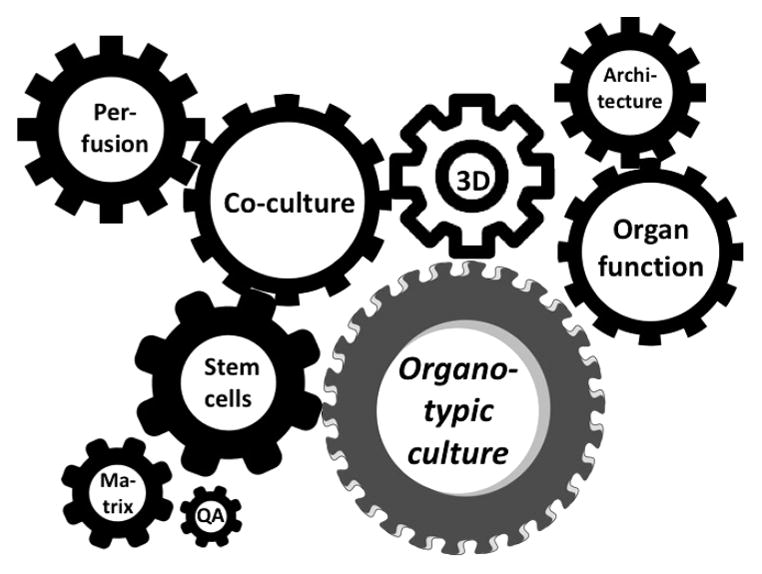
Synergy of different approaches to achieve organo-typic cultures.
2.1. Coculture
A lot of effort in cell culture is spent in the derivation of pure, often monoclonal cultures. This makes sense, when a single cell types’ response is assessed, but comes with the detriment of depriving cells from their natural environment, interactions, and coordinated responses.
Besides the direct mixture of different cell types, tissue slice culture, reaggregating tissues (following dissociation), or the generation of multiple cell types by codevelopment out of stem cells33 are opportunities to derive cocultures. All of these have their problems. The direct combination of cells typically requires access to the cells at the same time in appropriate quality and quantity. They will not necessarily come from the same donor and have other mismatches. Given the enormous complexity of many organs, e.g., more than 40 cell types of the lung,34 these mixes will usually be incomplete. The reproduction of tissue architecture is also challenging.35,36
The culture of tissue slices37–39 starts at least with some of this architecture, but isolation damage, outgrowth, and dedifferentiation can deteriorate this often fast. Slice thickness, limited by technical aspects such as access to oxygen and nutrients, limits how much of tissue architecture can be transferred into culture to begin with. The lack of blood perfusion is mainly responsible, though some techniques allow even perfusion of slices, parts of, or entire organs.40 The dissociation of tissues and their subsequent reaggregation and self-assembly41,42 come with damage to the cells and a subselection of some populations, which are more robust to the dissociation and the culture conditions.
Arguably the most physiological creation of a coculture is out of stem cells, replicating some embryonic developments. However, as we are typically not interested in creating embryos but organoids, these developments need to be directed, typically by growth factors, e.g., first moving them into pluripotent organ-specific stem cells. These are, however, very artificial conditions, forcing a certain development, not necessarily adequate for all desired cell types, and some cells do not originate from the organ stem cell reservoir but, for example, invade organs such as immune cells or cause angiogenesis leading to blood vessel formation.
Cocultures can show cellular interactions such as inflammatory responses, as we showed many years ago for hepatocyte/Kupffer cell cocultures.43,44 Other interactions include metabolic activation of toxicants (e.g., by cocultured liver cells), myelination of axons, the formation of the blood–brain barrier, and many others.
2.2. 3D Culture
Our tissues are 3D, and it seems logical that cultures reflecting this should be more physiologically similar to the intact organism. Cell–cell contacts are key for interactions as well as to induce differentiation, as we know from contact inhibition often observed when cultures become confluent. A lot of positive things have been shown for 3D cultures,45 among others (i) increased cell survival, (ii) increased differentiation, (iii) increased cell–cell interaction, or (iv) better reproduction of the complexity of the organ.
Many approaches to produce 3D cultures are emerging,46–50 but 3D cultures come at a price: Arguably, all cells in 2D are the same, though center positions vs edges likely differ quite a bit with respect to the height of the culture medium, for example. This is not the case for 3D cultures: A cell on the surface is very different from one in the center, quite evidently with respect to access to oxygen and nutrients again, but also, for example, shear forces. Many organoids will not be homogeneous, showing polarizations or more or less physiological structures forming. Not all organoids, even if produced in one batch, will be necessarily the same; this can include size, form, and cell composition among others. Especially for designing test systems, e.g., for toxicology or efficacy testing, this represents a major problem.
There is also a lot of effort necessary to keep cells in 3D: Cells usually try to attach and will grow out. Culture materials prohibiting that, such as Teflon (not translucent!), are limited. Hanging drop methods or keeping organoids in shakers/bioreactors under constant movement (not all cells like the associated shear) represent alternatives.
Because of the thickness and high scattering of this 3D models (preventing light from penetrating), many microscopical or spectrometrical measurements are not easily applicable to organoids. Techniques that can image thicker biological specimens at high resolution include confocal microscopy, multiphoton microscopy, and optical coherence tomography.51
The cell mass can also be limiting. The most important dimension in 3D-cultures is the 1-dimensional outside-in-path of nutrients and oxygen diffusion into the core of cell aggregates, and in the opposite direction, the inside-out-removal of metabolic end products and CO2. Oxygen and nutrient access by diffusion does typically limit organoid size to less than 300 μm. This will correspond to about 20,000 cells per organoid. In many instances more than one organoid can be used, but this starts complicating experimental setups.
The complexity of organoids and duration to produce them imply complex protocols, difficulties in standardizing, low reproducibility, opportunities for infections, as well as the requirement for the respective skills of personnel to do all this. In addition, the lack of detailed understanding of some human organs and tissues makes in vitro modeling difficult. This leads to the needs and opportunities of outsourcing these processes, and indeed, an industry producing organoids is forming.
