

Abstract
Through an shRNA screen we identified the protein arginine methyltransferase Prmt1 as a vulnerable intervention point in murine p53/Rb-null osteosarcomas, the human counterpart of which lacks effective therapeutic options. Depletion of Prmt1 in p53-deficient cells impaired tumor initiation and maintenance in vitro and in vivo. Mechanistic studies reveal that translation-associated pathways were enriched for Prmt1 downstream targets, implicating Prmt1 in translation control. In particular, loss of Prmt1 led to a decrease in arginine methylation of the translation initiation complex, thereby disrupting its assembly and inhibiting translation. p53/Rb-null cells were sensitive to p53-induced translation stress, and analysis of human cancer cell line data from Project Achilles further revealed that Prmt1 and translation-associated pathways converged on the same functional networks. We propose that targeted therapy against Prmt1 and its associated translation-related pathways offer a mechanistic rationale for treatment of osteosarcomas and other cancers that exhibit dependencies on translation stress response.
Keywords: Prmt1, translation, eIF4G1, p53, arginine methylation
INTRODUCTION
Eukaryotic translation initiation involves the assembly of translation initiation factors, eIF4G eIF4E, and eIF4A (termed eIF4F) on the mRNA. eIF4F interacts with other factors including eIF4B, PABP, and eIF3 to recruit a 43S ribosome complex to the mRNA to initiate translation. Translation initiation is rate-limiting and tightly controlled by multiple mechanisms, including the phosphorylation of eIF2 and/or hypophosphorylation of 4EBPs, among others. Under stress conditions, global protein synthesis is repressed to conserve energy, while selective translation of specific repair or prosurvival programs becomes active. Translation regulation of protein expression allows for an immediate response to stress imposed by the environment (1,2), thereby ensuring cell survival.
Cancer cells sustain the tumorigenic state by adapting to stress imposed by many hallmarks of cancer(3). An increasing number of studies suggest that cancer cells hijack mRNA translation machinery and selectively reactivate translation of cancer-associated genes to drive tumorigenesis and maintain tumor-associated phenotypes(2). Deregulation of ribosomal proteins and translation initiation factors, including the eIF4F complex, among others, have been shown to contribute to tumor development(4,5). Oncogenic signaling controlled by Myc, Ras, and PI3K-mTOR or loss of tumor suppressors such as PTEN and TP53 can lead to aberrant translation, thereby promoting selective translation of mRNAs that support oncogenesis(5,6). Given that translation deregulation is a critical nexus of cancer development, progression, and drug resistance, understanding the molecular underpinnings of translation control may reveal new therapeutic opportunities for the development of more effective anti-cancer drugs.
Inactivation of p53 and Rb tumor suppressors is frequently observed in many human cancers, including the most common pediatric bone cancer, osteosarcoma (OS). Studies of familial cancer syndromes indicate that germ-line p53/Rb mutations predispose individuals to develop OS among other cancer types. To investigate the underlying molecular mechanisms of OS development driven by p53 and/or Rb loss, we and others have developed an OS mouse model based on deletion of p53 and Rb in osteoblastic progenitor cells(7,8). The model confirms the requirement of p53 in OS tumor initiation and progression, and shows that combined deletion of p53 and Rb accelerates the disease. Furthermore, the model offers tumor-derived cell line-based platforms for genetic vulnerability screens. Indeed, using an shRNA-based genome-wide screening strategy, we have previously reported that the PI3K-mTOR pathway represents a vulnerability for p53/Rb-deficient cells(9). Given that p53 and PI3K-mTOR pathways feed into translation, we hypothesize that targeting translation factors and its regulators may effectively eradicate p53/Rb-deficient tumors.
Although phosphorylation of some components of the translation machinery has been widely recognized, emerging data suggest that protein arginine methylation of translation factors and their interacting partners occurs in certain cellular contexts, implicating a role of arginine methylation in translation regulation(10–13). Prmt1 is one of the major protein arginine methyltransferases that catalyzes mono-methylation and asymmetric dimethylation of arginine bearing substrates, including histones, estrogen receptor, RNA binding proteins, and numerous other non-histone substrates(14,15). Thus, Prmt1 is involved in regulating a wide range of cellular processes, including transcription, RNA processing, DNA damage response, and signal transduction. Aberrant expression of Prmt1 has been reported in several malignancies(15); however, how altered Prmt1 expression contributes to oncogenesis remains incompletely understood. Finally, Prmt1 or arginine methylation of its major substrate H4R3 correlates with poor clinical outcome in some cancers (16–18), suggesting that anti-cancer strategies designed to target Prmt1 or Prmt1-associated pathways may hold therapeutic potential.
Building on our previous work on identifying vulnerabilities of cancer cells, we have discovered that p53/Rb-deficient mOS cells are highly sensitive to translation inhibition. Among the top candidates identified by loss-of-function screens, we have identified Prmt1 as a vulnerability of p53/Rb-null mOS cells and a cooperating oncogenic driver of tumor initiation. Of particular note, quantitative proteomics analysis of substrates regulated by Prmt1 implicates its involvement in translation control. Further mechanistic studies reveal that Prmt1 regulates global translation at transcription, translation, and post-translation levels. Our work suggests that p53 and Prmt1 signaling pathways converge on translation regulation. Analysis of the Project Achilles dataset also implicates a correlation between Prmt1 dependency, translation-associated dependencies, and p53 status in human cancer cells. Taken together, these findings provide a rationale for targeting Prmt1 and translation-associated pathways in cancers that exhibit dependency on translation-stress response for survival.
MATERIALS AND METHODS
OS cell culture
Derivation and genotyping of the mOS cell lines from p53 and Rb conditional knockout mice were described previously(7,9). mOS cells were derived from tumor-bearing mice between 2010–2015 and tested for mycoplasma by PCR. Early passages (passages < 20) of mOS cells were used in the study between 2012-2016. Prmt1f/f mOS cell lines were derived from tumors, genotyped, and cultured in αMEM supplemented with 10% FBS, L-glutamine, and penicillin-streptomycin between 2012-2016. mOS cells to be used for SILAC experiments were cultured in light or heavy-MEM media for SILAC prepared according to manufacturer’s protocol (Thermo Scientific) between 2013-2016. All mOS cell lines were authenticated using genotyping protocols described here and by Walkley et.al(7). Human cancer cell lines were obtained from ATCC and cultured in DMEM supplemented with 10% FBS between 2012-2016. MC3T3-E1 (subclones 4 and 30) cells were purchased from ATCC between 2012-2016 and maintained in αMEM supplemented with 10% FBS, 1mM sodium pyruvate, L-glutamine, and penicillin-streptomycin. All cell lines from ATCC, including the MC3T3-E1 cells were tested for mycoplasma by PCR upon receipt between 2012-2016. All ATCC cell lines used in this study were early passage cells (passages <10) and exhibited the original morphology characterized by ATCC. We verified p53 and Rb protein expression by western blot. No additional authentication was performed.
Mice
Prmt1-null OS Model
Prmt1 conditional knockout mice were generated by blastocyst injection from Prmt1fl/fl ES cells imported from EUCOMM between 2009-2010. Prmt1f/f mice were crossed with Osx-Cre+ p53f/f, Rbf/f mice to generate Osx-Cre+ p53f/f, Rbf/f, Prmt1f/f mice.
Xenograft studies
OS cells were infected with virus and selected with puromycin (puro) for two days. Three days post-puro selection, 1 × 106 murine OS were injected subcutaneously into the flanks of NCRNU-M mice (Taconic). Tumor volume was measured using a caliper. The estimated volume in mm3 was calculated using the following formula: length × width × height. All proposed animal experiments were approved by the Boston Children’s Hospital (BCH) Institutional Animal Care and Use Committee.
Viral packaging, plasmids and cloning
Production of lenti-virus was carried out according to Luo et al.(19). Retroviral virus was produced using Platimun-E Retroviral Packaging Cell Line(20).
The following lenti-shRNA constructs were obtained from the Broad Institute of MIT and Harvard: TRCN0000072261 (Luciferase), TRCN0000018493 (Prmt1 #1), TRCN0000018492 (Prmt1 #2), TRCN0000316215 (Eif4g1 #1), TRCN0000096809 (Eif4g1 #2), TRCN0000310243 (PRMT1#1), TRCN0000035929 (PRMT1 #2), pLKO-TRC2 control vector was purchased from Sigma. MSCV-CreERT2 was a gift from Tyler Jacks (Addgene plasmid #22776) and pInducer20 was a gift from Stephen Elledge (Addgene plasmid # 44012). We purchased pENTR11 vector from Thermo Fisher Scientific.
