

Abstract
Environmental toxicants such as chemicals, heavy metals, and pesticides have been shown to promote transgenerational inheritance of abnormal phenotypes and/or diseases to multiple subsequent generations following parental and/or ancestral exposures. This study was designed to examine the potential transgenerational action of the environmental toxicant trichloroethane (TCE) on transmission of liver abnormality, and to elucidate the molecular etiology of hepatocyte cell damage. A total of thirty two healthy immature female albino mice were randomly divided into three equal groups as follows: a sham group, which did not receive any treatment; a vehicle group, which received corn oil alone, and TCE treated group (3 weeks, 100 μg/kg i.p., every 4th day). The F0 and F1 generation control and TCE populations were sacrificed at the age of four months, and various abnormalities histpathologically investigated. Cell death and oxidative stress indices were also measured. The present study provides experimental evidence for the inheritance of environmentally induced liver abnormalities in mice. The results of this study show that exposure to the TCE promoted adult onset liver abnormalities in F0 female mice as well as unexposed F1 generation offspring. It is the first study to report a transgenerational liver abnormalities in the F1 generation mice through maternal line prior to gestation. This finding was based on careful evaluation of liver histopathological abnormalities, apoptosis of hepatocytes, and measurements of oxidative stress biomarkers (lipid peroxidation, protein carbonylation, and nitric oxide) in control and TCE populations. There was an increase in liver histopathological abnormalities, cell death, and oxidative lipid damage in F0 and F1 hepatic tissues of TCE treated group. In conclusion, this study showed that the biological and health impacts of environmental toxicant TCE do not end in maternal adults, but are passed on to offspring generations. Hence, linking observed liver abnormality in the offspring to environmental exposure of their parental line. This study also illustrated that oxidative stress and apoptosis appear to be a molecular component of the hepatocyte cell injury.
Keywords: Apoptosis, Hepatocyte cell injury, Oxidative stress, Parental transmission
Introduction
In recent years, there is an increasing scientific evidence for the inheritance of environmentally induced abnormal phenotypes and/or diseases from parents to offspring (Skinner et al., 2010). These abnormalities may not be only limited to first generation of descendents but to subsequent generations either exposed (multigenerational inheritance) or unexposed (transgenerational inheritance) (Skinner et al., 2013; Nilsson and Skinner 2014, 2015).
Hence, the concept that today’s environmental exposures can significantly affect later generations has been widely accepted under the term “epigenetic inheritance” the transmission of phenotypic traits (e.g. molecular, cellular, organismal) that involve alterations in gene expression without changes in gene sequence (Nilsson and Skinner, 2014). Epigenetic inheritance has now been shown to be present in plants (Henderson and Jacobsen, 2007), nematode worms (Greer et al., 2011), flies (Ruden and Lu, 2008), rats (Manikkam et al., 2012), mice (Guerrero-Bosagna et al., 2012), and humans (Pembrey, 2010). Experimental studies in rodents indicate that exposure to environmental toxic substances during intrauterine life, postnatal life, early life, and/or germ cell exposures play a significant role in determining the mature phenotype and susceptibility to diseases in later life (Skinner et al., 2010, 2013). Epigenetic abnormalities/diseases in humans have been documented for reproductive tract abnormalities, brain and behavior abnormalities, immune system abnormalities (Anway et al., 2006a; Crews et al., 2007) as well as kidney (Anway et al., 2006b), ovarian, testis and prostate diseases (Salian et al., 2009; Manikkam et al., 2012; Nilsson et al., 2012; Al-Griw et al., 2015a).
A number of environmental factors have been shown to induce transgenerational inheritance of adult onset disease and phenotypic variations (Skinner et al., 2010). These include exposures to synthetic chemical agents (e.g. hydrocarbons, dioxin, trichloroethylene), pesticides (e.g. methoxychlor, permethrin, dichlorvos, vinclozolin), heavy metals (e.g. cadmium, mercury, arsenic, nickel), and plastics (e.g. bisphenol A, phthalates) (Aitken et al., 2004; Wong, 2010; Manikkam et al., 2012; Easley et al., 2015).
These environmental exposures can act on both somatic and gamete (sperm or egg) genomes inducing specific altered epigenetic patterns such as DNA methylation, histone modification, and miRNA alterations (Hou et al., 2012; Guerrero-Bosagna and Skinner, 2012; Nilsson and Skinner, 2015); that can be transmitted to future generations in the absence of direct environmental exposure (Hou et al., 2012; Skinner et al., 2013; Nilsson and Skinner, 2014). Therefore, the transmission of epigenetic variations rather than the direct induction of such variations is responsible for disease states through changes in gene expression and the establishment of heritable states of chromatin architecture (Kelly, 2014).
The transgenerational changes are specific and could be used as biomarkers of exposure and disease (Manikkam et al., 2012; Skinner et al., 2013). The current study was designed to investigate the transgenerational actions of a specific environmental toxicant trichloroethane (TCE), an industrial solvent and a degreasing agent, to induce alteration in a somatic cell integrity that correlates to the induction of abnormality in the hepatic tissue.
Recently, it has been reported that TCE exposure environmentally (Al-Griw et al., 2015a, 2015c, 2016) or occupationally (Bruckner et al., 2001) on a daily basis is associated with increased risk of infertility, low fetal weight and early neonatal neurobehavioral abnormalities (Al-Griw et al., 2015b), as well as autoimmunity (Wang et al., 2013).
Epigenetic inheritance has been studied extensively in the germ line through in utero exposure (Skinner, 2007; Manikkam et al., 2012; Skinner et al., 2013). Most studies expose the gestating female F0 generation mice to a toxicant at the time their embryo were undergoing sex determination, the F1 generation mice were exposed directly as a fetus. In addition, the germ cells present in the developing fetus were exposed directly. Furthermore, when F0 generation male is exposed to an environmental insult, his sperm is exposed directly. On the other hand, studies on the epigenetic changes in the somatic cells have been scarce. In addition, there are few studies (Skinner and Guerrero-Bosagna, 2009; Skinner et al., 2010) addressed the transgenerational aspects of the environmental toxicant exposures on the subsequent development of an adult somatic cells. Therefore, the purpose of this study was to investigate the ability of TCE to induce intragenerational inheritance of liver abnormalities in F1 generation mice (offspring) through direct exposure of immature female F0 generation mice (dams); and to elucidate the molecular components of TCE-induced hepatocyte cell injury.