2.3. Homeostatic Culture
The key to homeostatic culture is perfusion (e.g., by microfluidics). Microfluidics is born from the combination of microelectro-mechanical systems (MEMS) and fluidic channels and allows for the manipulation of small amounts of fluids on a micrometer scale.52 Perfusion can be applied to the entire culture device, can be done with fibers, or by using (acellular) blood vessels from organs, to mention only a few approaches. This can be done recirculating or as run-through. Both come with advantages and disadvantages. Strictly, only the run-through setup represents a constant environment, but obviously, secreted factors are washed out and cells cannot “condition” their medium. Recirculation is actually closer to static conditions or requires the replenishment of oxygen and nutrients in a continuous manner. There are technical solutions for this, ideally with sensors controlling this process, but they complicate the experimental setup even further. Advances in microfluidics and other engineering technologies such as microfabrication, material sciences, sensors, etc. allowed for the development of organ-on-chip or microphysiological systems.3 They often enable more than the homeostatic culture conditions, increasingly even accommodating several types of organoids, often referred to as human-on-chip approaches,53 though this term is still largely overselling what we actually have in hand. However, perfusion systems bring some challenges related to the technology of such a protein and test substance binding to the materials or accumulation of bubbles in the conducting tubes.
Perfusion of 2D cultures has been around for more than two decades, to which one of the authors contributed early on.54,55 It is a market with some commercial solutions to it.5 If 3D is complex, perfused 3D is even more so. For all perfusion, there are some challenges: The low cell mass vs the perfusion volume is often problematic; a homogeneous flow equally reaching all cells is too. The number of replicates is often limited by the number of pumps and parallel units available, but these are engineering challenges, and the grant support and increasing interest in these technologies promise more and more appropriate solutions.
2.4. Tissue Architecture and Functionality
A ball of cells, even of different cell types or perfused, is not yet a representation of an organ.56 This requires organ functionality, and this can often only be achieved by tissue like architecture (Figure 4). Sometimes, this architecture will self-assemble especially in stem-cell-derived systems. In other cases, we will have to create these by bioprinting or scaffolds to achieve in vivo like structures. Some organoids, such as the lung or gut,57,36 benefit from physical stretch. Impressive technical solutions were found, but they are even more complex than perfusion cultures, only leaving this normally to a few specialized laboratories and not allowing high throughput. However, commercialization is on the way and standardization, quality assurance, and broader availability can be expected.
Figure 4.
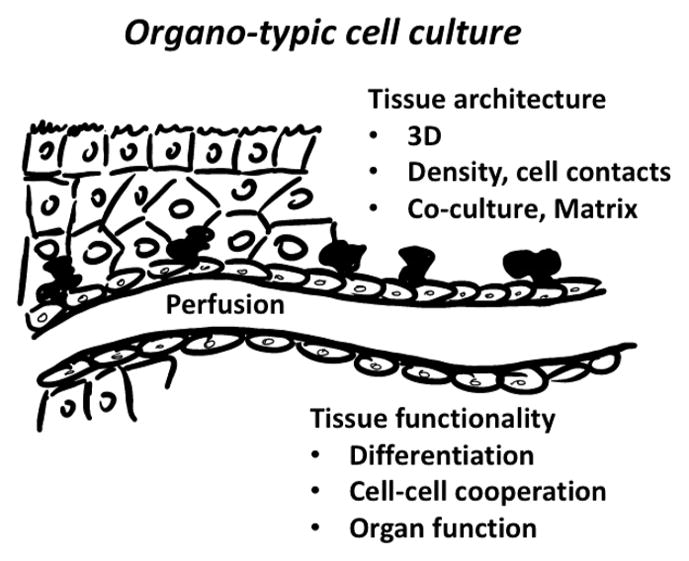
Some key features of tissues to be reproduced by organo-typic cultures.
3. FIT FOR PURPOSE CELL CULTURE
We have argued earlier for a utilitarian approach to cell models.58 Utilitarianism judges the moral worth of an action only by its resulting outcome: “The end justifies the means”. Cell culture most closely representing the physiology in question, appears desirable, but there are many reasons to go for more simple systems (feasibility, costs, and throughput). We might argue that we should use the highest complexity versus efficiency/throughput for a given purpose because we would expect better results. However, often some of the most successful test systems are the simplest, e.g., the Ames test in bacteria (still the most predictive mutagenicity test59) or the Limulus Amebocyte Lysate test for pyrogens. What the simple systems loose with regard to physiological resemblance they gain from the higher number of replicates, ease of interpretation of results, and reproducibility (reduction in systematic and random errors). The more components or work steps in a test system, the more things can go wrong and need to be controlled, as detailed above.
The utilitarian approach likely to be most successful is the one that uses the simplest test system that gets the job done. As Albert Einstein coined it: “Everything should be made as simple as possible, but not simpler”. For the identification of pathways of toxicity,60 this means that a system that shows a hazard manifestation will allow pathway identification (notably, not all pathways if alternative routes can lead to the same phenotype). However, it is likely to be easier to use a number of test systems, which together cover all the relevant reference toxicants, than trying to find a single test system that almost completely reflects human pathophysiology.
Cell-based assays should be tailored for specific needs. Robust predictions about the toxic potential of agents tested by cellular systems strictly depend on the principle that mechanistically defined perturbations induced in vitro match recognized adverse effects and responses at an organismal level. This relies on two major premises: the identification of function-specific responses and the validation of cell-based assays suitable for the detection of those responses.