Prmt1 and Eif4g1 were amplified from murine or human OS cDNAs using PCR primers that contain FLAG or HA tag sequences. The PCR products were cloned into pENTR11 vector and sequenced verified. Prmt1/Eif4g1 cDNAs were subsequently moved into pInducer20 using the Gateway cloning system (Thermo Fisher Scientific). The E144Q or the G80R Prmt1 mutants(21,22) and the R689A eIF4G1 mutants were generated using the Quickchange II XL site-directed mutagenesis kit (Stratagene), using their corresponding wild-type pENTR11-Prmt1/PRMT1/Eif4g1 plasmids as templates. The resulting pENTR11-Prmt1/Eif4g1 mutant clones were sequenced verified and moved into pInducer20 using the Gateway system.
shRNA screens
In vitro and in vivo shRNA screens were described previously (9). shRNA rankings were computed using the second best rank statistical method by RIGER-E(19). For the in vivo screen, the following shRNA clones targeting Prmt1 and control genes were used to generate the pooled lenti-virus for the screen: Prmt1 (TRCN0000274537, TRCN0000274580, TRCN0000018492), RFP (TRCN0000072209), GFP (TRCN0000072197), LacZ (TRCN0000072232), Luciferase (TRCN0000072253), and pLKO-empty (TRCN0000208001).
Immunoprecipitation
Whole cell lysates were prepared using NP-40 buffer (20mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA pH 8.0, 1% NP-40) supplemented with protease and phosphatase inhibitors (Roche). Equal amounts of protein for each condition were incubated with the indicated primary antibodies and Dynabeads Protein G (Thermo Fisher) overnight at 4°C. The samples were washed in NP-40 buffer 3 times, followed by protein elution using the manufacture’s protocol.
Western Blot
Whole cell lysates were prepared using RIPA buffer (150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50mM Tris, pH 8.0) supplemented with protease and phosphatase inhibitors (Roche). The amounts of proteins were quantified using DC™ Protein Assay (BioRad). Equal amounts of proteins were separated by SDS-PAGE and transferred to a PVDF membrane. Membrane were blocked in 5% Milk in TBST (Tris-Buffered Saline pH 7.6, 0.1 % Tween 20), followed by incubation with the indicated antibodies.
Antibodies and reagents
The following antibodies were obtained from Cell Signaling Technology: Mono-Methyl Arginine (R*GG; D5A12; #8711), Mono-Methyl Arginine (Me-R4-100; #8015), eIF4G1 (#2858), eIF4G1 (D6A6; #8701), eIF4E (C46H6; #2067), Phospho-eIF4E (Ser209; #9741), eIF4A (C32B4; #2013), eIF2alpha (D7D3; #5324), phospho-eIF2alpha (Ser51; #3398), PABP1 (#4992), GAPDH (14C10; #2118), HA-Tag (6E2; HRP Conjugate), mouse anti-rabbit IgG conformation specific (#5127), and rabbit IgG isotype control (#3900). We purchased Prmt1 antibodies from Millipore (#07-404) and Abcam (ab73246 and ab7027). p53 (#554147) and p21 (sc-6246) antibody were purchased from BD Pharmingen and Sana Cruz Biotechnology, Inc., respectively. Anti-GFP (ab6556) was obtained from Abcam. Anti-FLAG M2 antibody (F1804), Anti-FLAG M2 affinity gel (A2220), doxycycline (D9891) and 4-hydroxytamoxifen (H7904) were purchased from Sigma. We obtained cycloheximide (#239763) and G418 from Calbiochem and Corning, respectively.
Quantitative Proteomics
Enrichment of methyl-arginine containing peptides
Cell pellets (~ 200 μL packed cell volume) from SILAC-equilibrated cultures were re-suspended in 2 mL of lysis buffer (150 mM NaCl, 50 mM Tris pH 7.5, 10% Glycerol, 0.1% RapiGest) containing protease inhibitors (Roche) and benzonase (Sigma) and lysed end over end for 30 minutes at 4°C. Cellular debris were pelleted for 10 minutes at 14.000 rpm at 4°C. 5 mg of protein from each sample (shControl and shPrmt1) were combined. Cysteine residues were reduced for 30 minutes at 56°C with 10 mM dithiothreitol and alkylated for 20 minutes in the dark at room temperature with 22.5 mM iodoacetamide. Proteins were digested overnight at 37°C with 100 μg of trypsin. Digestion was continued for another 6 hours after adding an additional 100 μg of trypsin. Complete digestion was confirmed by analyzing a 4 μg protein-equivalent aliquot by PAGE followed by silver staining. Tryptic peptides were purified by reverse phase chromatography on a 100 mg SepPak tC18 96-well plate (Waters) and solubilized in 1 mL of immuno-affinity purification buffer (IAP buffer: 50 mM MOPS/NaOH, 10 mM Na2HPO4, 50 mM NaCl). Peptides containing arginine residues modified by mono-methylation, symmetric and asymmetric di-methylation were enriched by sequential immuno-affinity purification using 40 μL of anti-methyl arginine antibody-agarose bead conjugates for 2 hours at 4°C (PTMScan, Cell Signaling Technology). Immuno-precipitates were washed 3 times with 500 μL of IAP buffer, 3 times with 500 μL of water and eluted with 55 μL of 0.15% TFA for 10 minutes at room temperature. Enriched methylated peptides were purified by batch mode reverse phase chromatography (Poros 50 R2, AB Sciex) and re-digested for 2 hours at 37°C with 500 ng of trypsin in 20 μL of 5% acetonitrile, 50mM ammonium bicarbonate. Re-digested peptides were purified by batch mode reverse phase chromatography and vacuum concentrated before LC-MS/MS analysis.
Protein normalization
A small aliquot of the supernatant remaining after the final immuno-affinity purification step was desalted on SOLA plate (Thermo) and used to quantify total protein in samples treated with control and Prmt1 shRNAs.
Enrichment of endogenous eIF4G1
Cell pellets (~ 50 μL packed volume) from SILAC-equilibrated 1369 cultures (“Light”: + 4-hydroxy Tamoxifen, Prmt1-depleted; “Heavy”: + Ethanol, no depletion control) were re-suspended in 250 μL of lysis buffer (180 mM NaCl, 50 mM Tris pH 7.5, 10% Glycerol, 0.5% Igepal) containing protease inhibitors (Roche) and phosphatase inhibitor cocktail III (Sigma) and lysed on a vortex mixer for 30 minutes at 4°C. Cellular debris were pelleted for 10 minutes at 20,000 × g at 4°C and an equivalent amount of soluble proteins from each sample (shControl and shPrmt1) were combined. The sample was pre-cleared for 3 hours at 4°C using 20 μL (50% slurry) of Protein-A Sepharose beads (GE healthcare). Endogenous eIF4G1 was purified overnight at 4°C using 10 μL of anti-eIF4G1 antibody (Cell Signaling Technology) and precipitated using 20 μL on Protein-A Sepharose beads. Beads were washed three times using 500 μL of lysis buffer with Igepal reduced to 0.05% and once with water. Following disulfide bond reduction and cysteine alkylation with iodoacetamide, enriched eIF4G1 was directly digested on beads using 5 μg of trypsin, followed by LC-MS/MS analysis. A more detailed description of LC-MS/MS analyses and data processing is delineated in the supplementary method.
Prmt1f/f genotype & excision PCR primers:
A:5-′CTTGCCACACAAGCAGAAAG-3′
B:5′-GGAGTAAGCAGACAGCCGAG-3′
C:5′-TGTGTGGGAACTGTAGAGCG-3′
qRT-PCR Primers:
Human PRMT1 F: 5′-CTTCTCACTGCTTTCCGCCT-3′
Human PRMT1 R: 5′-GCCGCGAACTGCATCAT-3′
Human GAPDH F: 5′- AAT GAAGGGGTCATTGATGG-3′
Human GAPDH R: 5′-AAGGTGAAGGTCGGAGTCAA-3′
Immunohistochemistry
Bone tissues were prepared as described previously(7,23). Deparaffinized tissue sections were subjected to antigen retrieval in Biocare’s Decloaking Chamber (pH 6). The section was then incubated with primary Prmt1 antibody (ab73246; 1:200) for 1 hour at RT. Protein detection was performed using Dako’s Envision kit. For detection of GFP (ab6556), similar procedure was employed as described above, except that the antigen retrieval step was performed at pH 8 instead.
Proliferation & cell cycle analysis
1000-2000 cells were seeded in triplicates onto multiple 96-well plate or 24-well plates. Proliferation/viability was assessed by CellTiter-Glo Luminescent Cell Viability Assay (Promega) or by crystal violet staining (Sigma). For growth curves, relative luminescence unit (RLU) was calculated at various time points by normalizing to the luminescence value at day 0 (20 hours post-plating). For cell cycle analysis, OS cells were incubated with BrdU for 1 hour prior to collection and stained according to manufacturer’s protocol using the BrdU Flow Kit (BD Pharmingen).