Materials and Methods
Animal Studies and Breeding
The protocol of the study was approved by Libyan National Committee for Biosafety and Bioethics on 2016. All efforts were made to fulfill the ethical experimentation standards such as minimizing the pain during animal handling and experiments as well as reducing the number of animals used. A total of twenty four animals used in this study were immature female Swiss albino mice (F0), 18 to 21 days old, weighing 12 to 15g. They were breed in the animal house of the Zoology Department (Faculty of Science, University of Tripoli, Tripoli, Libya), housed under standard conditions of light (12 hour cycle) and temperature (26 ± 2°C). The animals were fed with a standard mouse diet and ad libitum tap water for drinking.
Animals were divided into three groups of eight mice each. The mice were administered twice weekly for three weeks intraperitoneal (i.p.) injections of TCE (100 μg/kg body weight every 4th day) or corn oil positive vehicle control, and the negative sham control did not receive any treatment. At ten weeks of age, treated F0 females were mated with fertility proofed control males. Mating was confirmed by the presence of vaginal plug, which was defined as gestation day-1 (GD-1). Once the vaginal plug was observed, F0 female mice were separated from males and individually caged. F0 pregnant dams were observed daily and total body weight (TBW) was measured daily to further confirm pregnancy. The dams were allowed to deliver naturally and the delivery day was designed as post-natal day 0 (PND-0).
After delivery, F1 litter weights, sex ratios, and percent of dead pups were recorded. Moreover, the size of each litter was standardized on PND-4 by eliminating extra pups through random selection within sex from litters with more than 10 pups to yield 10 pups, with five females and five males per litter. Control litters with 10 or fewer pups were not standardized. The treated female mice were designated as the F0 generation. The offspring of the F0 generation mice were the F1 generation. The control populations were larger than the TCE population due to the lower incidence of abnormality/disease in the control populations.
The increased number of control animals allowed for an increased ability to detect abnormality in the control populations that then allowed for more accurate statistical comparison of the control versus TCE populations. In consistence with our previous observations (Al-Griw et al., 2015c), we have found alterations in sex ratios, but not in litter size, in the F1 generations for the TCE, but not control animals.
Clinical Assessment
The clinical assessment included animal survival, TBW, weight gain/loss and locomotor activity. During the course of the exposure period, mice were observed twice daily for any abnormal clinical signs or behavior that may result from toxicity. Night deaths were recorded the next morning. Two independent observers confirmed the cause of death to exclude TCE-nonrelated mortality. A subset of F1 generation offspring of control and TCE populations was randomly selected to examine congenital malformations.
Tissue Harvest and Histopathology Processing
F0 and F1 generation mice (2 males and 2 females from each litter) were sacrificed at postnatal day 120 (four months) for tissue harvest. Body and organ (e.g. liver) weights were measured at dissection time. Livers were fixed in 10% formalin, and then processed for paraffin embedding by standard procedures for histopathology examination.
Six to eight micrometer tissue sections were made, stained and examined for histopathology. Portions of livers from control and TCE populations were stored at -20 °C for the measurment of oxidative stress biomarkers. Blood samples were also collected at the time of dissection, allowed to clot, centrifuged and serum samples stored for further analysis.
Histopathology
All histopathology was examined in randomly selected animals by three independent observers. H&E stain was used for general histopathological changes. For specific liver damage special stains: Mallory Trichrome (MTC) and Periodic Acid-Schiff stain (PAS) were used. Liver histopathology criteria included the presence of a vacuole, steatosis and inflammatory infiltrates and ’other’ abnormalities including fibrotic and hepatocyte alterations (cell with vacuolated and pale-staining cytoplasm) as well as alterations in nuclear morphology.
A cut-off was established to declare a tissue ‘diseased’ based on the mean number of histopathological abnormalities plus two standard deviations from the mean of control tissues. This number was used to classify mice into those with and without liver abnormality in each population. A mouse tissue section was finally declared ’diseased’ only when two of the three observers marked the same tissue section ’diseased’. The proportion of mice with obesity was obtained by counting those that had these conditions out of all the animals evaluated. The number of animals per litter (litter representation) mean ± SEM used for the control versus TCE population comparisons for each specific abnormality was found not to be statistically different (P > 0.05). Therefore, no litter representation differences or litter bias was detected for any of the specific abnormality assessed.
Scoring of Cell Death
To assess cell death, images of H&E-stained liver tissues were opened in ImageJ software (version 1.45) and a manual count was performed. Cell counts were expressed as a percentage of the number present for each treatment group and an overall percentage obtained by averaging the data for all cells within a treatment group. The hepatocellular apoptotic cells were then quantified. Hepatocytes undergoing apoptosis were identified by morphological criteria such as cell shrinkage, chromatin condensation and margination, and apoptotic bodies (Gujral et al., 2001). However, hepatocytes undergoing necrosis were determined using the criteria such as increased eosinophilia, cell swelling and lysis, loss of architecture, karyolysis, and karyorrhexis (Gujral et al., 2002). The percent of cell death was estimated by evaluating the number of microscopic fields with dead cells compared to the entire histologic section.
The scoring scale was set from 0 (worst) to 5 (best), with the following criteria: (0): no tissue damage; (1): mild; (2): mild to moderate; (3): moderate; (4): moderate to severe; and (5): severe. For each liver tissue, the eight scores were averaged, and this average was considered as a replicate.
Lipid Peroxidation Measurment
Lipid peroxidation levels in the livers from F0 and F1 generation control and TCE populations was determined spectrophotometrically as a concentration of final lipid peroxidation products, which in reaction with thiobarbituric acid (TBARS) form colour complex (thiobarbituric acid-reactive substances; TBARS). In brief, liver tissues were homogenized with a tissue homogenizer (IKA, RW 20.n, Germany) in ice-cold 10% (w/v) phosphate-buffered saline (PBS) solution. After centrifugation, a 500 µL aliquot of liver homogenate samples were added to 2 ml of TCA-TBA HCL reagent (thioarbituric acid 0.37%, 0.24 N HCL and 15% TCA) and then boiled at 100 °C for 15 min, and allowed to cool. After centrifugation (Sigma 2K15, Germany) at 3000 rpm for 10 min, the supernatant was removed and the absorbance was read at 532 nm. The calibration curve was obtained using different concentrations of 1, 1, 3, 3-tetramethoxypropane as standard to determine the concentration of TBA-MDA (malondialdehyde; MDA) adducts in the samples (Zhang et al., 2004).