Cell-based assays developed for toxicity testing and other areas should capture the complexity underlying selected processes and convey the relevant biological information into readouts to account for the mechanistic bases of their occurrence, thereby embodying the specificity of a recognizable functional perturbation. The development of procedures tailored to detect specific functional alterations will be a driver for the establishment of effective suites of cell-based assays for toxicity testing that support mechanistic-based risk assessment and for their use as alternatives to animal experimentation. In fact, it is not only the model itself and how it is cultured (2D or 3D) but also most importantly the mode through which data are achieved and collected. In this respect, successful examples are recent European consortia DETECTIVE and Predict-IV.61–63
4. QUALITY ASSURANCE, REPORTING STANDARDS, AND VALIDATION
Adequate quality assurance of any test system is necessary to safeguard the reproducibility and reliability of results. GCCP64–68 has over the last two decades pioneered the development of guidance in this field. Not applying GCCP in laboratories significantly increases the risk of generating erroneous data, withdrawal of publications, loss of scientific reputation, failed patent applications, wasted resources, laboratory worker infections, and exposure of host institutes to legal liability.69 Validation of cell models has pioneered such quality assurance, but much work still needs to be done to create a culture of evidence-based science. The 21st century cell culture technologies discussed here pose many challenges for this.
4.1. Good Cell Culture Practices (GCCP)
Efforts between 1996 and 2005 (summarized earlier70,17) led to the development of GCCP guidance.66 In brief, parallel initiatives (1996 in Berlin under the auspices of the German Society for Cell and Tissue Culture and 1999 in Bologna at the Third World Congress on Alternatives and Animal Use in the Life Sciences) led to a declaration toward GCCP:47
“The participants… call on the scientific community to develop guidelines defining minimum standards in cell and tissue culture, to be called Good Cell Culture Practice… should facilitate the interlaboratory comparability of in vitro results… encourage journals in the life sciences to adopt these guidelines…”
A GCCP task force was then established, which produced two reports.65,66 The six GCCP principles are:66 (1) Establishment and maintenance of a sufficient understanding of the in vitro system and of the relevant factors, which could affect it; (2) assurance of the quality of all materials and methods, and of their use and application, in order to maintain the integrity, validity, and reproducibility of any work conducted; (3) documentation of the information necessary to track the materials and methods used, to permit the repetition of the work, and to enable the target audience to understand and evaluate the work; (4) establishment and maintenance of adequate measures to protect individuals and the environment from any potential hazards; (5) compliance with relevant laws and regulations, and with ethical principles; and (6) provision of relevant and adequate education and training for all personnel, to promote high quality work and safety.
At the time of the GCCP report, most of the approaches of 21st century cell culture discussed here were in their infancy. Induced pluripotent stem cells, which are currently transforming human cell culture, were not even yet available. The advent of human embryonic in 1998 and induced pluripotent stem cells in 2006 has had a significant impact on human cell culture models. First, it promises to overcome the problems of the availability of human primary cells, though a variety of commercial providers nowadays make almost all relevant human cells available in reasonable quality but at costs that are challenging, at least for academia. However, for human pluripotent stem cells we do not yet have optimal protocols to achieve fully functional differentiation of any cell type. This will probably be achieved given time and effort, but many of the nonphysiologic conditions taken from traditional cell culture contribute to the problems here. Originally, hPSC cultures were thought to be genetically stable, but we increasingly learn about their limitations in that respect too.71–73 Other limitations are costs of culture and complex differentiation protocols, which may require months of labor, media, and supplements. The risk of infection also rises for these long and complex procedures. Despite these challenges and risks, we still we cannot obtain pure cell types, often requiring cell sorting, which requires detachment of cells disrupting the culture conditions and physiology.
GCCP guidance was developed before the broad use of human stem cells. We attempted an update in a workshop:74 “Human embryonic stem cell (hESC) technology for toxicology and drug development: summary of current status and recommendations for best practice and standardization. The Report and Recommendations of an ECVAM Workshop”.
Another important development was the bionengineering leading to organo-typic cultures (also known as organoids, spheroids, microphysiological systems, 3D cultures, organ-on-chip, perfusion cultures, etc.).5 The novel types of tests will represent additional challenges as to standardization of design and generation of optimized culture systems and devices. The systems are considerably more complex than traditional in vitro approaches, involving 3D constructs,45 various cell types, and other engineering.
Together, this further prompted a revision of the earlier GCCP guidance. A number of organizations (such as NIH NCATS, FDA, EPA, ECVAM, ASCCT, ATTC, NIH, and UK Stem Cell Bank) therefore teamed up and organized two workshops in 2015 in the US and Europe65,75 in order to form the International GCCP Collaboration to ultimately develop GCCP 2.0.
4.2. Reporting Standards for in Vitro Systems
GCCP already addresses aspects of reporting but is not explicitly giving checklists or any detailed guidance. In order to fill this gap, CAAT started an initiative, which over the last five years developed such detailed guidance (Daneshian et al., in preparation). Leist et al.76 made early suggestions for guidance as to the publication of in vitro journal articles. A CAAT workshop was held in March 2012 in San Francisco, and a taskforce was formed to further this work. These activities are currently unitedly forming the GCCP 2.0.76 Notably, they have not at this stage expanded to the new approaches of 21st century cell culture, a gap, which shall be filled by the GCCP Collaboration.
Recently developed reporting standards for animal studies (the ARRIVE guidance77) have been adopted by more than 350 journals, though implementation seems to be slow.78 Still it represents a tremendous advance, and strategies for implementation and enforcement will be needed. Noteworthy, ARRIVE is not the only framework, e.g., the Gold Standard Publication Checklist has been proposed.79 This is the role model on which the in vitro reporting standards are based.
An important contribution to the quality of in vitro work and its reporting is the development of cell culture standard operating procedures for general procedures and for specific cultures giving more detail than those available from cell banks. They give guidance for establishing methods but also improve reporting of experiments, where methods can be referenced and only deviations from these protocols noted. Some of this has pioneered in the field of alternatives to animal experiments to ease finding alternatives and to change to cell culture methods. The offers are steadily expanding by journals (including video-based journals), commercial providers, and various governmental and nongovernmental groups. Some prominent examples include DB-ALM (https://ecvam-dbalm.jrc.ec.europa.eu) by the European Center for the Validation of Alternative Methods (ECVAM), which is a public, factual database service that provides evaluated information on development and applications of advanced and alternative methods to animal experimentation in biomedical sciences and toxicology, both in research and for regulatory purposes; the ZEBET database (http://www.bfr.bund.de/en/zebet_database_on_alternatives_to_animal_experiments_on_the_internet__animalt_zebet_-1508.html) on alternatives to animal experiments on the Internet, which provides scientists from industry, universities, and public authorities with information on alternative methods in a database developed for that specific purpose; Nature Protocols (http://www.nature.com/nprot/index.html) is an online journal of laboratory protocols for bench researchers; Springer Protocols (http://www.springerprotocols.com) is the largest subscription-based online database of reproducible laboratory protocols in the biomedical and life sciences; and ThermoFisher Scientific (formerly Gibco) Methods & Cell Culture Protocols (https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/cell-culture-protocols.html) provides a number of basic protocols.
