

Abstract
The relationship between RNA-binding proteins, particularly TAR DNA binding protein 43 (TDP-43), and neurodegeneration is an important area of research. TDP-43 is involved in so many cellular processes that perturbation of protein homeostasis can lead to countless downstream effects. Understanding what leads to this disease-related protein imbalance and the resulting cellular and molecular effects will help to develop targets for disease intervention, whether it be prevention of protein accumulation, or addressing a secondary effect of protein accumulation. Here we review the current literature of TDP-43 and TDP-43 pathologies, the effects of TDP-43 overexpression and disruption of synaptic proteins through its binding of messenger RNA, leading to synaptic dysfunction. This review highlights some of the still-limited knowledge of the protein TDP-43 and how it can contribute to disease.
Introduction
Transactivation response DNA binding protein of 43 kDa (TDP-43) is a 414 amino acid protein of the heterogenous nuclear ribonucleoprotein (hnRNP) family involved in the regulation of thousands of genes via DNA/RNA binding, RNA splicing and protein/protein interactions (Baralle, Buratti, & Baralle, 2013; Chaudhury, Chander, & Howe, 2010). The protein contains two RNA-recognition motifs (RRM1 and RRM2), which can bind nucleic acids, and a glycine-rich region at the C-terminal. TDP-43 contains a nuclear localization sequence (NLS) and a nuclear export signal (NES), which allow it to shuttle between the nucleus and cytoplasm. TDP-43 has a specific affinity for binding to GU-rich regions of RNA, and RRM1 is specifically involved in this binding (Buratti & Baralle, 2001). The ability of TDP-43 to bind RNA is essential for all of its roles in RNA processing such as mRNA transport, pre-mRNA splicing, and mRNA stability (Baralle, Buratti, & Baralle, 2013; Polymenidou, et al., 2011). Because of the many roles and targets of TDP-43, cells are very sensitive to alterations in TDP-43 levels, and TDP-43 expression is tightly regulated (Cohen, Lee, & Trojanowski, 2011). TDP-43 is able to self-regulate using a negative feedback mechanism in which the protein binds to its own 3′ untranslated region (UTR), leading to TDP-43 instability and degradation (Ayala, et al., 2010; Polymenidou, et al., 2011).
TDP-43 was first identified in 1995 as a repressor of HIV-1 gene expression, working by blocking assembly of transcription complexes (Ou, Wu, Harrich, Garcia-Martinez, & Gaynor, 1995). It has also been implicated in the pathogenesis of cystic fibrosis caused by skipping of cystic fibrosis transmembrane conductance regulator (CFTR) exon 9 from the mature mRNA (Buratti & Baralle, 2001). TDP-43 is ubiquitously expressed in many tissues and is conserved across species, underlining its importance in normal cellular function (Ayala, et al., 2005; Cohen, Lee, & Trojanowski, 2011).
TDP-43 is encoded by the TARDBP gene, located on chromosome 1 (Ou, Wu, Harrich, Garcia-Martinez, & Gaynor, 1995). Mutations in TARDBP are associated with familial amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). Most ALS-associated TARDBP mutations are in the glycine-rich region of the C-terminal (Cohen, Lee, & Trojanowski, 2011). This C-terminal region is the site of protein-protein binding (Buratti, et al., 2005) and is likely responsible for the tendency of TDP-43 to aggregate (Baralle, Buratti, & Baralle, 2013; Chen, et al., 2010; Saini & Chauhan, 2011). Mutated forms of TDP-43 have been shown to have a greater tendency to aggregate than wild-type (WT) forms (Dewey, et al., 2012; Liu-Yesucevitz, et al., 2010). A 2009 review by Pesiridis et al. examines the type and location of known TARDBP mutations. Of the 70 known mutations at that time, most occurred in exon 6, which encodes about 60% of the total protein and contains the glycine-rich region (Pesiridis, Lee, & Trojanowski, 2009). There are 28 missense mutations, 21 intronic mutations, seven 5′ UTR mutations, 6 synonymous mutations, 5 3′UTR mutations, 2 benign missense mutations, and 1 nonsense mutation (Pesiridis, Lee, & Trojanowski, 2009). TARDBP mutations have also been found in FTLD-ALS and FTLD patients, but are much rarer (Benajiba, et al., 2009; Borroni, et al., 2009; Borroni, et al., 2010; Janssens & Van Broeckhoven, 2013; Kovacs, et al., 2009).