Nitric Oxide Measurment
Nitric oxide in the livers from F1 generation control and TCE populations was determined as described previously (Xu et al., 2011) with some modification. The liver samples were homogenized in cold 0.9% saline. The homogenates were then centrifuged at 10,000 rpm for 5 min at 4 °C. 1 ml of the supernatant was mixed with an equal volume of Greiss reagent containing 1% sulphanilamide and 0.1% naphthylethylenediamine in 5% phosphoric acid. The mixture was then allowed to stand at room temperature for 30 min. The absorbance of the mixture was measured against the corresponding blank solutions at 546 nm. Sodium nitrate solution was used to obtain a standard curve.
Isolation and Measurment of Total Protein
Livers from F0 and F1 generation control and TCE populations were homogenized in sodium phosphate buffer pH 7.4. The homogenate was then centrifuged at 6000 x g at 4 °C for 15 min, and the supernatants collected were used for protein assay (Goa, 1953; Bradford, 1976). Bovine serum albumin was used as a standard.
Protein Carbonyl Measurment
Carbonyl content in the in the livers from F1 generation control and TCE populations was quantitated by the protein carbonyl assay as previous described (Wang et al., 2013).
Microscopy
The liver cytoarchitecture and cell death scoring were observed and imaged using a low-power objective under a light microscope (Leica, Germany).
Statistical analysis
Data are expressed as means ± SEM. A computerized Kolmogorov-Smirnov test was used to determine whether the data fitted a normal distribution. Statistical analysis was performed using one-way ANOVA followed by a post-hoc test for multiple comparisons within SPSS 20.0 for Windows. Two tailed Student’s t test was used when only two independent groups were compared. P-values less than 0.05 were considered statistically significant.
Result
The present study provides experimental evidence for intragenerational transmission of liver abnormality. The results of this study show that exposure to the environmental toxicant TCE promoted adult onset liver abnormalities in F0 female mice as well as unexposed F1 generation offspring (Fig. 1). It is the first study to report a transgenerational liver abnormality in the F1 generation mice through maternal line prior to gestation. Transgenerational inheritance signifies that environmental toxic effects manifested in the exposed generation also appear in the unexposed future generation(s). This finding was based on careful evaluation of liver histopathological abnormalities, apoptosis of hepatocytes, and measurements of oxidative stress biomarkers (lipid peroxidation, nitric oxide and protein carbonylation) in control and TCE groups (as explained below). There was an increase in liver histopathological abnormalities, cell death, and oxidative lipid damage in F0 and F1 hepatic tissues. Therefore, these results indicate exposure of immature F0 female mice to TCE transgenerationally transmitted liver histopathology to their F1 generation offspring implying transgenerational transmission.
Fig. 1.
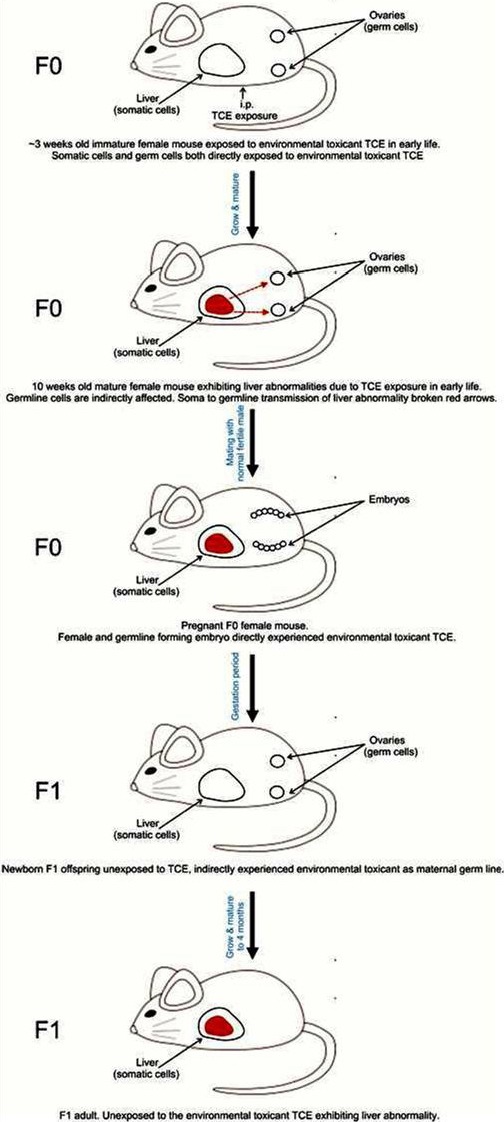
Schematic early life exposures to the environmental toxicant TCE induce liver abnormality which leads to liver abnormality in later life. This is passed on to F1 generation indicating intragenerational inheritance of liver abnormalities and perhaps soma to germline transmission. In this study offspring (F1 generation) have never been exposed to environmental toxicant. F0 immature female somatic cells (e.g. ovaries) and germ cells (e.g. oocytes) were directly exposed to environmental toxicant in early life. TCE exposure caused epigenetic variations in the germline indirectly (red broken arrows) through its effect on somatic liver cells.
Liver Abnormality and Histopathology
Liver abnormalities were characterized by the presence of several histopathological lesions which included: disturbed parenchyma architecture of the hepatic lobules, infiltration of mononuclear cells (macrophages and lymphocytes) around central veins and in portal areas, some dilated congested blood vessels; severe cytoarchitectural distortions of the hepatocytes accompanied by steatosis and inflammatory infiltrates, and complete loss of nucleus (Fig. 2A) was found throughout hepatic tissue of TCE treated F0 and F1 mice. The incidence of liver abnormality in F0 and F1 of control and TCE l populations is presented in Figure 2B. There was a statistically significant increase in liver abnormality in TCE F0 generation compared to control F0 generation (Fig. 2B).