4.3. Validation
Formal validation is typically applied only to regulatory tests and especially alternatives to current animal tests.80–83 At this moment, no test based on the 21st century technologies discussed here has been proposed for formal validation, but this might change soon. The complexity of these models will make this a challenge, though arguably already the very successful validation of skin models for skin irritation and corrosion that employ an organo-typic culture exists. It was the commercial availability of these models which allowed for the validation, and similarly, the new approaches will have to be brought out of individual academic laboratories to allow for standardization and broad availability.
5. CONCLUSIONS
The age of organoid culture has only just begun. Technical opportunities come with challenges. Many technologies are synergistic and only in combination will achieve a truly organo-typic culture. This level of complexity is clearly not needed for all in vitro work; on the contrary, there is good reason to use the simplest system possible. However, many of the complex questions we pose to our models require in vivo-like responses and thus physiological behavior, i.e., organ functionality, perhaps even inter-organ functionality. The rigorous quality assurance and reporting of the new approaches can accelerate their implementation into routine use, making them fit for purpose, increasing reliability and reproducibility. Their commercial availability and the resulting market forces84 will be important for moving them into validation and use.
Acknowledgments
Funding
The work referenced here was supported by National Institute for Environmental Health Sciences [grant number R01 ES020750] and National Center for Advancing Translational Sciences [grant number U18TR000547] at the National Institutes of Health.
ABBREVIATIONS
- ARRIVE
Animal Research: Reporting of In Vivo Experiments Guidelines
- ASCCT
American Society for Cellular and Computational Toxicology
- ATCC
American Type Culture Collection
- BLV
bovine leukemia virus
- BVDV
bovine viral diarrhea virus
- CAAT
Center for Alternatives to Animal Testing
- DB-ALM
Database for Alternative Methods by ECVAM
- DSMZ
German Collection of Microorganisms and Cell Cultures GmbH (Deutsche Sammlung von Mikroorganis-men and Zellkulturen GmbH)
- ECACC
European Collection of Cell Cultures
- ECVAM
European Centre for the Validation of Alternative Methods
- EPA
Environmental Protection Agency
- FDA
Food and Drug Authority
- GCCP
Good Cell Culture Practice
- hESC
human embryonic stem cells
- ISCBI
International Stem Cell Banking Initiative
- JCRB
Japanese Collection of Research Bioresources Cell Bank
- LUHMES
Lund Human Mesencephalic cell line
- MCF-7
breast cancer cell line from Michigan Cancer Foundation
- MEMS
micro-electro-mechanical systems
- NIH NCATS
National Institutes of Health, National Center for Advancing Translational Sciences
- PCV1
porcine circovirus 1
- PSC
pluripotent stem cell
- RPTEC/TERT1
telomerase-immortalized human renal epithelial cell line
- RIKEN
Rikagaku Kenkyūsho, Japanese Institute of Physical and Chemical Research
- STR
short tandem repeat microsatellites
- ZEBET
Centre for Documentation and Evaluation of Alternative Methods to Animal Experiments of the German Federal Institute for Risk Assessment (BfR)
Biographies
David Pamies, M.S., Ph.D. is Research Associate at the Center for Alternatives to Animal Testing (CAAT) at Johns Hopkins Bloomberg School of Public Health, Baltimore and director of the Good Cell Culture Practice (GCCP) 2.0 collaboration. He has developed a 3D brain microphysiological system in vitro model from human iPSC, which promises broad use as a novel method in (developmental) neurotoxicity and neurological pathogenesis. He received his doctorate in Bioengineering (specialty Toxicology) from Miguel Hernandez University in 2012, carried out in part at the European Commission’s Center for the Validation of Alternative Methods (ECVAM).
Thomas Hartung, MD, Ph.D., is Professor of Toxicology (Chair for Evidence-based Toxicology), Pharmacology, Molecular Microbiology, and Immunology at Johns Hopkins Bloomberg School of Public Health, Baltimore, and University of Konstanz, Germany; he also is Director of their Centers for Alternatives to Animal Testing (CAAT) with the portal AltWeb. CAAT hosts the secretariat of the Evidence-based Toxicology Collaboration, the Good Cell Culture Practice Collaboration, the Green Toxicology Collaboration, etc. As PI, he heads the Human Toxome project, an NIH Transformative Research Grant. He is the former Head of the European Commission’s Center for the Validation of Alternative Methods (ECVAM), Ispra, Italy.
Footnotes
Notes
The authors declare the following competing financial interest(s): The authors have contributed to a provisional patent filed by Johns Hopkins University for some of the technologies mentioned in this paper (to produce brain organoids), which was licensed to Organome, LLC. Thomas Hartung is a cofounder of Organome, LLC. David Pamies is a consultant at Organome.
References
- 1.http://www.understandinganimalresearch.org.uk/animals/numbers-animals/.