TDP-43 pathology
While mutations in TARDBP are associated with a small percentage of ALS and FTLD, almost all cases of these diseases have TDP-43 pathology in the affected brain and spinal cord regions. Ubiquitin-positive, tau- and α-synuclein-negative inclusions were known to be a hallmark of ALS and what was known as FTLD-U (now FTLD-TDP), but the identity of the misfolded disease protein in these inclusions was unknown. Neumann et al. were the first to identify TDP-43 as the constituent protein of these ubiquitin-positive protein inclusions in the brains of ALS and FTLD patients (Neumann, et al., 2006). They showed that pathologic TDP-43 was hyperphosphorylated, ubiquitinated, and cleaved into 25 and 35 kDa C-terminal fragments (Neumann, et al., 2006). Though normally localized to the nucleus, under pathological conditions, TDP-43 is lost from the nucleus of some neurons and glia and forms cytoplasmic protein aggregates. There is still debate as to whether this mislocalization leads to disease via a loss of TDP-43 nuclear function mechanism or by a toxic gain of cytoplasmic function, though these are not mutually exclusive (Zufiria, et al., 2016). While TDP-43 is the primary protein effected in ALS and FTLD, there is also secondary TDP-43 pathology in several other neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD) (Chen-Plotkin, Lee, & Trojanowski, 2010). TDP-43 aggregates have also been found in a small (~3%) number of aged control patients, indicating that TDP-43 aggregation may have a role in aging (Nakashima-Yasuda, et al., 2007; Wilson, Dugger, Dickson, & Wang, 2011).
TDP-43 protein aggregation
The role of TDP-43 aggregates in disease pathogenesis is an important focus of research. TDP-43 has been found to be present in a type of protein aggregation called stress granules (SGs) (Cohen, Lee, & Trojanowski, 2011). SGs are non-membrane bound cytoplasmic structures which contain translation initiation proteins (Reineke & Lloyd, 2013) and are thought to be the site of stalled translation initiation and function to repress protein translation (Monahan, Shewmaker, & Pandey, 2016). SGs form in response to various cellular stresses, including oxidative, mitochondrial, and proteasomal stress (Monahan, Shewmaker, & Pandey, 2016). SGs are thought to prevent translation of unwanted proteins, such as housekeeping proteins, so that other proteins can be made to protect the cell in response to stress (Mazroui, Di Marco, Kaufman, & Gallouzi, 2007; Monahan, Shewmaker, & Pandey, 2016). In addition to translation initiation factors, SGs contain polyadenylated mRNAs, small ribosomal subunits, and RNA binding proteins, such as TDP-43 (Monahan, Shewmaker, & Pandey, 2016). The protein T-cell intracellular antigen-1 (TIA-1) is involved in early formation of SGs (Monahan, Shewmaker, & Pandey, 2016). Polyglutamine-rich regions of TIA-1 promote prion-like TIA-1 aggregation, providing a scaffold for SG formation (Gilks, et al., 2004). TDP-43 controls the expression of many different mRNAs, and its presence in SGs is likely part of its role in regulating translation (Ratti & Buratti, 2016). SG formation is a reversible process so that the cell can respond to stress when needed but resume normal protein production once a stressor is removed (Gilks, et al., 2004). When TDP-43 is mislocalized to the cytoplasm, as in TDP-43 proteinopathies, this could lead to inappropriate interaction with and incorporation into SGs (Monahan, Shewmaker, & Pandey, 2016). TDP-43 may be responsive to stress-responsive, colocalizing with SGs after various insults (Cohen, Lee, & Trojanowski, 2011; Colombrita, et al., 2009). Mutated TDP-43 has a stronger aggregation capacity than wild-type TDP-43, localizing to SGs faster, and forming more and larger granules (Dewey, et al., 2012; Liu-Yesucevitz, et al., 2010). RNA binding proteins undergo liquid-liquid phase separation into protein-rich droplets, which can then become protein aggregates. Mutations can cause disruption in phase separation, leading to functional defects and increased propensity to aggregate (Conicella, Zerze, Mittal, & Fawzi, 2016; Sun & Chakrabartty, 2017)