Fig. 2.
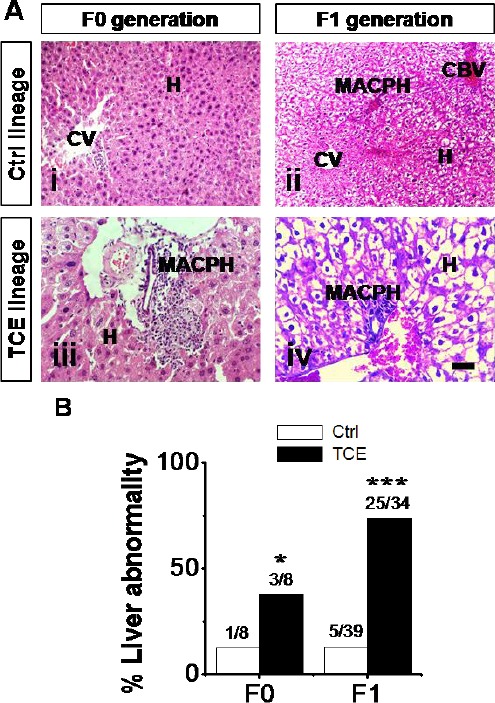
Control (Ctrl) and TCE F0 and F1 generation adult-onset liver abnormality. (A) Micrographs H&E–stained sections show liver abnormality in F0 (panel iii) and F1 (panel iv) generation TCE animals compared to F0 (panel i) and (panel ii) Ctrl populations. Hepatocytes (H), central vein (CV), congested blood vessels (CBV), and macrophages (MACPH). Scale bar = 100 μm, 40X. (B) Percentages of mice with liver abnormality and number of diseased mice/total number of mice. Ctrl and TCE livers were examined with the mean ± SEM presented and asterisks (*) indicating a statistically significant difference (P ≤ 0.05) vs. TCE-treated mice. Student’s t-tests. *P ≤ 0.05, ***P ≤ 0.001.
To further study the effect of TCE on liver histopathology, liver sections from F0 and F1 control and TCE groups were stained with two special stains Mallory’s trichrome (MTC) and Periodic Acid-Schiff stain (PAS). The F0 and F1 control population showed normal liver architecture. Whereas the F1 generation mice of TCE populations showed destructed nuclei of hepatocytes, vacuolated cytoplasm; completely loss of the hepatocytes, mild pericellular fibosis is present in lobule and periportal distribution and is associated with portal–portal linkage, massive accumulation of monocellular phagocytic cells around the blood vessels. There was also deposition of collagen fibers, the cytoplasmic vacuolation were still observed (Fig. 3).
Fig. 3.
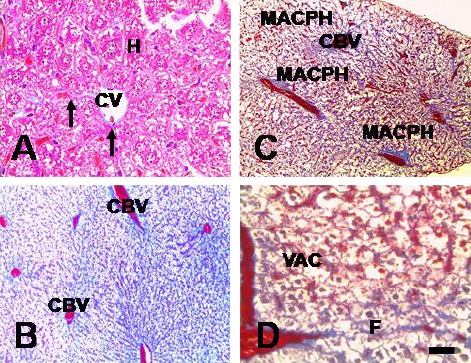
Ctrl and TCE F1 generation adult-onset liver abnormality. Micrographs of MTC–stained liver sections show liver abnormality in F1 generation TCE populations (panel C and D) compared to that in F1 Ctrl population (panel A and B). Hepatocytes (H), central vein (CV), congested blood vessels (CBV), macrophages (MACPH), and vacuolated cytoplasm (VAC). Scale bar = 100 μm, 40X. To observe the location of hepatic glycogen granules and the glycogen density of hepatocytes, PAS stain was used.
In contrast to control populations, TCE F1 generation showed a remarkable reduction in the glycogen storage as well as carbohydrate content (Fig. 4).
Fig. 4.
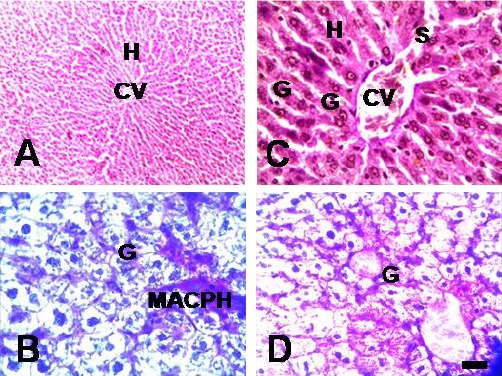
Ctrl and TCE F1 generation adult-onset liver abnormality. Micrographs of PAS–stained sections show liver abnormality in F1 generation TCE populations (panel C and D) compared to Ctrl F1 generation (panel A and B). Hepatocytes (H), central vein (CV), congested blood vessels (CBV), macrophages (MACPH), glycogen (G), and vacuolated cytoplasm (VAC). Scale bar = 100 μm, 63X.
Hepatocyte Nuclear Alterations
The percent of hepatocellular apoptosis was determined by strict morphological criteria. There was morphological evidence of apoptosis for individual hepatocyte cells in F0 and F1 generation of TCE population compared to control population. Nuclear morphological abnormality was characterized by prominent chromatin condensation, DNA fragmentation, and the formation of apoptotic bodies. This was accompanied by other degenerative changes (Fig. 5A), but no such changes were found in control populations. The quantitative analysis showed that the F0 and F1 generations of TCE populations had an increased percent of apoptotic hepatocyte cells compared to F0 and F1 generations control population (Fig. 5B).
Fig. 5.