- 2.Daneshian M, Busquet F, Hartung T, Leist M. Animal use for science in Europe. ALTEX. 2015;32:261–274. doi: 10.14573/altex.1509081. [DOI] [PubMed] [Google Scholar]
- 3.Rovida C, Asakura C, Daneshian M, Hofman-Huether H, Leist M, Meunier L, Reif D, Rossi A, Schmutz M, Valentin JP, Zurlo J, Hartung T. Toxicity testing in the 21st century beyond environmental chemicals. ALTEX. 2015;32:171–181. doi: 10.14573/altex.1506201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suter-Dick L, Alves PM, Blaauboer BJ, Bremm KD, Brito C, Coecke S, Flick B, Fowler P, Hescheler J, Ingelman-Sundberg M, Jennings P, Kelm JM, Manou I, Mistry P, Moretto A, Roth A, Stedman D, van de Water B, Beilmann M. Stem cell-derived systems in toxicology assessment. Stem Cells Dev. 2015;24:1284–1296. doi: 10.1089/scd.2014.0540. [DOI] [PubMed] [Google Scholar]
- 5.Marx U, Andersson TB, Bahinski A, Beilmann M, Beken S, Cassee FR, Cirit M, Daneshian M, Fitzpatrick S, Frey O, Gaertner C, Giese C, Griffith L, Hartung T, Heringa MB, Hoeng J, de Jong WH, Kojima H, Kuehnl J, Luch A, Maschmeyer I, Sakharov D, Sips AJAM, Steger-Hartmann T, Tagle DA, Tonevitsky A, Tralau T, Tsyb S, van de Stolpe A, Vandebriel R, Vulto P, Wang J, Wiest J, Rodenburg M, Roth A. Biology-inspired microphysiological system approaches to solve the prediction dilemma of substance testing. ALTEX. 2016;33:272–321. doi: 10.14573/altex.1603161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hartung T. Toxicology for the twenty-first century. Nature. 2009;460:208–212. doi: 10.1038/460208a. [DOI] [PubMed] [Google Scholar]
- 7.Hartung T, Leist M. Food for thought··· on the evolution of toxicology and phasing out of animal testing. ALTEX. 2008;25:91–102. doi: 10.14573/altex.2008.2.91. [DOI] [PubMed] [Google Scholar]
- 8.Leist M, Hartung T, Nicotera P. The dawning of a new age of toxicology. ALTEX. 2008;25:103–114. [PubMed] [Google Scholar]
- 9.Hartung T. A toxicology for the 21st century: mapping the road ahead. Toxicol Sci. 2009;109:18–23. doi: 10.1093/toxsci/kfp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.FDA. [last accessed 9 Aug 2016];PART 1271—Human cells, tissues, and cellular and tissue-based products, updated 2015. 2001 available at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=1271.
- 11.EU. Commission Directive 2006/17/EC of 8 February 2006 implementing Directive 2004/23/EC of the European Parliament and of the Council as regards certain technical requirements for the donation, procurement and testing of human tissues and cells. [last accessed 9 Aug 2016];Off J Eur Union, L 38/40. 2006 available at http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2006:038:0040:0052:EN:PDF.
- 12.CBAT. Donation of Starting Material for Cell-Based Advanced Therapies: a SaBTO Review. Advisory Committee on the Safety of Blood Tissues and Organs, Department of Health; London, UK: 2014. [last accessed 9 Aug 2016]. available at: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/326823/Cellular_Therapy.pdf. [Google Scholar]
- 13.Scholz D, Pöltl D, Genewsky A, Weng M, Waldmann T, Schildknecht S, Leist M. Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J Neurochem. 2011;119:957–971. doi: 10.1111/j.1471-4159.2011.07255.x. [DOI] [PubMed] [Google Scholar]
- 14.Aschauer L, Limonciel A, Wilmes A, Stanzel S, Kopp-Schneider A, Hewitt P, Lukas A, Leonard MO, Pfaller W, Jennings P. Application of RPTEC/TERT1 cells for investigation of repeat dose nephrotoxicity: A transcriptomic study. Toxicol In Vitro. 2015;30:106–116. doi: 10.1016/j.tiv.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 15.Frattini A, Fabbri M, Valli R, De Paoli E, Montalbano G, Gribaldo L, Pasquali F, Maserati E. High variability of genomic instability and gene expression profiling in different HeLa clones. Sci Rep. 2015;5:15377. doi: 10.1038/srep15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleensang A, Vantangoli M, Odwin-DaCosta S, Andersen ME, Boekelheide K, Bouhifd M, Fornace AJ, Jr, Livi CB, Madnick S, Maertens A, Zhao L, Rosenberg M, Yager JD, Hartung T. Genetic variability in a frozen batch of MCF-7 cells invisible in routine authentication affecting cell function. Sci Rep. 2016;6:28994. doi: 10.1038/srep28994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marx V. Cell-line authentication demystified. Nat Methods. 2014;11:483–488. doi: 10.1038/nmeth.2932. [DOI] [PubMed] [Google Scholar]
- 18.Andrews PW, Arias-Diaz J, Auerbach J. Consensus guidance for banking and supply of human embryonic stem cell lines for research purposes. Stem Cell Rev Rep. 2009;5:301–314. doi: 10.1007/s12015-009-9085-x. [DOI] [PubMed] [Google Scholar]
- 19.Coecke S, Ahr H, Blaauboer BJ, Bremer S, Casati S, Castell J, Combes R, Corvi R, Crespi CL, Cunningham ML, Elaut G, Eletti B, Freidig A, Gennari A, Ghersi-Egea JF, Guillouzo A, Hartung T, Hoet P, Ingelman-Sundberg M, Munn S, Janssens W, Ladstetter B, Leahy D, Long A, Meneguz A, Monshouwer M, Morath S, Nagelkerke F, Pelkonen O, Ponti J, Prieto P, Richert L, Sabbioni E, Schaack B, Steiling W, Testai E, Vericat JA, Worth A. Metabolism: a bottleneck in in vitro toxicological test development. Altern Lab Anim. 2006;34:49–84. doi: 10.1177/026119290603400113. [DOI] [PubMed] [Google Scholar]
- 20.Hartung T. Food for thought··· on cell culture. ALTEX. 2007;24:143–152. doi: 10.14573/altex.2007.3.143. [DOI] [PubMed] [Google Scholar]
- 21.Hartung T. Look Back in anger – what clinical studies tell us about preclinical work. ALTEX. 2013;30:275–291. doi: 10.14573/altex.2013.3.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drexler HG, Uphoff CC, Dirks WG, Macleod RAF. Mix-ups and mycoplasma: the enemies within. Leuk Res. 2002;26:329–333. doi: 10.1016/s0145-2126(01)00136-9. [DOI] [PubMed] [Google Scholar]
- 23.Drexler HG, Uphoff CC. Mycoplasma contamination of cell cultures: incidence, sources, effects, detection, elimination, prevention. Cytotechnology. 2002;39:75–90. doi: 10.1023/A:1022913015916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye F, Chen C, Qin J, Liu J, Zheng C. Genetic profiling reveals an alarming rate of cross-contamination among human cell lines used in China. FASEB J. 2015;29:4268–4272. doi: 10.1096/fj.14-266718. [DOI] [PubMed] [Google Scholar]
- 25.http://www.hpacultures.org.uk/services/celllineidentityverification/misidentifiedcelllines.jsp.