Dewey et al. suggested two different pathways for TDP-43 protein aggregation: the “independent model” where TDP-43 aggregation occurs independently of SGs and the “precursor model” where SG formation acts as a seed for TDP-43 aggregation (Dewey, et al., 2012). When SGs persist, due to prolonged stress for example, they can become aberrant aggregates, unable to be reversed or removed (Monahan, Shewmaker, & Pandey, 2016; Ratti & Buratti, 2016). This alteration in SG homeostasis could play a role in the process of TDP-43 protein aggregation, leading to aberrant protein inclusions. SGs may represent a link between normal TDP-43 function and the pathological accumulation of TDP-43 inclusions. Studies of ALS/FTLD patients, as well as cell culture experiments, have shown that TDP-43 colocalizes with TIA-1, suggesting that TDP-43 aggregates are actually SGs (Liu-Yesucevitz, et al., 2010). However, we found that re-localization of TDP-43 in the cytoplasm is protective (Chen, et al., 2014; Hebron, et al., 2013), suggesting that TDP-43 aggregation in the cytosol may prevent aberrant TDP-43 binding and reduce its pathologic effects. In addition, reduction of soluble and nuclear TDP-43 leads to neuronal protection, independent of protein solubility (Chen, et al., 2014).
Another type of cytoplasmic ribonucleoprotein structures is the processing (P)-body. P-bodies have a similar structure to SGs and are where mRNA decapping and degradation takes place (Dewey, et al., 2012; Sheth & Parker, 2003). Neuronal P-body-like structures called transport ribonucleoproteins (tRNPs) are moved along neurons to the dendrites and are involved in local translation at the dendrite (Barbee, et al., 2006; Dewey, et al., 2012). TDP-43 has been found to localize to both P-bodies and tRNPs (Wang, Wu, Chang, & Shen, 2008), suggesting a role for TDP-43 in local translation in dendrites.
TDP-43 pathology in ALS and FTLD
ALS and FTLD have many pathological hallmarks in common and are thought to be on a spectrum of the same disorder (Neumann, et al., 2006). While ALS, a type of motor neuron disease (MND) and FTLD can be viewed as distinct diseases, there are many cases of overlap in which ALS presents with dementia, or where FTLD is accompanied by MND. The comorbidity of ALS and FTLD is about 50% of patients (Janssens & Van Broeckhoven, 2013).
ALS is the most common form of MND. It is a progressive neurodegenerative disease affecting upper and lower motor neurons. The motor neurons undergo cell death, leading to muscle denervation, atrophy, and loss of voluntary movement (Zufiria, et al., 2016). Patients become paralyzed and usually die of respiratory failure. According to the ALS Association, the age of onset ranges from 40–70, with the average being 55 years. The incidence of ALS is about 2 per 100,000 people per year, and the prevalence is about 5–7 per 100,000 people. ALS occurs about 1.56 times more often in men than women. Death due to respiratory failure usually occurs within 5 years of diagnosis (Da Cruz & Cleveland, 2011).
About 5–10% of ALS cases are inherited, or familial (fALS) forms (Renton, Chio, & Traynor, 2014). The first link between genes and ALS came with the discovery of superoxide dismutase 1 (SOD1) mutations in fALS (Rosen, et al., 1993). SOD1 mutations account for about 12% of all fALS cases (Renton, Chio, & Traynor, 2014). The next gene to be associated with fALS was TARDBP, the gene encoding TDP-43 (Chio, et al., 2010; Sreedharan, et al., 2008). Other genes associated with fALS include: Fused in sarcoma (FUS), Optineurin (OPTN), Valosin-containing protein (VCP), ubiquilin 2 (UBQLN2), sequestosome 1 (SQSTM1), and profilin 1 (PFN1) (Renton, Chio, & Traynor, 2014). In 2011, a GGGGCC hexanucleotide repeat expansion (HRE) in chromosome 9 open reading frame 72 (C9ORF72) was found to be associated with fALS (Renton, et al., 2011; DeJesus-Hernandez, et al., 2011). This pathogenic HRE is the most common cause of fALS, accounting for about 40% of cases (Renton, Chio, & Traynor, 2014). Interestingly, all familial ALS patients, with the exception of SOD1 mutations, have TDP-43 pathology in the brain and spinal cord (Mackenzie, et al., 2007). About 90% of ALS cases are sporadic, meaning they have no familial history of disease. All sporadic ALS cases have TDP-43 protein aggregates in affected brain and spinal cord areas. Because most cases of MND and FTLD are not directly due to a genetic mutation, but do exhibit TDP-43 pathology, it is important to understand how wild-type TDP-43 may contribute to disease pathogenesis.