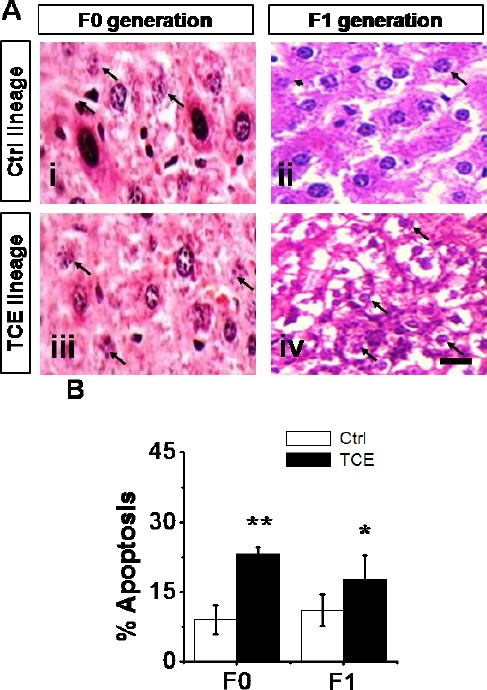
Hepatocellular apoptosis in Ctrl and TCE F0 and F1 generation. (A) Micrographs H&E–stained sections show hepatocellular apoptosis in F0 (panel iii) and F1 (panel iv) generation TCE population compared to F0 (panel i) and (panel ii) Ctrl population. Scale bar = 100 μm, 63X. (B) Percentages of hepatocellular apoptosis. Ctrl and TCE livers were examined with the mean ± SEM presented and asterisks (*) indicating a statistically significant difference (P ≤ 0.05) vs. TCE-treated mice. Student’s t-tests. *P ≤ 0.05, **P ≤ 0.01.
Lipid Peroxidation in the Livers of the F0 and F1 Generations
Lipid peroxidation determination in hepatic tissues of control and TCE F0 and F1 generations was performed by the TBARS method. Levels of liver MDA formation in the four months old F0 and F1 generations of TCE population significantly increased compared to F0 and F1 generations of control, respectively, (Fig. 6), but no difference between sham and vehicle controls. Observations indicate that there were major F1 generation toxicological effects from the indirect TCE exposure.
Fig. 6.
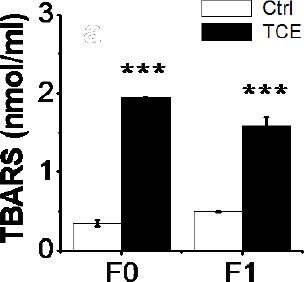
Oxidative stress biomarker TBARS levels (nmol/ml) in Ctrl and TCE F0 and F1 generations. Ctrl and TCE liver TBARS levels were examined with the mean ± SEM presented and asterisks (*) indicating a statistically significant difference (P ≤ 0.05) vs. TCE-treated mice. Student’s t-tests. ***P ≤ 0.001.
Protein Carbonylation and Nitric Oxide in the Livers of the F1 Generation
Protein carbonyl content is not only a biomarker of oxidative stress, but also provides evidence of oxidative protein damage (Morgan et al., 2005). To evaluate the extent of protein oxidation in liver, protein carbonyls in this major organ, which is targets of TCE, was also analyzed. Our data showed that carbonyl protein content in the liver was significantly increased in TCE F1 generation compared to that in control F1 generation (Fig. 7A), suggesting increased protein oxidation (carbonylation) as a result of indirect exposure to TCE.
Fig. 7.
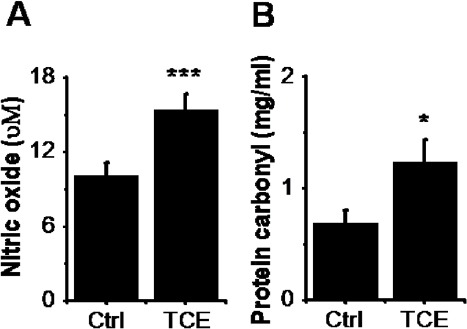
Oxidative stress biomarkers in Ctrl and TCE F1 generations. (A) Nitric oxide (μM) in the livers of Ctrl and TCE F1 generations. (B) Protein carbonyl content (mg/ml) in the liver of Ctrl and TCE F1 generation. Liver oxidative stress biomarkers were examined in Ctrl and TCE groups with the mean ± SEM presented and asterisks (*) indicating a statistically significant difference (P ≤ 0.05) vs. control. Student’s t-tests. *P ≤ 0.05, ***P ≤ 0.001.
Then we have assessed the status of inflammation in F1 generation livers by measuring nitric oxide levels in livers concerning markers of inflammation, levels of nitric oxide in livers were strongly increased in TCE F1 generation compared to that in control population F1 generation (Fig. 7B). These data correlated well with the increased oxidative lipid damage in F0 and F1 hepatic tissues. Observations indicate F0 generation parents exposed to TCE intragenerationally transmitted liver histopathology to their unexposed F1 generation descendants. Taken together, these observations indicate that maternal generation directly exposed to TCE transgenerationally transmitted liver abnormality to their unexposed F1 generation descendants.
Discussion
The present study demonstrates that maternal exposure to the environmental toxicant TCE promoted the transmission of abnormal liver phenotypes between generations (intragenerational inheritance) based on nontoxic pharmacological dose 0.1% of the oral LD50 for TCE. This study provides evidence of the activation and/or inhibition of different proteins involved in oxidative stress and cell damage in an animal model of a chemically induced hepatotoxicity. Liver histology, biomarkers of oxidative stress and nuclear alterations related to molecular and cellular mechanisms of hepatocyte cell damage were measured in control and TCE F0 and F1 generations. Oxidative stress biomarker levels (e.g. lipid peroxidation, protein carbonyl, and nitric oxide), indicative of hepatic tissue damage and hepatocyte cell injury, as well as hepatocellular apoptosis were significantly increased in the TCE F0 and F1 generations compared to that in control, reaching values similar to those previously reported in other models of progressive cirrhosis induced in rat by repeated injections of diethyl nitrosamine (DEN) (Guiu et al., 2012).