- 26.Hay RJ, Macy ML, Chen TR. Mycoplasma infection of cultured cells. Nature. 1989;339:487–488. doi: 10.1038/339487a0. [DOI] [PubMed] [Google Scholar]
- 27.Pinheiro de Oliveira TF, Fonseca AA, Camargos MF, de Oliveira AM, Pinto Cottorello AC, Souza ADR, de Almeida IG, Heinemann MB. Detection of contaminants in cell cultures, sera and trypsin. Biologicals. 2013;41:407–414. doi: 10.1016/j.biologicals.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Kumar A, Yerneni LK. Semi-automated relative quantification of cell culture contamination with mycoplasma by photoshop-based image analysis on immunofluorescence preparations. Biologicals. 2009;37:55–60. doi: 10.1016/j.biologicals.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 29.Armstrong SE, Mariano JA, Lundin DJ. The scope of mycoplasma contamination within the biopharmaceutical industry. Biologicals. 2010;38:211–213. doi: 10.1016/j.biologicals.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 30.Baker M. 1,500 scientists lift the lid on reproducibility. Nature. 2016;533:452–454. doi: 10.1038/533452a. [DOI] [PubMed] [Google Scholar]
- 31.Hartung T, Stephens M, Hoffmann S. Mechanistic validation. ALTEX. 2013;30:119–130. doi: 10.14573/altex.2013.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andersen M, Betts K, Dragan Y, Fitzpatrick S, Goodman JL, Hartung T, Himmelfarb J, Ingber DE, Jacobs A, Kavlock R, Kolaja K, Stevens JL, Tagle D, Taylor DL, Throckmorton D. Developing microphysiological systems for use as regulatory tools - challenges and opportunities. ALTEX. 2014;31:364–367. doi: 10.14573/altex.1405151. extended online version available at http://www.altex.ch/resources/altex_2014_3_Suppl_Andersen.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paschos NK, Brown WE, Eswaramoorthy R, Hu JC, Athanasiou KA. Advances in tissue engineering through stem cell-based co-culture. J Tissue Eng Regener Med. 2015;9:488–503. doi: 10.1002/term.1870. [DOI] [PubMed] [Google Scholar]
- 34.Franks TJ, Colby TV, Travis WD, Tuder RM, Reynolds HY, Brody AR, et al. Resident cellular components of the human lung: current knowledge and goals for research on cell phenotyping and function. Proc Am Thorac Soc. 2008;5:763–766. doi: 10.1513/pats.200803-025HR. [DOI] [PubMed] [Google Scholar]
- 35.D’Arcangelo E, McGuigan AP. Micropatterning strategies to engineer controlled cell and tissue architecture in vitro. BioTechniques. 2015;58:13–23. doi: 10.2144/000114245. [DOI] [PubMed] [Google Scholar]
- 36.Oliveira SM, Reis RL, Mano JF. Towards the design of 3D multiscale instructive tissue engineering constructs: Current approaches and trends. Biotechnol Adv. 2015;33:842–855. doi: 10.1016/j.biotechadv.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 37.Parrish AR, Gandolfi AJ, Brendel K. Precision-cut tissue slices: applications in pharmacology and toxicology. Life Sci. 1995;57:1887–1901. doi: 10.1016/0024-3205(95)02176-j. [DOI] [PubMed] [Google Scholar]
- 38.Gahwiler BH, Capogna M, Debanne D, McKinney RA, Thompson SM. Organotypic slice cultures: a technique has come of age. Trends Neurosci. 1997;20:471–477. doi: 10.1016/s0166-2236(97)01122-3. [DOI] [PubMed] [Google Scholar]
- 39.Fisher RL, Vickers AE. Preparation and culture of precision-cut organ slices from human and animal. Xenobiotica. 2013;43:8–14. doi: 10.3109/00498254.2012.728013. [DOI] [PubMed] [Google Scholar]
- 40.Huang Y, Williams JC, Johnson SM. Brain slice on a chip: opportunities and challenges of applying microfluidic technology to intact tissues. Lab Chip. 2012;12:2103–2117. doi: 10.1039/c2lc21142d. [DOI] [PubMed] [Google Scholar]
- 41.Layer PG, Robitzki A, Rothermel A, Willbold E. Of layers and spheres: the reaggregate approach in tissue engineering. Trends Neurosci. 2002;25:131–134. doi: 10.1016/s0166-2236(00)02036-1. [DOI] [PubMed] [Google Scholar]
- 42.Kelm JM, Fussenegger M. Microscale tissue engineering using gravity-enforced cell assembly. Trends Biotechnol. 2004;22:195–202. doi: 10.1016/j.tibtech.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 43.Hartung T, Wendel A. Endotoxin-inducible cytotoxicity in liver cell cultures - I. Biochem Pharmacol. 1991;42:1129–1135. doi: 10.1016/0006-2952(91)90298-j. [DOI] [PubMed] [Google Scholar]
- 44.Hartung T, Wendel A. Endotoxin-inducible cytotoxicity in liver cell cultures - II: demonstration of endotoxin-tolerance. Biochem Pharmacol. 1992;43:191–196. doi: 10.1016/0006-2952(92)90277-p. [DOI] [PubMed] [Google Scholar]