FTLD is the pathological process, marked by degeneration of frontal and temporal lobes, that underlies the disorder frontotemporal dementia (FTD). FTD is the second most common form of dementia for patients under 65, second only to Alzheimer’s disease (Neary, et al., 1998). The average age of onset is 58, with a survival of 6–11 years (Gotzl, Lang, Haass, & Capell, 2016). The estimated prevalence is 15–22 per 100,000 people and the incidence is 2.7–4.1 per 100,000 people per year (Onyike & Diehl-Schmid, 2013). FTD is a heterogenous disorder that can be categorized based on symptoms (Cardarelli, Kertesz, & Knebl, 2010; Gotzl, Lang, Haass, & Capell, 2016), while FTLD can be categorized based on the histopathology found post mortem. The most common form of FTD is the behavioral variant (bvFTD), which is characterized by changes such as disinhibition, impulsiveness, loss of social awareness, and loss of personality (Neary, et al., 1998; Josephs, et al., 2011). The other two variants of FTD are language variants: semantic dementia (SD) and primary non-fluent aphasia (PNFA). SD is characterized by language comprehension deficits, while PNFA is characterized by difficulty in producing speech (Neary, et al., 1998).
FTLD subtypes are identified by identification of abnormal protein aggregation in the brain. About 35–45% of cases have aggregation of the microtubule associated protein tau (FTLD-tau), about 5–10% have aggregation of FUS protein, and 45–60% of cases have aggregation of TDP-43 (FTLD-TDP) (Gotzl, Lang, Haass, & Capell, 2016). FTLD-TDP can be further subcategorized based on detailed histopathology (Mackenzie & Neumann, 2017). Type A has compact neuronal cytoplasmic inclusions (NCI), short and think dystrophic neurites (DN), and lentiform neuronal intranuclear inclusions (NII) in the upper cortical layers (Mackenzie & Neumann, 2017). These cases are either sporadic or due to a GRN mutation and have bvFTD or SD, but not ALS (Mackenzie & Neumann, 2017). Type B has diffuse granular NCI and few DN in all layers, with wispy thread and dot pathology (Mackenzie & Neumann, 2017). These cases are either sporadic or familial with a C9ORF72 mutation and have a combination of FTD and ALS. And Type C has long think DN and few NCI in all layers of the cortex (Mackenzie & Neumann, 2017). These cases are sporadic and have SD and not ALS (Mackenzie & Neumann, 2017).
About 60% of FTLD cases are sporadic, or have no known family history of the disease, and about 40% are familial and caused by a genetic mutation. There have only been a few cases of familial FTLD being caused by a mutation in the TARDBP gene (Chio, et al., 2010). About 25% of familial FTD is due to the C9ORF72 HRE (Renton, Chio, & Traynor, 2014).