Many studies reported that exposure to environmental insults may promote the transition of abnormal phenotypes between generations (Jones et al., 1996; Anway et al., 2005). The results of this study support our findings from previous studies in mice (Al-Griw et al., 2015b) by examining a variety of different abnormalities states in four-month-old male mice and characterizing the transgenerational changes in the F1 generation sperms (Al-Griw et al., 2015a). Early exposure to TCE reduces fertility and negatively affects pregnancy outcomes across multiple generations (Al-Griw et al., 2015c). Alterations in sperm quality and testicular tissue architecture in the F1 generation offspring were observed after direct TCE exposure of the F0 generation parents. Other toxic effects of direct exposure to TCE included increase in relative liver weight and histopathological lesions (Xia and Yu, 1992; Al-Griw et al., 2016), weight loss (NTP, 2000; EPA, 2003), and kidney damage (NTP, 2000). Other studies showed evidence for the neurobehavioral and developmental teratogenicity of intermittent prenatal TCE exposure to high concentrations of TCE in rats (Jones et al., 1996; Coleman et al., 1999; Al-Griw et al., 2015b). Exposure of F0 generation gestating rats to environmental toxicant bisphenol-A (BPA) caused decreased fertility in F3 generation males (Salian et al., 2009). Vinclozolin exposure resulted in testis disease, prostate disease, kidney disease, immune system abnormalities, tumors, uterine hemorrhage during pregnancy and polycystic ovarian disease (Anway et al., 2005; Guerrero-Bosagna et al., 2012; Guerrero-Bosagna and Skinner, 2012; Nilsson et al., 2012); and alterations in the methylation patterns of imprinted genes in sperm of F3 generation male mice.
The transgenerational phenomena of toxic effects involve the transmission of abnormal phenotypes independently of direct exposure (Anway et al., 2006a, b). Two possible mechanisms induce abnormal phenotype through transgeneration of toxic effects, the first is alteration in genetic material structure such as DNA sequence change, the second one includes the changing in epigenetic information such as histone acetylation and methylation, or DNA methylation (Rakyan and Whitelaw, 2003; Skinner, 2007; Nadea, 2009). Animals studies show that parental exposure (F0 generation) to a variety of environmental factors can lead to observable effects (including both genetic and epigenetic) in the somatic cells of their offspring over several generations that are not attributable to the inheritance of a simple mutation through the parental germ line. Interestingly, we also demonstrate intragenerational inheritance of somatic (hepatocyte) effects induced by TCE in mice. This was accompanied by a significant decrease in the hepatocyte integrity. Perhaps, these findings indirectly support circumstantial evidence that hereditary information may transfer from soma to the germline. It has been reported that several environmental exposures can act on both somatic and germ cell genome replication and transcription, influencing the establishment and/or maintenance of specific epigenetic patterns (Guerrero-Bosagna and Skinner, 2012) that are transmitted to future generations in the absence of continued environmental exposures. The environmental exposures can cause heritable modifications (e.g. phenotypic variations) in the germline directly or indirectly through their primary effects on the soma (Jablonka, 2012; Sharma, 2013). Factors such as RNAs and hormones, including neurohormones and neuropeptides, have previously been considered to potentially mediate soma to germline communication in epigenetic inheritance (Jablonka, 2012). Cossetti et al. (2014) suggested that exosomes (extracellular vesicles) carrying various RNAs shed by somatic cells are carriers of flow of information from somatic cells to gametes (germ cells). It has been revealed that a flow of hereditary information can be transferred from the soma to the germline, escaping the principle of the Weismann barrier (Weismann, 1993) which postulates that somatically acquired genetic variations cannot be transferred to the germline (Cossetti et al., 2014). In consistent, we have demonstrated transgenerational inheritance of somatic effects induced by TCE in mice in the absence of continued TCE exposure. We have found that the vast majority of the livers in the F0 generation (parents) carried a hepatocyte (somatic) cell defect of increased apoptosis. This increased apoptosis persisted into the F1 generation (offspring), and in outcrossed offspring, exhibiting non-Mendelian genetic inheritance and affecting 90% of the mouse populations. Because the female F0-generation mice were directly exposed to the toxicant TCE before gestation, the F1-generation animals were not exposed directly. In addition, the germ cells present in the F0 generation were exposed directly to TCE. These exposed germ cells created the offspring (F1 generation). Therefore, the first generation without direct environmental exposure is the F1 generation, and this is the first generation said to exhibit transgenerational inheritance of disease susceptibility. Together, these findings suggest transgenerational inheritance of somatic effects.
In conclusion, environmental and/or occupational exposure to toxic substances (i.e. chemicals, pesticides, plastics, and heavy metals) continues to be a major worldwide public health concern. Many of these environmental toxicants are widely spread and difficult to degrade in the environment (Shi et al., 2008). It is estimated that approximately 24% of human diseases are caused by exposure to environmental toxicants (Hou et al., 2012); with the possibility of these diseases to be transmitted to future generations in the absence of direct exposure. In this study, we have shown that direct exposure of maternal generation to environmental toxicant TCE can promote inheritance of adult-onset liver abnormality. Associated with the occurrence of this transgenerational abnormality may be genetics/epigenetic alterations in mouse hepatocyte cell DNA. This transgenerational inheritance may be useful as early stage biomarkers of compound exposure and adult onset abnormality/disease. Although not designed for risk assessment, these findings have implications for the human populations that are exposed to a variety of toxicants. Especially to those who are experiencing declines in fertility and increases in adult onset abnormality/disease, with a potential to transmit them to future offspring generations. The degree that environmentally induced transgenerational inheritance of phenotypic variations and/or adult-onset disease is implicated in human disease etiology remains unknown. However, since the majority of chronic diseases have increased dramatically over the past decades, environmental exposures and transgenerational epigenetics will likely be a component of disease etiology to seriously consider in the future (Skinner et al., 2013). A more thorough and mechanistic understanding of the molecular etiology of disease, including the role of environmental epigenetics, is anticipated to provide insights into new diagnostics and therapeutics for specific diseases.
Conflict of interest
The authors declare that they have no competing interests.
Acknowledgements
This investigation was supported in part by the Division of Developmental Biology, Zoology Department, Faculty of Science, University of Tripoli, Tripoli, Libya.