Polysome profiling
10 million cells were grown to 70-80% confluence in multiple 15 cm2 plates. Cells were washed once with 5ml 100mg/ml cyclohexamide/PBS and scraped into 1ml 100μg/ml cyclohexamide/PBS. Next, cells were pelleted for 10 minutes, at 300xg, 4˚C and then resuspended in 300μl polysome lysis buffer (5mM Tris pH 7.4, 2.4 mM MgCl2, 1.5mM KCl, 2μM DTT, 0.5% Triton-X, 0.5% sodium deoxycholate and 1x cOmplete protease inhibitor (Roche)). Lysates were cleared for 2 minutes at 12000xg, 4˚C. 50μl of cleared lysate was retained for total RNA extraction and 250μl of cleared lysate was loaded onto 12ml 10-50% sucrose gradients (prepared in 15mM Tris pH 7.4, 15mM MgCl2, 150mM NaCl) and spun 2 hours at 40,000rpm, 4˚C on an SW40Ti rotor (Beckman Coulter). 0.5ml fractions were collected immediately following centrifugation, using a BioComp Gradient Master instrument.
RNA-seq Analysis
RNAs from cytoplasmic, pooled light (<3 ribosomes) polysome fractions, and pooled heavy (>3 ribosomes) polysome fractions were precipitated using Trizol-LS (Thermo Scientific). 500 μg of RNAs were used to prepare cDNA libraries using the IIlumina Truseq v2 kit. The libraries were quantified and sequenced using Illumina Nextseq sequencer.
The fastq files were aligned to mm10 using STAR (24) with default parameters, followed by counting the aligned reads in the genomic transcripts annotations from GenomicFeatures (25) using Rsamtools (26).The differentially expressed gene analysis was performed using DESeq (27), with the threshold adjusted p-value <0.05, fold-change >1.5. The change in transcriptional activity due to Prmt1 KO was presented as the log2 fold-change of the gene expression in total mRNAs isolated from the KO as compared to the control. Enrichment of gene sets were performed using the default GSEAPreranked algorithm, GO gene sets, and hallmark gene sets(28).
The change in translational activity due to Prmt1 KO was quantified as the ratio between the efficiently translated (associated with heavy polysomes) and the poorly translated (associated with light polysomes) mRNAs. Translation efficiency was defined as the difference between the Log2 ratios of the KO and the control. Transcripts that were down or up regulated by more than 1.5 fold were analyzed to determine any functional enrichment using DAVID bioinformatics Resources 6.8.(29).
The Gene Expression Omnibus (GEO) accession number for the RNA-seq data reported in this article is GSE93309.
Analysis of Project Achilles dataset
The PRMT1 gene dependency scores for 501 cell lines were used to rank all gene dependencies from the Achilles v2.20.1 dataset using the RNMI (rescaled normalized mutual information) metric in the PARIS algorithm (30,31). The ranked list of gene dependencies most to least associated with PRMT1 dependency was subsequently analyzed using Gene Set Enrichment Analysis (GSEA v2.2.2, http://www.broadinstitute.org/gsea/) and the c5 genesets (GO terms) from MSigDb (28). GSEA was run using default settings, using the ‘GseaPreranked’ tool, except that data was not collapsed to gene symbols (this was done prior to GSEA). ATLANTIS model is described in detail in the publication by Tsherniak A. et al.(31)
RESULTS
Genome-scale shRNA screening in vitro reveals vulnerability of p53/Rb-null OS cells to Prmt1 inhibition
To discover potential vulnerabilities of p53/Rb-null cancer cells, we previously performed a genome-scale shRNA screen in vitro using p53/Rb-null murine tumor derived OS cells (9). The screen surveyed approximately 8000 genes for their roles in p53/Rb-null cell proliferation. Many of the top ranking candidates represent pathways associated with translation, development, cell cycle, and adhesion. This led us to speculate that p53/Rb-null cells may rely on one or more of these pathways for survival.
To validate the screen, we focused our investigation on Prmt1, which was one of the top 50 candidates. We found that Prmt1-targeting shRNAs were significantly depleted upon growth of p53/Rb-null cells, implicating Prmt1 in proliferation or cell survival in vitro (Figure 1A). We verified the specificity of the Prmt1-targeting shRNAs and their effects on p53/Rb-null cell proliferation by an shRNA-mediated knockdown strategy. Compared with the control, independent Prmt1-targeting shRNAs depleted >80 % of Prmt1 protein level and was accompanied by growth arrest (Figure 1B and 1C). Consistent with this observation, cell cycle analysis showed that depletion of Prmt1 led to an increase in the percentage of apoptotic (sub G0) cells, while the percentage of proliferating cells (S-phase) was significantly decreased in Prmt1 knockdown cells (Figure 1D). Both p53/Rb-null and p53-null/Rb-wt mOS cells were sensitive to Prmt1 depletion, suggesting that Rb tumor suppressor protein does not play a major role in conferring Prmt1 resistance. Similarly, depletion of Prmt1 in human OS also led to growth arrest and death (Supplementary Figures 1A and 1B).
Figure 1. shRNA screening identifies Prmt1 as an essential gene for tumor-derived p53-deficient mOS cells.
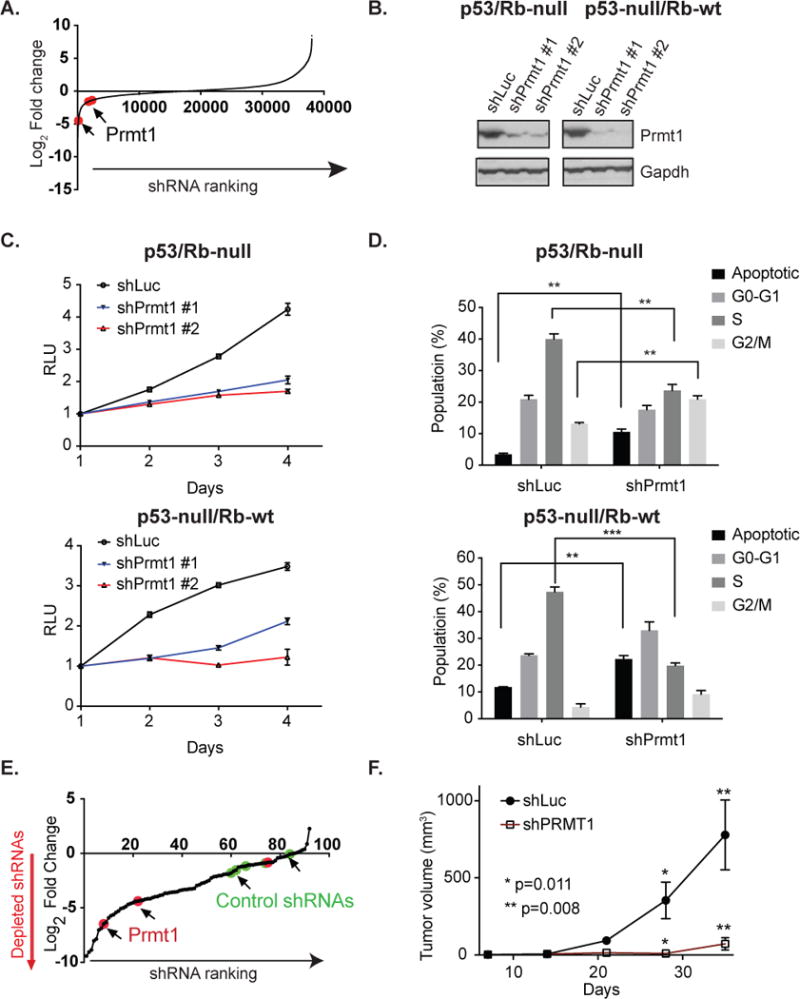
(A) Log2 fold change in shRNA abundance for p53/Rb-null mOS cell line at the end of the genome-scale in vitro shRNA screen relative to the initiatial reference pool. Prmt1-targeting shRNAs are highlighted in red. (B) Western blot analysis of Prmt1 expression in control (shLuc) and Prmt1 knockdown p53/Rb-null and p53-null/Rb-wt mOS cells. (C) Proliferation of p53/Rb-null and p53-null/Rb-wt mOS cell lines infected with non-targeting shRNA (shLuc) and Prmt1-targeting shRNAs. (D) Cell cycle analysis of p53/Rb-null and p53-null/Rb-wt mOS cells infected with control and Prmt1-targeting shRNA. The mean and standard deviation of triplicate samples are shown and t-tests were performed to determine the statistical significance between samples. ** p<0.01. *** p<0.001. (E) Log2 fold change in shRNA abundance for mOS xenografts relative to the initiation reference pool. shRNAs targeting Prmt1 are highlighted in red, while non-targeting control shRNAs are highlighted in green. (F) Growth of p53/Rb-null xenografts established using control shRNA (n=8) and Prmt1-targeting shRNA (n=8) infected mOS cells. The data are represented as mean + s.e.m. P values for the last time points are shown.