- 45.Alépée N, Bahinski T, Daneshian M, De Wever B, Fritsche E, Goldberg A, Hansmann J, Hartung T, Haycock J, Hogberg H, Hoelting L, Kelm JM, Kadereit S, McVey E, Landsiedel R, Leist M, Lübberstedt M, Noor F, Pellevoisin C, Petersohn D, Pfannenbecker U, Reisinger K, Ramirez T, Rothen-Rutishauser B, Schäfer-Korting M, Zeilinger K, Zurich MG. State-of-the-art of 3D cultures (organs-on-a-chip) in safety testing and pathophysiology – a t4 report. ALTEX. 2014;31:441–477. doi: 10.14573/altex1406111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hogberg HT, Bressler J, Christian KM, Harris G, Makri G, O’Driscoll C, Pamies D, Smirnova L, Wen Z, Hartung T. Toward a 3D model of human brain development for studying gene/environment interactions. Stem Cell Res Ther. 2013;4(Suppl 1):S4. doi: 10.1186/scrt365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang YS, Aleman J, Arneri A, Bersini S, Piraino F, Shin SR, Dokmeci MR, Khademhosseini A. From cardiac tissue engineering to heart-on-a-chip: beating challenges. Biomed Mater. 2015;10:034006. doi: 10.1088/1748-6041/10/3/034006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jastrzebska E, Tomecka E, Jesion I. Heart-on-a-chip based on stem cell biology. Biosens Bioelectron. 2016;75:67–81. doi: 10.1016/j.bios.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 50.Kim HJ, Li H, Collins JJ, Ingber DE. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc Natl Acad Sci U S A. 2016;113:E7–15. doi: 10.1073/pnas.1522193112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Graf BW, Boppart SA. Imaging and analysis of three-dimensional cell culture models. Methods Mol Biol. 2010;591:211–227. doi: 10.1007/978-1-60761-404-3_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Whitesides GM. The origins and the future of microfluidics. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 53.Marx U, Walles H, Hoffmann S, Lindner G, Horland R, Sonntag F, Klotzbach U, Sakharov D, Tonevitsky A, Lauster R. Human-on-a-chip” developments: a translational cutting-edge alternative to systemic safety assessment and efficiency evaluation of substances in laboratory animals and man? Altern Lab Anim. 2012;40:235–257. doi: 10.1177/026119291204000504. [DOI] [PubMed] [Google Scholar]
- 54.Koppelstaetter C, Jennings P, Ryan MP, Morin JP, Hartung T, Pfaller W. Assessment of a new cell culture perfusion apparatus for in vitro chronic toxicity testing. Part 1: Technical description. ALTEX. 2004;21:51–60. [PubMed] [Google Scholar]
- 55.Jennings P, Koppelstaetter C, Pfaller W, Morin JP, Hartung T, Ryan MP. Assessment of a new cell culture perfusion apparatus for in vitro chronic toxicity testing. Part 2: Toxicological evaluation. ALTEX. 2004;21:61–66. [PubMed] [Google Scholar]
- 56.Hartung T. 3D - a new dimension of in vitro research. Adv Drug Delivery Rev. 2014;69:vi. doi: 10.1016/j.addr.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huh D, Leslie DC, Matthews BD, Fraser JP, Jurek S, Hamilton GA, Thorneloe KS, McAlexander MA, Ingber DE. A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice. Sci Transl Med. 2012;4:159ra147. doi: 10.1126/scitranslmed.3004249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rossini GP, Hartung T. Towards tailored assays for cell-based approaches to toxicity testing. ALTEX. 2012;29:359–372. doi: 10.14573/altex.2012.4.359. [DOI] [PubMed] [Google Scholar]
- 59.Kirkland D, Aardema M, Henderson L, Müller L. Evaluation of the ability of a battery of three in vitro genotoxicity tests to discriminate rodent carcinogens and non-carcinogens. Mutat Res, Genet Toxicol Environ Mutagen. 2005;584:1–256. doi: 10.1016/j.mrgentox.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 60.Kleensang A, Maertens A, Rosenberg M, Fitzpatrick S, Lamb J, Auerbach S, Brennan R, Crofton KM, Gordon B, Fornace AJ, Jr, Gaido K, Gerhold D, Haw R, Henney A, Ma’ayan A, McBride M, Monti S, Ochs MF, Pandey A, Sharan R, Stierum R, Tugendreich S, Willett C, Wittwehr C, Xia J, Patton GW, Arvidson K, Bouhifd M, Hogberg HT, Luechtefeld T, Smirnova L, Zhao L, Adeleye Y, Kanehisa M, Carmichael P, Andersen EM, Hartung T. Pathways of Toxicity. ALTEX. 2014;31:53–61. doi: 10.14573/altex.1309261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jennings P. The future of in vitro toxicology. Toxicol In Vitro. 2015;29:1217–1221. doi: 10.1016/j.tiv.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 62.Mueller SO, Dekant W, Jennings P, Testai E, Bois F. Comprehensive summary–Predict-IV: a systems toxicology approach to improve pharmaceutical drug safety testing. Toxicol In Vitro. 2015;30(1 Pt A):4–6. doi: 10.1016/j.tiv.2014.09.016. [DOI] [PubMed] [Google Scholar]
- 63.Wilmes A, Bielow C, Ranninger C, Bellwon P, Aschauer L, Limonciel A, Chassaigne H, Kristl T, Aiche S, Huber CG, Guillou C, Hewitt P, Leonard MO, Dekant W, Bois F, Jennings J. Mechanism of cisplatin proximal tubule toxicity revealed by integrating transcriptomics, proteomics, metabolomics and biokinetics. Toxicol In Vitro. 2015;30(1 Pt A):117–127. doi: 10.1016/j.tiv.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 64.Gstraunthaler G, Hartung T. Bologna declaration toward Good Cell Culture Practice. Altern Lab Anim. 1999;27:206. [Google Scholar]
- 65.Hartung T, Balls M, Bardouille C, Blanck O, Coecke S, Gstraunthaler G, Lewis D. Report of ECVAM task force on good cell culture practice (GCCP) Altern Lab Anim. 2002;30:407–414. doi: 10.1177/026119290203000404. [DOI] [PubMed] [Google Scholar]