References
- Aitken R.J, Koopman P, Lewis S.E.M. Seeds of concern. Nature. 2004;432:48–52. doi: 10.1038/432048a. [DOI] [PubMed] [Google Scholar]
- Al-Griw M.A, Al-Azreg S.A, Bennour E.M, El-Mahgiubi S.A.M, Al-Attar A.R, Salama N.M, Elnfati A.S. Fertility and Reproductive Outcome in Mice Following Trichloroethane (TCE) Exposure. Am. J. Life Sci. Res. 2015c;3:293–303. [Google Scholar]
- Al-Griw M.A, Al-Ghazeer R.O, Al-Azreg S.A, Bennour E.M. Cellular and Molecular Etiology of Hepatocyte Injury in a Murine Model of Environmentally Induced Liver Abnormality. Open Vet. J. 2016;6(3):150–157. doi: 10.4314/ovj.v6i3.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Griw M.A, Maamar M.S, Salama N.M, Algadi L.N, Elnfati A.S, Bennour E.M. Maternal Exposure of Mouse to Low-Dose of Trichloroethane is Associated with Increased Birth Weight and Early Neonatal Neurobehavioral abnormalities. Am. J. Biol. Life Sci. 2015b;3:206–210. [Google Scholar]
- Al-Griw M.A, Salama N.M, Treesh S.A, Elnfati A.H. Transgenerational Genetic Effect of Trichloroethane (TCE) on Phenotypic Variation of Acrosomal Proteolytic Enzyme and Male Infertility Risk. Int. J. Genet. Genomics. 2015a;3(5):43–49. [Google Scholar]
- Anway M.D, Cupp A.S, Uzumcu M, Skinner M.K. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Sci. 2005;308:1466–1469. doi: 10.1126/science.1108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anway M.D, Leathers C, Skinner M.K. Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease. Endocrinology. 2006a;147:5515–5523. doi: 10.1210/en.2006-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anway M.D, Memon M.A, Uzumcu M, Skinner M.K. Transgenerational effect of the endocrine disruptor vinclozolin on male spermatogenesis. J. Androl. 2006b;27:868–879. doi: 10.2164/jandrol.106.000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M.M. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Bruckner J.V, Kyle G.M, Luthra R, Acosta D, Mehta S.M, Sethuraman S, Muralidhara S. Acute, short-term, and subchronic oral toxicity of 1,1,1-trichloroethane in rats. Toxicol. Sci. 2001;60:363–372. doi: 10.1093/toxsci/60.2.363. [DOI] [PubMed] [Google Scholar]
- Coleman C.N, Mason T, Hooker E.P. Developmental effects of intermittent prenatal exposure to 1,1,1-trichloroethane in the rat. Neurotoxicol. Teratol. 1999;21:699–708. doi: 10.1016/s0892-0362(99)00035-5. [DOI] [PubMed] [Google Scholar]
- Cossetti C, Lugini L, Astrologo L, Saggio I, Fais S, Spadafora C. Soma-to-Germline Transmission of RNA in Mice Xenografted with Human Tumour Cells: Possible Transport by Exosomes. PLoS One. 2014;9:e101629. doi: 10.1371/journal.pone.0101629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews D, Gore A.C, Hsu T.S, Dangleben N.L, Spinetta M. transgenerational epigenetic imprints on mate preference. Proc. Natl. Acad. Sci. U. S. A. 2007;104:5942–5946. doi: 10.1073/pnas.0610410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easley I.V, Bradner J.M, Mosera A, Rickmana C.A, McEachina Z.T, Merritt M.M, Hansenc J.M, Caudle W.M. Assessing reproductive toxicity of two environmental toxicants with a novel in vitro human spermatogenic model. Stem Cell Res. 2015;14:347–355. doi: 10.1016/j.scr.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EPA U. S. interpretation of body weight data;Health Effects Division (HED) Guidance Document #G2003.01. Prepared by the HED Toxicology Science Advisory Council, Health Effects Division, Office of Pesticide Programs (OPP), July 1. 2003 [Google Scholar]
- Goa J. A Micro Biuret Method for Protein Determination Determination of Total Protein in Cerebrospinal Fluid. Scand. J. Clin. Lab. Invest. 1953;5:218–222. doi: 10.3109/00365515309094189. [DOI] [PubMed] [Google Scholar]
- Greer E.L, Maures T.J, Ucar D, Hauswirth A.G, Mancini E. transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature. 2011;479:365–371. doi: 10.1038/nature10572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Bosagna C, Covert T, Haque M.M, Settles M, Nilsson E.E. Epigenetic Transgenerational Inheritance of Vinclozolin Induced Mouse Adult Onset Disease and Associated Sperm Epigenome Biomarkers. Reprod. Toxicol. 2012;34:694–707. doi: 10.1016/j.reprotox.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Bosagna C, Skinner M.K. Environmentally induced epigenetic transgenerational inheritance of phenotype and disease. Mol. Cell Endocrinol. 2012;354:3–8. doi: 10.1016/j.mce.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiu B, Deschamps F, Boulin M, Boige V, Malka D, Ducreux M, Hillon P, de Baère T. Serum gamma-glutamyl-transferase indepen-dently predicts outcome after transarterial chemoembolization ofhepatocellular carcinoma: external validation. Cardiovasc. Intervent. Radiol. 2012;35:1102–1108. doi: 10.1007/s00270-011-0293-9. [DOI] [PubMed] [Google Scholar]
- Gujral J.S, Bucci T.J, Farhood A, Jaeschke H. mechanism of Cell Death During Warm Hepatic Ischemia-Reperfusion in Rats: Apoptosis or Necrosis? Histol. 2001;33:397–405. doi: 10.1053/jhep.2001.22002. [DOI] [PubMed] [Google Scholar]
- Gujral J.S, Knight T.R, Farhood A, Bajt M.L, Jaeschke H. mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol. Sci. 2002;67:322–328. doi: 10.1093/toxsci/67.2.322. [DOI] [PubMed] [Google Scholar]
- Henderson I.R, Jacobsen S.E. Epigenetic inheritance in plants. Nature. 2007;447:418–424. doi: 10.1038/nature05917. [DOI] [PubMed] [Google Scholar]
- Hou L, Zhang X, Wang D, Baccarelli A. Environmental chemical exposures and human epigenetics. Int. J. Epidemiol. 2012;41:79–105. doi: 10.1093/ije/dyr154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonka E. epigenetic inheritance and plasticity: the responsive germline. Prog. Biophys. Mol. Biol. 2012;111(2-3):99–107. doi: 10.1016/j.pbiomolbio.2012.08.014. [DOI] [PubMed] [Google Scholar]