Prmt1 is essential for tumorigenicity of p53/Rb-null OS cells in vivo
To assess the dependence of in vivo tumor formation on Prmt1, we employed a pooled, shRNA screening approach in vivo to assess multiple shRNAs for their role in tumor formation. Specifically, we introduced a pool of Prmt1-targeting and control shRNAs into p53/Rb-null mOS cells, which were then used to establish tumor xenografts. In confirmation of the in vitro results, we found that the majority of Prmt1 shRNAs were depleted in tumors as compared to the control shRNAs (Figure 1E). To validate the in vivo screen, we infected p53/Rb-null mOS cells with Prmt1-targeting or control shRNAs. Knockdown of Prmt1 impaired murine xenograft formation, supporting a role of Prmt1 in promoting in vivo tumorigenicity (Figure 1F).
Prmt1 is required for tumor initiation in p53/Rb-null OS mice
Deletion of p53 and Rb leads to OS development with high penetrance in mice (7,8). To assess if Prmt1 expression is required for OS initiation, we generated Osx-Cre+; p53flox/flox; Rbflox/flox; Prmt1flox/flox conditional knockout (hereinafter, Prmt1f/f OS) to permit combined inactivation of p53, Rb, and Prmt1 in committed osteoblast progenitor cells. We confirmed that depletion of Prmt1 in Osx+ (GFP+) osteoblasts (Supplementary Figure 2A). Prmt1f/f OS mice developed normally. MicroPET/CT imaging showed that Prmt1wt and Prmt1f/+ OS mice had earlier tumor onset than Prmt1f/f OS mice (Supplementary Figure 2B). Indeed, deletion of Prmt1 significantly extended the lifespan of the mice (Figure 2A). Heterozygous loss of Prmt1 did not extend survival, as compared to wild-type mice. These findings indicate that loss of Prmt1 inhibits tumor development driven by p53/Rb loss.
Figure 2. Prmt1 is required for tumor initiation driven by p53/Rb loss.
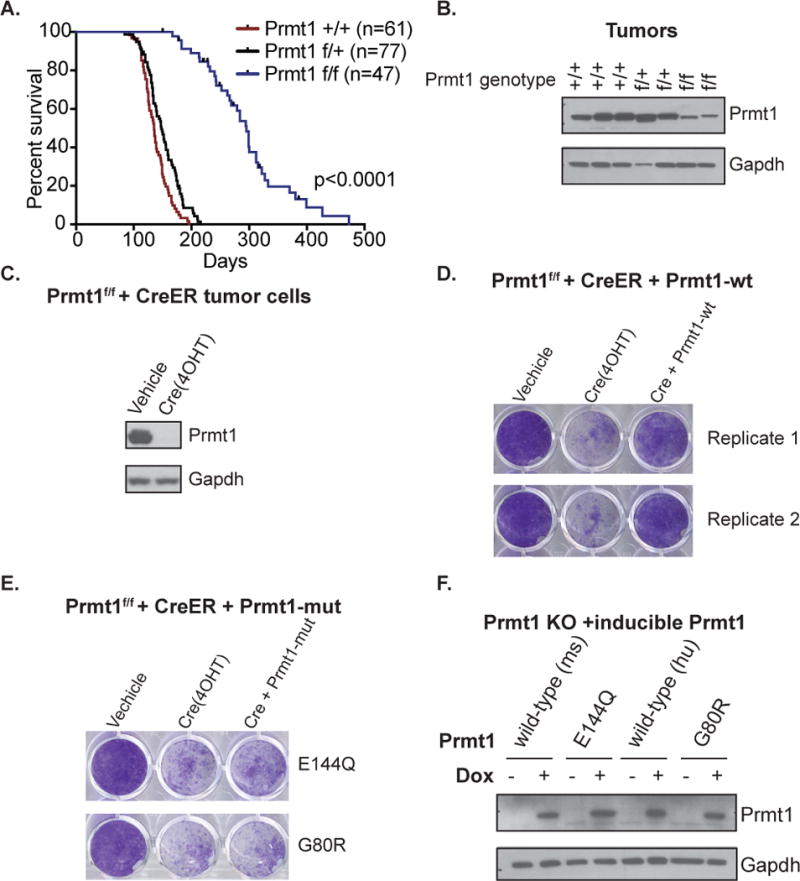
(A) Conditional knockout Prmt1f/f mice were crossed to OsxCre+, p53f/f; Rbf/f to generate OsxCre+, p53f/f; Rbf/f; Prmt1f/f mice. Kaplan-Meier survival plot and summary of median survival of Prmt1+/+, Prmt1+/f, and Prmt1f/f OS mice. Logrank test was performed for comparison. *p<0.0001 (B) Western blot analysis of Prmt1 protein expression in Prmt1+/+, Prmt1 +/f, and Prmt1f/f p53/Rb-null OS tumors. Gapdh expression serves as the loading control. (C) Western blot analysis of Prmt1 protein expression in Cre-ER expressing tumor derived Prmt1f/f p53/Rb-null mOS cell lines following vehicle (Ethanol) or tamoxifen (4OHT) treatment. (D) Proliferation assessment of conditional Prmt1 KO (Prmt1f/f CreER) cells expressing a dox-inducible wild-type Prmt1 following vehicle, 4OHT, or both 4OHT and dox treatment by crytal violet staining. (E) Proliferation assessment of conditional Prmt1 KO (Prmt1f/f CreER) cells expressing a dox-inducible catalytic point mutant Prmt1 (E144Q or G80R) following vehicle, 4OHT, or both 4OHT and dox treatment by crytal violet staining. Representative images of the three replicate experiments are shown.
(F) Western blot analysis of doxycycline-inducible murine wild-type Prmt1 (mPrmt1), Prmt1 catalytic mutant (E144Q), human wild-type Prmt1 (hPrmt1), and Prmt1 catalytic mutant (G80R) in 4OHT treated Prmt1 conditional knockout p53/Rb-null mOS cells (Prmt1f/f CreER).
To assess whether Prmt1 was deleted in tumors arising in Prmt1f/f OS mice, we isolated DNA and protein from Prmt1f/f OS tumors. The tumors retained residual Prmt1 protein expression and a non-excised Prmt1 allele (Figure 2B and Supplementary Figure 2C). Thus, residual protein was detected in Prmt1f/f OS tumors, either due to tumor cells that escaped Cre-mediated excision or due to a leaky requirement for Prmt1.
We derived cell lines from Prmt1f/f OS tumors. These mOS tumor cell lines retained Prmt1 expression. Upon expression of tamoxifen-inducible Cre (CreER), the cells underwent Prmt1 inactivation and growth arrest (Figures 2C, 2D, and 2E,). Thus, residual Prmt1 detectable in the tumors reflects incomplete excision and the generation of Prmt1+ tumors. Taken together, these findings provide persuasive evidence that Prmt1 is essential for initiation of OS in vivo.
Cell growth and survival depend on catalytically active Prmt1
Prmt1 mediates cellular processes by catalyzing protein arginine methylation. We next sought to determine whether the methyltransferase activity is required for the function of Prmt1 in sustaining p53/Rb-null cell growth. We introduced a single amino acid substitution in the S-adenosyl-methionine binding domain of the wild-type murine Prmt1 (E144Q) and human PRMT1 (G80R). These Prmt1 mutants were previously shown to be defective in catalytic activity (21,22,32). We expressed a dox-inducible wild-type Prmt1 or catalytically inactive Prmt1 mutants in Prmt1f/f OS cells harboring CreER (Figures 2D, 2E, and 2F). We then assessed the ability of the mutants to rescue proliferative defects of Prmt1 conditional knockout cells. Upon Cre activation, Prmt1f/f OS cells lost Prmt1 expression and underwent growth arrest as expected (Figures 2D and 2E). Inducible wild-type murine or human Prmt1, but not the catalytically-inactive mutants (E144Q mPrmt1 or G80R hPRMT1), substantially rescued proliferation of conditional Prmt1 knockout cells (Figures 2D, 2E, and 2F). Thus, we conclude that the methyltransferase activity of Prmt1 is required to sustain proliferation and survival of Prmt1 deficient cells.
Quantitative proteomics identify Prmt1 substrates and associated pathways
We employed a SILAC (stable isotope labeling by amino acids in cell culture)-based quantitative proteomics strategy to identify substrates methylated by Prmt1 in p53/Rb-null mOS cells (Figure 3A). Control and Prmt1 knockdown cells were labeled with culture media containing either heavy (13C6 15N4 and 13C6) or light L-arginine and L-Lysine. Proteins from equal numbers of control and Prmt1 knockdown cells were extracted for subsequent tryptic digestion and antibody-based enrichment of arginine-methylated peptides. The abundance of isotopically-encoded methylated peptides was determined across biological replicates by multidimensional LC-MS/MS (liquid chromatography-tandem mass spectrometry) analysis(33,34) (Supplementary Table 1). We also analyzed immuno-purification supernatants to measure differences in protein abundance between control and Prmt1-depleted cells and to normalize methylated peptides ratios. This analysis further allowed us to confirm the specific depletion of Prmt1 in the knockdown cells as compared to the control, and ascertain that abundance of several other protein arginine methyltransferases remained unchanged (Supplementary Figure 3A). Of note, the majority of the R-methylated peptides purified using methyl-R specific antibodies did not change in abundance upon Prmt1 knockdown (Figure 3B). Prmt1 depletion resulted in marked down-regulation of 110 and 237 R-bearing peptides purified using admR and mmR antibodies, respectively (Figures 3B and 3C).