- 66.Coecke S, Balls M, Bowe G, Davis J, Gstraunthaler G, Hartung T, Hay R, Merten OW, Price A, Schechtman L, Stacey G, Stokes W. Guidance on Good Cell Culture Practice. Altern Lab Anim. 2005;33:261–287. doi: 10.1177/026119290503300313. [DOI] [PubMed] [Google Scholar]
- 67.Balls M, Coecke S, Bowe G, Davis J, Gstraunthaler G, Hartung T, Hay R, Merten OW, Price A, Schechtman LM, Stacey G, Stokes W. The importance of good cell culture practice (GCCP) ALTEX. 2006;23:270–273. [Google Scholar]
- 68.Stacey GN, Hartung T. Availability, standardization and safety of human cells and tissues for drug screening and testing. In: Marx U, Sandig V, editors. Drug Testing In Vitro: Breakthroughs and Trends in Cell Culture Technology. Wiley-VCH Verlag; Weinheim, Germany: 2007. pp. 231–250. [Google Scholar]
- 69.Pamies D, Martin U, Bal-Price A, Stacey G, Jennings P, Grillari R, Schwamborn J, Ellinger-Ziegelbauer C, Hartung T. Good Cell Culture Practice: human (stem) cells and organoids. ALTEX 2016 [Google Scholar]
- 70.Hartung T, Zurlo J. Alternative approaches for medical countermeasures to biological and chemical terrorism and warfare. ALTEX. 2012;29:251–260. doi: 10.14573/altex.2012.3.251. [DOI] [PubMed] [Google Scholar]
- 71.Mitalipova MM, Rao RR, Hoyer DM, Johnson JA, Meisner LF, Jones KL, Dalton S, Stice SL. Preserving the genetic integrity of human embryonic stem cells. Nat Biotechnol. 2005;23:19–20. doi: 10.1038/nbt0105-19. [DOI] [PubMed] [Google Scholar]
- 72.Lund RJ, Narva E, Lahesmaa R. Genetic and epigenetic stability of human pluripotent stem cells. Nat Rev Genet. 2012;13:732–744. doi: 10.1038/nrg3271. [DOI] [PubMed] [Google Scholar]
- 73.Steinemann D, Gohring G, Schlegelberger B. Genetic instability of modified stem cells - a first step towards malignant transformation? Am J Stem Cells. 2013;2:39–51. [PMC free article] [PubMed] [Google Scholar]
- 74.Adler S, Allsopp T, Bremer S, Buzanska L, Drake R, Frandsen U, Gribaldo L, Gmeiner K, Harkness L, Hartung T, Healy L, Hescheler J, Knaut H, Mandenius C-F, Pazos P, Price A, Pellizzer C, Pera M, Reubinoff B, Stojanov T, Strehl R, Stummann T, Trosko J, Hasiwa M, Stacey G. hESC Technology for Toxicology and Drug Development: Summary of Current Status and Recommendations for Best Practice and Standardization. The Report and Recommendations of an ECVAM Workshop; 2007. [last accessed 11 July 2016]. available at https://eurl-ecvam.jrc.ec.europa.eu/about-ecvam/archive-publications/publication/hESC_%20010711.pdf. [Google Scholar]
- 75.Pamies D, Bal-Price A, Simeonov A, Tagle D, Allen D, Gerhold D, Yin D, Pistollato F, Inutsuka T, Sullivan K, Stacey G, Salem H, Leist M, Daneshian M, Vemuri MC, McFarland R, Coecke S, Fitzpatrick SC, Lakshmipathy U, Mack A, Wang WB, Sekino Y, Kanda Y, Hartung T. Good Cell Culture Practice for stem cells and stem-cell-derived models. ALTEX. 2016 doi: 10.14573/altex.1607121. [DOI] [PubMed] [Google Scholar]
- 76.Leist M, Efremova L, Karreman C. Food for thought··· considerations and guidelines for basic test method descriptions in toxicology. ALTEX. 2010;27:309–317. doi: 10.14573/altex.2010.4.309. [DOI] [PubMed] [Google Scholar]
- 77.Kilkenny CC, Browne WJW, Cuthill ICI, Emerson MM, Altman DGD. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baker D, Lidster K, Sottomayor A, Amor S. Two years later: journals are not yet enforcing the ARRIVE guidelines on reporting standards for pre-clinical animal studies. PLoS Biol. 2014;12:e1001756. doi: 10.1371/journal.pbio.1001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hooijmans C, de Vries R, Leenaars M, Ritskes-Hoitinga M. A gold standard publication checklist to improve the quality of animal studies, to fully integrate the Three Rs, and to make systematic reviews more feasible. Altern Lab Anim. 2010;38:167–182. doi: 10.1177/026119291003800208. [DOI] [PubMed] [Google Scholar]
- 80.Hartung T, Bremer S, Casati S, Coecke S, Corvi R, Fortaner S, Gribaldo L, Halder M, Hoffmann S, Roi AJ, Prieto P, Sabbioni E, Scott L, Worth A, Zuang V. A modular approach to the ECVAM principles on test validity. ATLA - Altern Lab Anim. 2004;32:467–472. doi: 10.1177/026119290403200503. [DOI] [PubMed] [Google Scholar]
- 81.OECD. [last accessed 11 July, 2016];Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment, OECD Series on Testing and Assessment, Guidance Document 34. 2005 available at http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?doclanguage=en&cote=env/jm/mono(2005)14.
- 82.Hartung T. Food for thought··· on validation. ALTEX. 2007;24:67–72. doi: 10.14573/altex.2007.2.67. [DOI] [PubMed] [Google Scholar]
- 83.Leist M, Hasiwa M, Daneshian M, Hartung T. Validation and quality control of replacement alternatives – current status and future challenges. Toxicol Res. 2012;1:8. [Google Scholar]
- 84.Bottini AA, Hartung T. Food for thought··· on economics of animal testing. ALTEX. 2009;26:3–16. doi: 10.14573/altex.2009.1.3. [DOI] [PubMed] [Google Scholar]