- Jones H.E, Kunko P.M, Robinson S.E. Developmental consequences of intermittent and continuous prenatal exposure to 1,1,1-trichloroethane in mice. Pharmacol. Biochem. Behavior. 1996;55:635–646. doi: 10.1016/s0091-3057(96)00288-2. [DOI] [PubMed] [Google Scholar]
- Kelly W.G. Transgenerational epigenetics in the germline cycle of Caenorhabditis elegans. Epigenetics Chromatin. 2014;7:1–17. doi: 10.1186/1756-8935-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manikkam M, Guerrero-Bosagna C, Tracey R, Haque M.M, Skinner M.K. Transgenerational actions of environmental compounds on reproductive disease and identification of epigenetic biomarkers of ancestral exposures. PLoS One. 2012;7:e31901. doi: 10.1371/journal.pone.0031901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan P.E, Sturgess A.D, Davies M.J. Increased levels of serum protein oxidation and correlation with disease activity in systemic lupus erythematosus. Arthritis Rheum. 2005;52:2069–2079. doi: 10.1002/art.21130. [DOI] [PubMed] [Google Scholar]
- Nadea J.H. Transgenerational genetic effects on phenotypic variation and disease risk. Hum. Mol. Genet. 2009;18:R202–R210. doi: 10.1093/hmg/ddp366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson E, Larsen G, Manikkam M, Guerrero-Bosagna C, Savenkova M. Environmentally Induced Epigenetic Transgenerational Inheritance of Ovarian Disease. PLoS One. 2012;7:e36129. doi: 10.1371/journal.pone.0036129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson E.E, Skinner M.K. Transgenerational Epigenetics. Pullman, WA, USA: Washington State University; 2014. Definition of Epigenetic Transgenerational Inheritance and Biological Impacts; pp. 11–16. [Google Scholar]
- Nilsson E.E, Skinner M.K. Environmentally induced epigenetic transgenerational inheritance of disease susceptibility. Transl. Res. 2015;165:12–17. doi: 10.1016/j.trsl.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NTP. NTP technical report on the toxicity studies of 1,1,1-trichloroethane (CAS no.71-55-6) administered in microcapsules in feed to F344/N rats and B6C3F1 mice. Public Health Service, U.S. Department of Health and Human Services;NTP Toxicity Report Series. No. 41. National Institute of Environmental Health Sciences, Research Triangle Park, NC, and National Technical Information Service, Springfield, VA; PB2001-100. 2000. p. 476. online: http://ntp.niehs.nih.gov/ntp/htdocs/ST_rpts/tox041.pdf .
- Pembrey M.E. Male-line transgenerational responses in humans. Hum. Fertil. (Camb) 2010;13:268–271. doi: 10.3109/14647273.2010.524721. [DOI] [PubMed] [Google Scholar]
- Rakyan V, Whitelaw E. transgenerational epigenetic inheritance. Curr. Biol. 2003;13:R6. doi: 10.1016/s0960-9822(02)01377-5. [DOI] [PubMed] [Google Scholar]
- Ruden D.M, Lu X. hsp90 affecting chromatin remodeling might explain transgenerational epigenetic inheritance in Drosophila. Curr. Genomics. 2008;9:500–508. doi: 10.2174/138920208786241207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salian S, Doshi T, Vanage G. Impairment in protein expression profile of testicular steroid receptor coregulators in male rat offspring perinatally exposed to Bisphenol A. Life Sci. 2009;85:11–18. doi: 10.1016/j.lfs.2009.04.005. [DOI] [PubMed] [Google Scholar]
- Sharma A. transgenerational epigenetic inheritance: Focus on soma to germline information transfer. Prog. Biophys. Mol. Biol. 2013;113:439–446. doi: 10.1016/j.pbiomolbio.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Shi X, Zhou S, Wang Z, Zhou Z, Wang Z. CYP1A1 and GSTM1 polymorphisms and lung cancer risk in Chinese populations: A meta-analysis. Lung Cancer. 2008;59:155–163. doi: 10.1016/j.lungcan.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Skinner M.K. Endocrine disruptors and epigenetic transgenerational disease etiology. Pediatr. Res. 2007;61:48R–50R. doi: 10.1203/pdr.0b013e3180457671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.K, Guerrero-Bosagna C. environmental signals and transgenerational epigenetics. Epigonomics. 2009;1:111–117. doi: 10.2217/epi.09.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.K, Manikkam M, Guerrero-Bosagna C. epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol. Metab. 2010;21:214–222. doi: 10.1016/j.tem.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.K, Manikkam M, Tracey R, Guerrero-Bosagna C, Haque M, Nilsson E.E. Ancestral dichlorodiphenyltrichloroethane (DDT) exposure promotes epigenetic transgenerational inheritance of obesity. BMC Medicine. 2013;11:1–16. doi: 10.1186/1741-7015-11-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Wang J, Ma H, Ansari G.A.S, Khan M.F. N-Acetylcysteine protects against trichloroethene-mediated autoimmunity by attenuating oxidative stress. Toxicol. Appl. Pharmacol. 2013;273:189–195. doi: 10.1016/j.taap.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weismann A. the germ-plasm: a theory of heredity Charles Scribner’s Sons. New York, USA: Electronic Scholarly Publishing; 1993. [Google Scholar]
- Wong E.W. Cell junctions in the testis as targets for toxicants. In: Richburg JH, Hoyer P, editors. Comprehesive toxicology. Academic Press; 2010. pp. 167–188. [Google Scholar]
- Xia L, Yu T. A study of the relationship between hepatotoxicity and free radical induced by 1,1,2-trichloroethane and l,1,1-trichloroethane in rat. Biomed. Environ. Sci. 1992;5:303–313. [PubMed] [Google Scholar]
- Xu Y, Zhao H, Zhang M, Li C.J, Lin X.Z, Sheng J, Shi W. Variations of antioxidant properties and NO scavenging abilities during fermentation of tea. Int. J. Mol. Sci. 2011;12:4574–4590. doi: 10.3390/ijms12074574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.T, Zheng Q.S, Pan J, Zheng R.L. Oxidative damage of biomolecules in mouse liver induced by morphine and protected by antioxidants. Basic Clin. Pharmacol. Toxicol. 2004;95:53–58. doi: 10.1111/j.1742-7843.2004.950202.x. [DOI] [PubMed] [Google Scholar]