Figure 3. Quantitative SILAC proteomics reveals novel Prmt1 substrates and associated pathways.
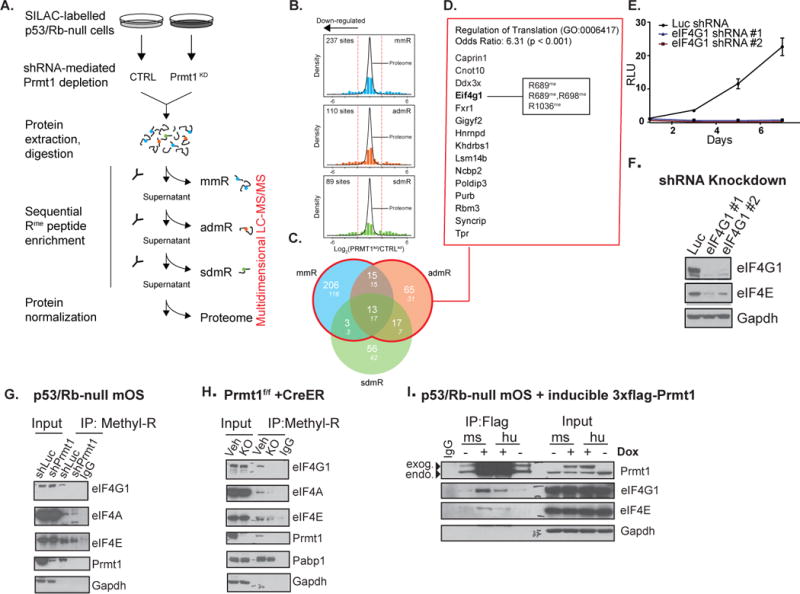
(A) Experimental work-flow for identifying R-methylated proteins in SILAC labeled control and Prmt1 knockdown p53/Rb-null mOS. (B) Histogram distribution of the Log2 ratio measured for peptides enriched using antibodies against monomethyl (“mmR”), asymmetric (“admR”) and symmetric dimethyl (“sdmR”) arginine residues (top to bottom). The kernel density plot of the Log2 ratio for peptides in the supernatant (“proteome”) is overlaid atop each distribution. The red dotted lines indicate the location of the mean +/– 3 standard deviations. (C) Venn diagram of the overlap between down-regulated methylated sites across the three immuno-affinity purifications. (D) Proteins with down-regulated mono-methyl and asymmetric di-methyl arginine residues following Prmt1 depletion are significantly enriched for factors that regulate translation. (E) Proliferation of p53/Rb-null mOS cells infected with control shRNA or two independent shRNAs against Eif4g1. (F) Western blot analysis of eIF4G1 and eIF4E in the control and eIF4G1 knockdown p53/Rb-null cells. (G) Immunoprecipitation of R-methylated proteins using methyl-R-specific antibody followed by western blot with the indicated antibodies in control and Prmt1 knockdown p53/Rb-null cells. (H) Immunoprecipitation of R-methylated proteins using methyl-R-specific antibody followed by western blot with the indicated antibodies in vehicle control (Prmt1f/f CreER) and 4OHT treated conditional Prmt1 knockout cells. (I) Flag-IP of Flag-Prmt1 in p53/Rb-null cells expressing a dox-inducible Flag-Prmt1 followed by western blot with the indicated antibodies against components of the translation initiation complex.
We next analyzed gene ontology (GO) term enrichment across the sets of putative Prmt1 protein substrates identified in our SILAC proteomic data. This analysis showed that each set was enriched for distinct annotation, with an overall preponderance of terms related to mRNA/RNA processing, stability metabolism, binding, and splicing (Supplementary Figure 3B). In particular, we observed an enrichment of the GO-term “Regulation of translation” among mono-methylated and asymmetric di-methylated putative Prmt1 substrates corroborating our experimental findings for genetic vulnerabilities of p53/Rb-null cells in the shRNA screen (Figure 3D and Supplementary Figure 3B). Of note, we found that one of the translation-associated genes identified, the eukaryotic translation initiation factor 4 gamma 1 (Eif4g1), was essential for p53/Rb-null cell proliferation. Indeed, shRNA-mediated knockdown of eIF4G1 in p53/Rb-null cells dramatically impaired growth of p53/Rb-null cells (Figure 3E). eIF4G1 depletion also led to down-regulation of eIF4E, which is another essential component of translation initiation complex (Figure 3F). Taken together, these data implicate Prmt1-mediated translation regulation via eIF4G1 methylation as critical to cell growth and survival of p53/Rb-null cells.
Prmt1 methylates R689 and/or R698 of eIF4G1
To validate the proteomics findings and identify arginine residues that were specifically modified by Prmt1, we conducted a more detailed analysis on the R methylation status of eIF4G1. SILAC-based proteomics identified three potentially Prmt1-regulated methyl-R sites on eIF4G1 (Figure 3D). Comparison of mass spectrometry MS/MS data for endogenous and synthetic eIF4G1 peptides confirmed single mono-methylation of R689, as well as double mono-methylation of both R689 and R698 (Supplementary Figures 4A and 4B).
Figure 4. Prmt1 regulates global translation initiation.
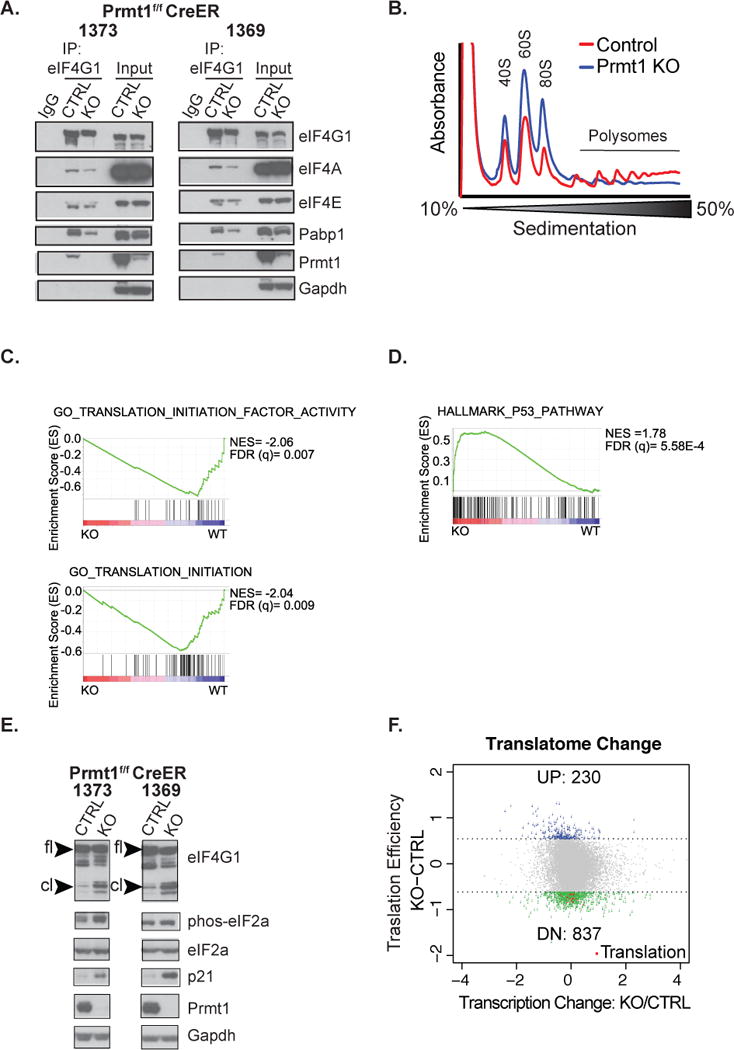
(A) Assessment of eIF4G1 interaction with other members of the translation initiation complex and Prmt1 by immunoprecipitation in two independent vehicle-treated control and 4OHT-treated Prmt1f/f CreER mOS cells (1373 & 1369). (B) Polysome analysis of RNAs isolated from control (Red) and Prmt1f/f CreER mOS treated with 4OHT (Blue). The positions of free ribosomal subunits (40S, 60S) monosomes (80S), and polysomes are indicated. Representative trace of one out of three biological replicates is shown. (C & D) GSEA analysis of the pre-ranked list of differentially expressed genes in the Prmt1f/f CreER mOS control as compared to the conditional knockout. Normalized enrichment score (NES) and False discovery rate FDR are both indicated. (E) Western blot analysis of indicated proteins in vehicle treated control and 4OHT-treated Prmt1f/f CreER mOS cells. “fl” indicates full-length protein, whereas “cl” denotes cleaved protein. (F) Analysis of mRNA abundance associated with the “heavy” polysome relative to the “light” polysome following Prmt1 depletion in Prmt1f/f CreER mOS. The translation efficiency (Y axis) is defined as the difference between the Log2 ratios of the “heavy” to “light” in the conditional KO versus the control. X-axis denotes Log2 fold change of genes affected at the transcriptional level in the conditional knockout (KO) as compared to the control (CTRL). Blue and green dots designate transcripts that are up regulated or down regulated at the translation level, respectively. Red dots highlight down regulated transcripts associated with translation GO processes.
To further verify that the R689 site was indeed regulated by Prmt1, we designed a secondary SILAC-based proteomics approach to quantitatively measure the methyl-R689 status of eIF4G1 using the Prmt1f/f OS CreER system. For this experiment, cultures from vehicle control and tamoxifen-treated Prmt1 conditional knockout cells were labeled with “heavy” and “light” SILAC media, respectively. Equal cell numbers of control and conditional knockout cells were combined, followed by enrichment of eIF4G1 using eIF4G1 specific antibodies. Peptides from enriched eIF4G1 were subsequently analyzed by LC-MS/MS to determine their relative abundance in the control as compared to the conditional knockout. Consistent with the original proteomics screen, loss of Prmt1 led to a dramatic decrease in R689 mono-methylation (Supplementary Figure 4C). Moreover, in vitro methyl transferase assay also confirmed that eIF4G1 is a substrate of Prmt1 (Supplementary Figure 4D).
R689 site of eIF4G1 contributes to protein stability
We next engineered p53/Rb-null cells to stably express HA-tagged wild-type eIF4G1 or the corresponding HA-tagged R689A eIF4G1 mutant to evaluate the function of R689 site. We performed a cycloheximide chase assay to determine the relative stability of wild-type eIF4G1 versus R689A eIF4G1 mutant. We observed that the full-length and the cleaved product of the wild-type protein were more stable over time, whereas the R689A eIF4G1 mutant exhibited a shorter half-life, suggesting that the R689 site contributes to the stability of eIF4G1 (Supplementary Figure 4E).
Prmt1 methylates the translation initiation complex
Emerging evidence suggests that some components of the translation initiation complex are arginine-methylated(11,12). In light of our proteomics findings, we hypothesized that Prmt1-mediated methylation of the translation initiation complex might play a key role in promoting oncogenic activities of p53/Rb-null cells. To test this hypothesis, we evaluated the effect of Prmt1 loss on R-methylated eIF4G1, eIF4A, eIF4E protein levels by methyl-R-specific immunoprecipitation, followed by western blot analysis of eIF4G1, eIF4A, eIF4E protein levels. We found that shRNA-mediated Prmt1 knockdown and Cre-mediated knockout of Prmt1 following treatment with 4OHT led to a dramatic decrease in R-methylated eIF4G1, eIF4A, and eIF4E, whereas Pabp1, a Carm1 target, was unaffected (13) (Figures 3G and 3H).
We next investigated whether Prmt1 physically interacts with the translation initiation complex. We generated p53/Rb-null cells that stably expressed dox-inducible flag-tagged Prmt1. Following doxycycline treatment, we performed a flag-tagged Prmt1 pulldown, followed by western blot analysis of eIF4G1 and eIF4E in the co-immunoprecipitates. We observed that Prmt1 co-immunoprecipitated with eIF4G1 and eIF4E (Figure 3I). To confirm this interaction, we also performed an eIF4G1 pull-down and found that Prmt1 co-immunoprecipitated with eIF4G1, along with eIF4E, and eIF4A (Figure 4A). Taken together, these experiments indicate that Prmt1 interacts with and methylates components of the translation initiation complex to regulate their activities.
Prmt1 regulates global translation in p53/Rb-null cells
Since translation initiation appeared as one of the major pathways affected following Prmt1 depletion, we next investigated if global mRNA translation was impaired upon Prmt1 depletion. We employed polysome profiling to quantify the mRNA translation activities of the conditional Prmt1 knockout in comparison to the control cells using the Prmt1f/f OS CreER system. We found that loss of Prmt1 led to a reduction in the level of polysome-associated mRNAs and an accumulation of poorly-translated/free mRNAs, consistent with a translation defect phenotype (Figure 4B).
We further elucidated transcriptional changes following Prmt1 knockout by RNA sequencing (RNA-seq) analysis. Prmt1 depletion led to significant down regulation of 1228 genes and up regulation of 878 genes at the transcription level (Fold Change=1.5, p<0.05; Supplementary Figure 5A). The RNA-seq data confirmed deletion of Prmt1 in the conditional knockout versus the control, as well as deletion of p53 and Rb in all samples (Supplementary Figure 5B and data not shown). Gene set enrichment analysis (GSEA) of differentially expressed genes revealed that translation-related gene sets were among those which was significantly down-regulated upon Prmt1 depletion (Figure 4C, Supplementary Figure 5C, and Supplementary Table 2). In addition, since Prmt1 and p53 appear to cooperate in transcription activation(35), we also assessed the effect of Prmt1 loss on p53 target genes by GSEA analysis. Interestingly, although our cells do not express p53, p53 target genes appeared to be induced upon Prmt1 loss (Figure 4D, Supplementary Figure 5C, Supplementary Table 3). In particular, loss of Prmt1 led a marked increase in Cdkn1a/p21, a transcriptional target of p53 (Figure 4E and supplementary Figure 5C). These data implicate a role for Prmt1 in transcriptional regulation of translation-related genes and p53 target genes.
We next sought to identify genes that were regulated by Prmt1 at the translation level. To this end, we analyzed Prmt1-dependent changes in the “heavy” (> 3 ribosomes) polysome-associated mRNA profiles relative to the changes observed in the “light” (< 3 ribosomes) polysome-associated mRNA profiles by RNA-seq. All biological replicates clustered in their respective groups (Supplementary Figure 5D). In total, we found 837 genes were significantly down regulated and 230 genes were significantly up regulated in the “heavy” polysomes as a result of Prmt1 loss (Figure 4F). Since Prmt1 regulates many genes at the transcriptional level, we excluded genes that were transcriptionally regulated, and then performed functional analysis on the remaining genes using DAVID (29). Among the biological processes identified, translation-associated GO terms were enriched in the list of down-regulated genes (Figure 4F, Supplementary Figure 5E, and Supplementary Table 4). There were no significantly enriched processes associated with up–regulated genes. In aggregate, these data implicate Prmt1 in control of growth/survival in part through regulating translation-associated genes at both transcription and translation levels.
Prmt1 regulates eIF4F integrity and assembly
To gain further molecular insight into the impact of Prmt1 loss on the translation initiation complex, we examined the interaction between eIF4G1 and its interacting partners by co-immunoprecipitation in conditional Prmt1 knockout cells relative to control. Although Prmt1 depletion had very minor effects on eIF4G-eIF4E interaction, we observed a marked decrease in eIF4G1 association with eIF4A and Pabp1 in independent p53/Rb-null cell lines (Figure 4A). In addition, Prmt1 knockout increased cleavage of eIF4G1 protein. However, other markers of stress-induced translation such as phospho-eIF2a were not consistently evident (Figure 4E). Together, these observations implicate Prmt1 in modulating the assembly and integrity of the translation initiation complex.
P53-deficient mOS cells are vulnerable to p53-mediated translation inhibition
In light of our RNA-seq data and other studies that suggest a role of p53 in translation control, we next investigated whether loss of p53 contributed to translation dependencies in the p53 deficient cells we have employed. We established p53/Rb-null and p53-null/Rb-wt tumor derived cells that stably expressed a dox-inducible p53 protein. Upon induction of p53 expression, proliferation of p53 deficient cells was greatly reduced (Figure 5A). In addition to induction of Cdkn1a/p21 protein expression, p53 activation was also accompanied by translation inhibition, as evidenced by an increase in phospho-eIF2a, and eIF4G1 cleavage, reminiscent of the Prmt1 knockout phenotype (Figure 5B). These observations suggest that p53 and Prmt1 downstream targets may converge on translation-related pathways.
Figure 5. p53 deficient mOS cells are sensitive to p53-induced translation stress.
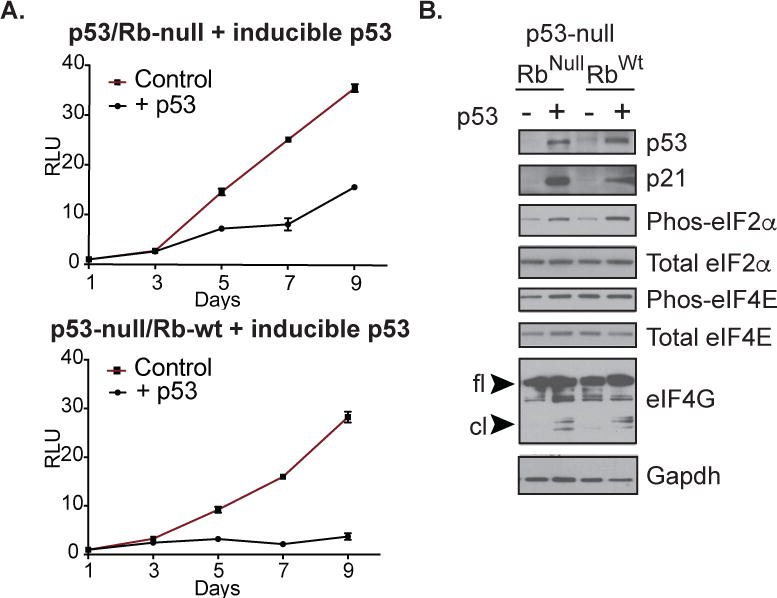
(A) Proliferation of p53/Rb-null and p53-null/Rb-wt expressing a dox-inducible p53 following treatment with vehicle control or doxycycline (B) Western blot analysis of the indicated proteins in vehicle control or dox-treated p53/Rb-null and p53-null/Rb-wt cells that express a dox-inducible p53. “fl” indicates full-length protein, whereas “cl” denotes cleaved protein.
Prmt1 dependency correlates with translation-associated dependencies and p53 mutation in human cancer cells
In recent years, the Achilles data set has facilitated the discovery of biomarkers associated with context-specific dependencies in human tumor cell lines(30). To investigate Prmt1 dependency in the context of human cancer cell lines, we interrogated the panel of cells contained within the shRNA-screen-based Project Achilles dataset (https://portals.broadinstitute.org/achilles). We hypothesize that cell lines that have preferential dependency on Prmt1 should exhibit similar dependency on genes belonging to the same functional gene network. Using the PARIS (Probability analysis by Ranked Information Score) algorithm, we identified other genetic dependencies that correlate with Prmt1 dependency(30,36). Interestingly, GSEA analysis of the ranked list of correlated genes revealed that Prmt1 dependency positively correlates with dependencies on translation-associated gene sets, as well as other Prmt1-regulated processes including many that are associated with RNA processing (Figure 6A and Supplementary Table 5). We also used a newly developed random-forest based method called ATLANTIS to identify biomarkers that are associated with preferential dependency on PRMT1(31). Interestingly, we found that p53 mutational status correlates with PRMT1 dependency across human cell lines of multiple lineages (Figure 6B). Taken together, these data imply that PRMT1, translation, and p53 mutation are involved in common genetic networks that may constitute an Achilles heel of many human cancer cells.
Figure 6. Prmt1 dependency correlates with translation-associated dependencies and p53 mutation in human cancers.
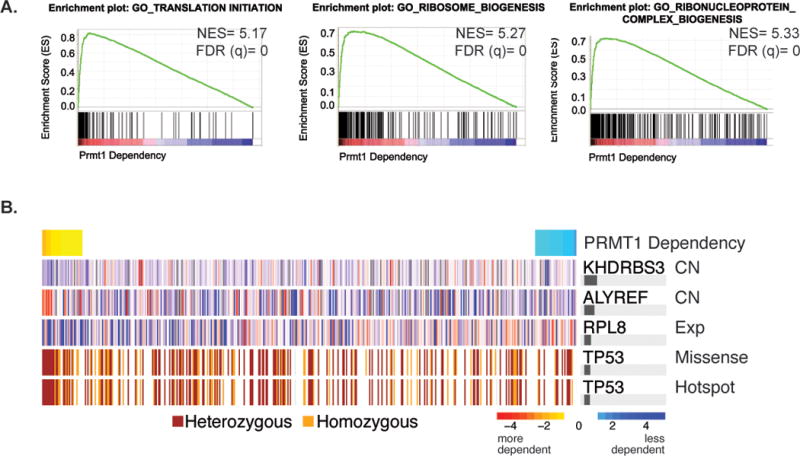
(A) GSEA analysis of the ranked list of gene dependencies associated with PRMT1 dependencies across 501 human cancer cell lines from the Achilles dataset. Black bars at the bottom of the figure indicate the location of genes positively correlated with Prmt1 dependency and the green curve indicates the running enrichment score for the gene set. Normalized enrichment score (NES) and False discovery rate (FDR) are shown. (B) ATLANTIS model for PRMT1 using known physical interactors as features. PRMT1 dependency is shown from most to least dependent cell line in columns. The top five predictive markers are shown in the successive rows; Copy number (CN) and expression (Exp) values are z scores (high to low, red to blue). Horizontal bars on the right indicate the relative contribution to the model’s out of bag R2.
DISCUSSION
Our search for genetic dependencies of p53/Rb-null mOS cells has led to the discovery of the important role of Prmt1 in p53/Rb-null cell growth and survival. We show that Prmt1 is required for the tumorigenicity of established p53/Rb-null cells and tumorigenesis of p53 and Rb-null cells in vivo, while not affecting normal bone development. In addition, embryonic stem cells derived from Prmt1 mutant are viable(37). Thus, Prmt1 is not essential in all cell types but rather exhibits cell context-specific requirements. Further, analysis of the Project Achilles dataset reveals correlation of Prmt1 dependency with translation-related dependencies and p53 mutation, suggesting common pathways. Interestingly, emerging data suggest that translation associated genes are potential targets of gain-of-function mutant p53(38). Hence, p53 mutant cells may be more dependent on translation-associated pathways and/or Prmt1-mediated translation pathways for growth or survival. Future studies are needed to elucidate the relationship between Prmt1 dependency, translation-associated dependency, and the p53 status of the cancer cells.
At a mechanistic level, various isoforms of Prmt1 methylate diverse arginine-bearing substrates in the cytoplasm and/or the nucleus to promote tumorigenicity (39). Our proteomics study identified some substrates of Prmt1 that have been previously described, as well as novel substrates of Prmt1, including some components of the translation initiation complex. Consistent with previous reports, R-methylation of translation factors by Prmt1 or other Prmt family of proteins has been demonstrated in human cell lines and mouse tissues(10–13). However, the biological consequence is incompletely understood. Here, we have found that depletion of Prmt1 in p53/Rb-null cells results in marked loss of global translation and destabilization of the translation initiation complex. Using RNA-seq analysis and pathway discovery tools, we found that Prmt1 regulates translation-related gene sets at both transcription and translation levels. The types of mRNAs that display a strong Prmt1 dependency remain to be determined, as some translation factors exhibit context-dependent translation activities. For example, select translational programs display a strong dependency on eIF4G1 or eIF4E in cancer (40–42). Furthermore, pathway analysis of the RNA-seq data indicates that other oncogenic and tumor suppressor networks, including Myc, E2F, Kras, and p53, are affected as a result of Prmt1 loss. Given that other studies have shown that mRNA translation serves as a node of convergence of many oncogenic and tumor suppressor pathways, the cumulative effects of Prmt1 loss on transcription, translation, and post-translational modification may contribute to the translation phenotype observed in p53/Rb-null cells(5,43).
In addition to Prmt1-associated translation dependency, p53/Rb-null mOS cells are vulnerable to translation inhibition via eIF4G1-mediated or p53-induced translation blockade. It is noteworthy that Prmt1, eIF4G1, or p53 signaling all converge on translation. However, each of these individual factors exerts a different context-dependent biological effect. For example, depletion of eIF4G1 leads to down-regulation of some of its associated translation factors, whereas depletion of Prmt1 does not. Hence, it is possible that eIF4G1 may play a broader role in translation control, whereas Prmt1-mediated methylation may serve to fine-tune specific translation programs. Future experiments will be needed to delineate the role of Prmt1, eIF4G1 and p53 in regulating cancer-specific translation program.
Hyper-activation of translation has been implicated in tumorigenesis, including some p53-deficient tumors(5,6,44). This in turn leads to cancer-specific dependency on translation and pathways that feed into the translation machinery. Although we have not observed a consistent increase in Prmt1 nor eIF4G1 expression in cancer cells as compared to normal cells (Supplementary Figure 6), published gene expression analysis of p53/Rb-deficient skin cells exhibits an increased in Eif4g1 expression(45). Thus, it remains to be determined whether dependency on Prmt1 and translation-associated pathways is due to hyper-activation of translation in p53/Rb-null cells.
Using OS model driven by p53/Rb-loss, we have identified Prmt1 as an essential oncogene and a regulator of translation. Our findings indicate that Prmt1 stabilizes the translation machinery and regulates translation activity to promote tumor initiation and maintenance. Taken together, these findings implicate that therapeutic agents targeting Prmt1 and/or its-associated translation machinery may hold promise for treating cancer that are addicted to translation and upstream pathways that feed into the translation network.
Supplementary Material
Acknowledgments
We thank Melanie Hamblen, Frank Godinho, Nihal Terzi Cizmecioglu, Sidinh Luc, Minh Nguyen, Partha Das, Kimberly Kim, Radhika Mathur, Dong-Yin Yuk, Ying Xie, and other members of the Orkin laboratory for helpful discussion and technical assistance. We thank Joseph Card for help with sample preparation and William Alexander for help with computational analysis. We also thank the Center for Cancer Computational Biology at Dana-Farber Cancer Institute, the Specialized Histopathology Services Core at Brigham and Women’s Hospital (Teresa Bowman and Christine Unitt), the Rodent Histopathology Core (Roderick Bronson), and BCH Small Animal Imaging Laboratory (Erin Snay) for various technical support and analysis.
FINANCIAL SUPPORT: J.H.Hsu was supported by the Young Investigator Grant from the Alex’s Lemonade Stand Foundation, fellowship awards from the National Cancer Center, Friends of Dana-Farber, and the David Abraham Fellowship fund. S.H. Orkin was supported in part by Alex’s Lemonade Stand Innovation Award and National Cancer Institute Grant (U01CA105423). This work was supported in part by U01 CA176058 and U01 CA199253 (W.C. Hahn.).
ABBREVIATIONS LIST
- OS
osteosarcoma
- Prmt1
protein arginine methyl transferase 1
- R
arginine
- KO
knock-out
- Osx
osterix
- SILAC
stable isotope labeling with amino acids in cell culture
- 4OHT
4-hydroxytamoxifen
- Dox
doxycycline
- mmR
mono-methyl arginine
- admR
asymmetric dimethyl arginine
- sdmR
symmetric dimethyl arginine
Footnotes
All authors declare no potential conflict of interest.
References
- 1.Spriggs KA, Bushell M, Willis AE. Translational regulation of gene expression during conditions of cell stress. Mol Cell. 2010;40:228–37. doi: 10.1016/j.molcel.2010.09.028. [DOI] [PubMed] [Google Scholar]
- 2.Truitt ML, Ruggero D. New frontiers in translational control of the cancer genome. Nat Rev Cancer. 2016;16:288–304. doi: 10.1038/nrc.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. Targeting the translation machinery in cancer. Nat Rev Drug Discov. 2015;14:261–78. doi: 10.1038/nrd4505. [DOI] [PubMed] [Google Scholar]
- 5.Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer. 2010;10:254–66. doi: 10.1038/nrc2824. [DOI] [PubMed] [Google Scholar]
- 6.Marcel V, Catez F, Diaz JJ. p53, a translational regulator: contribution to its tumour-suppressor activity. Oncogene. 2015;34:5513–23. doi: 10.1038/onc.2015.25. [DOI] [PubMed] [Google Scholar]
- 7.Walkley CR, Qudsi R, Sankaran VG, Perry JA, Gostissa M, Roth SI, et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev. 2008;22:1662–76. doi: 10.1101/gad.1656808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berman SD, Calo E, Landman AS, Danielian PS, Miller ES, West JC, et al. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc Natl Acad Sci U S A. 2008;105:11851–6. doi: 10.1073/pnas.0805462105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perry JA, Kiezun A, Tonzi P, Van Allen EM, Carter SL, Baca SC, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U S A. 2014;111:E5564–73. doi: 10.1073/pnas.1419260111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wooderchak WL, Zang T, Zhou ZS, Acuna M, Tahara SM, Hevel JM. Substrate profiling of PRMT1 reveals amino acid sequences that extend beyond the “RGG” paradigm. Biochemistry. 2008;47:9456–66. doi: 10.1021/bi800984s. [DOI] [PubMed] [Google Scholar]
- 11.Guo A, Gu H, Zhou J, Mulhern D, Wang Y, Lee KA, et al. Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol Cell Proteomics. 2014;13:372–87. doi: 10.1074/mcp.O113.027870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larsen SC, Sylvestersen KB, Mund A, Lyon D, Mullari M, Madsen MV, et al. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci Signal. 2016;9:rs9. doi: 10.1126/scisignal.aaf7329. [DOI] [PubMed] [Google Scholar]
- 13.Lee J, Bedford MT. PABP1 identified as an arginine methyltransferase substrate using high-density protein arrays. EMBO Rep. 2002;3:268–73. doi: 10.1093/embo-reports/kvf052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicholson TB, Chen T, Richard S. The physiological and pathophysiological role of PRMT1-mediated protein arginine methylation. Pharmacological research: the official journal of the Italian Pharmacological Society. 2009;60:466–74. doi: 10.1016/j.phrs.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 15.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37–50. doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- 16.Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, et al. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435:1262–6. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- 17.Mathioudaki K, Papadokostopoulou A, Scorilas A, Xynopoulos D, Agnanti N, Talieri M. The PRMT1 gene expression pattern in colon cancer. British journal of cancer. 2008;99:2094–9. doi: 10.1038/sj.bjc.6604807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valentijn LJ, Koster J, Haneveld F, Aissa RA, van Sluis P, Broekmans ME, et al. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:19190–5. doi: 10.1073/pnas.1208215109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, et al. Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci U S A. 2008;105:20380–5. doi: 10.1073/pnas.0810485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063–6. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Cheng X. Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure. 2003;11:509–20. doi: 10.1016/s0969-2126(03)00071-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Huang ZQ, Xia L, Feng Q, Erdjument-Bromage H, Strahl BD, et al. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science. 2001;293:853–7. doi: 10.1126/science.1060781. [DOI] [PubMed] [Google Scholar]
- 23.Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133:3231–44. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- 24.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, et al. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9:e1003118. doi: 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morgan MPH, Obenchain V, Hayden N. Rsamtools: Binary alignment (BAM), FASTA, variant call (BCF), and tabix file import. R package version 1.24.0. 2016 [Google Scholar]
- 27.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 30.Cowley GS, Weir BA, Vazquez F, Tamayo P, Scott JA, Rusin S, et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci Data. 2014;1:140035. doi: 10.1038/sdata.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS et al. Defining a Cancer Dependency Map. Cell. 2017 doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McBride AE, Weiss VH, Kim HK, Hogle JM, Silver PA. Analysis of the yeast arginine methyltransferase Hmt1p/Rmt1p and its in vivo function. Cofactor binding and substrate interactions. J Biol Chem. 2000;275:3128–36. doi: 10.1074/jbc.275.5.3128. [DOI] [PubMed] [Google Scholar]
- 33.Ficarro SB, Zhang Y, Carrasco-Alfonso MJ, Garg B, Adelmant G, Webber JT et al. Online nanoflow multidimensional fractionation for high efficiency phosphopeptide analysis. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.O111.011064. O111 011064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou F, Lu Y, Ficarro SB, Adelmant G, Jiang W, Luckey CJ, et al. Genome-scale proteome quantification by DEEP SEQ mass spectrometry. Nat Commun. 2013;4:2171. doi: 10.1038/ncomms3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117:735–48. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 36.Shao DD, Tsherniak A, Gopal S, Weir BA, Tamayo P, Stransky N, et al. ATARiS: computational quantification of gene suppression phenotypes from multisample RNAi screens. Genome Res. 2013;23:665–78. doi: 10.1101/gr.143586.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pawlak MR, Scherer CA, Chen J, Roshon MJ, Ruley HE. Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol Cell Biol. 2000;20:4859–69. doi: 10.1128/mcb.20.13.4859-4869.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature. 2015;525:206–11. doi: 10.1038/nature15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goulet I, Gauvin G, Boisvenue S, Cote J. Alternative splicing yields protein arginine methyltransferase 1 isoforms with distinct activity, substrate specificity, and subcellular localization. J Biol Chem. 2007;282:33009–21. doi: 10.1074/jbc.M704349200. [DOI] [PubMed] [Google Scholar]
- 40.Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–13. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Badura M, Braunstein S, Zavadil J, Schneider RJ. DNA damage and eIF4G1 in breast cancer cells reprogram translation for survival and DNA repair mRNAs. Proc Natl Acad Sci U S A. 2012;109:18767–72. doi: 10.1073/pnas.1203853109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Truitt ML, Conn CS, Shi Z, Pang X, Tokuyasu T, Coady AM, et al. Differential Requirements for eIF4E Dose in Normal Development and Cancer. Cell. 2015;162:59–71. doi: 10.1016/j.cell.2015.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boussemart L, Malka-Mahieu H, Girault I, Allard D, Hemmingsson O, Tomasic G, et al. eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature. 2014;513:105–9. doi: 10.1038/nature13572. [DOI] [PubMed] [Google Scholar]
- 44.Petroulakis E, Parsyan A, Dowling RJ, LeBacquer O, Martineau Y, Bidinosti M, et al. p53-dependent translational control of senescence and transformation via 4E-BPs. Cancer Cell. 2009;16:439–46. doi: 10.1016/j.ccr.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 45.Martinez-Cruz AB, Santos M, Lara MF, Segrelles C, Ruiz S, Moral M, et al. Spontaneous squamous cell carcinoma induced by the somatic inactivation of retinoblastoma and Trp53 tumor suppressors. Cancer Res. 2008;68:683–92. doi: 10.1158/0008-5472.CAN-07-3049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.