TDP-43 and RNA-related functions
Genes that are mainly involved in RNA/DNA metabolism are closely associated with the ALS phenotype (Hardy & Rogaeva, 2014). The binding domains of TDP-43 allow for control of many aspects of RNA metabolism, including pre-mRNA splicing, microRNA (miRNA) regulation, and mRNA regulation (Ratti & Buratti, 2016). TDP-43 involvement in pre-mRNA splicing was first described in 2001 as a regulator of CFTR splicing (Buratti & Baralle, 2001). TDP-43 was then found to regulate pre-mRNA splicing in human ALS/FTLD and TDP-43 mouse models, including many synaptic proteins (Polymenidou, et al., 2011; Lagier-Tourenne, et al., 2012). TPD-43 is also involved in processing of microRNA, small, non-coding RNAs that function as regulators of RNA silencing and translational repression (Bartel, 2004). TDP-43 controls RNA integrity and loss of TDP-43 has been shown to cause dysregulation of many miRNAs (Ratti & Buratti, 2016). TDP-43 also has roles in the cytoplasm, including stability, transport, and translation of mRNA. The function of TDP-43 in mRNA stability is likely via many binding regions in the 3′UTR (Sephton, et al., 2011; Colombrita, et al., 2012). mRNAs stabilized by TDP-43 include Vascular Endothelial Growth Factor A (VEGFA), Granulin (GRN), and Interleukin-6 (IL-6) (Colombrita, et al., 2012; Lee, et al., 2015). TDP-43 has been shown to be actively transported along axons of primary motor neurons in RNA granules, which can transport mRNAs to and from the synapse (Fallini, Bassell, & Rossoll, 2012; Alami, et al., 2014; Ratti & Buratti, 2016). TDP-43 has also been shown to be involved in translation of mRNA via association with RNA granules which can regulate translation (Ratti & Buratti, 2016). TDP-43 may also be involved in local translation in the synapse, translocating into dendrites in an activity-dependent manner (Wang, Wu, Chang, & Shen, 2008). Recently, we showed that TDP-43 controls mRNA expression of synaptic proteins such as synapsin I, leading to alterations in protein levels of synaptic proteins (Heyburn, et al., 2016). Together, the role of TDP-43 in regulation of gene expression is wide-reaching and disruption of TDP-43 in disease can affect all of these cellular processes, leading to cellular pathology. Because TDP-43 and other MND-FTLD-related proteins such as FUS and C9ORF72 have important roles in RNA metabolism, this process is thought to be important in disease pathogenesis of TDP-43 proteinopathies.
Because neurons are so polarized, with long distances between synapses and cell bodies, especially in long motor neurons, local translation at the synapse is important for normal neuronal function. Neurons are able to respond to changes at the synapse without having to produce and transport mRNAs and proteins from the soma to the synapse. There is currently some evidence that TDP-43 plays an important role at the synapse. TDP-43 regulates RNA processing of many genes that encode synaptic proteins, including presynaptic markers like synaptotagmin, and glutamate transporters and receptors (Polymenidou, et al., 2011; Honda, et al., 2014). Under normal conditions, TDP-43 is found in the dendrites to colocalize with RNA granules and the dendritic marker PSD-95 (Wang, Wu, Chang, & Shen, 2008). TDP-43 also plays a role in mRNA transport and local translation in dendritic spines (Wang, Wu, Chang, & Shen, 2008; Sephton & Yu, 2015). In response to neuronal stimulation, there is increased localization of TDP-43 to dendritic spines (Wang, Wu, Chang, & Shen, 2008), indicating that TDP-43 is activity-responsive in postsynaptic neurons. Studies using Drosophila neuromuscular junctions revealed that TDP-43 is necessary for synaptic growth, formation and pruning (Godena, et al., 2011; Lin, Cheng, & Shen, 2011). Another study using a Drosophila model shows that the Drosophila homolog of TDP-43, TBPH binds to and regulates the expression of the mRNA of syntaxin 1A, which is a vesicular protein involved in vesicular fusion and exocytosis (Romano, et al., 2014). Our group has found that TDP-43 controls the expression of the synaptic proteins synapsin I and synaptotagmin (Heyburn, et al., 2016). Overexpression of TDP-43 in neurons leads to decreased expression of these proteins, providing further evidence of the importance of TDP-43 for synaptic function (Heyburn, et al., 2016).
We recently reported that TDP-43 overexpression did not change the number of dendritic spines in the mouse brain, but that it does lead to impaired tricarboxylic acid (TCA) cycle function, as evidenced by a reduction in the TCA cycle intermediates succinate and citrate, and to increased oxidative stress, as evidenced by increased lactate production (Hebron, Chen, Miessau, Lonskaya, & Moussa, 2014; Heyburn, et al., 2016). The TCA cycle occurs in mitochondria, which are present in high concentrations at the pre-synaptic terminal (Lee & Peng, 2008). We did find, however, that reduction of soluble and nuclear TDP-43 significantly increased dendritic spine numbers and restored TCA cycle function (Chen, et al., 2014; Heyburn, et al., 2016), suggesting that pre-synaptic mitochondrial integrity may contribute to maintenance of glutamate metabolism and could affect brain plasticity by increasing excitatory post-synaptic spine density.
The protein granules with which TDP-43 associates regulate the distribution, translation, and degradation of mRNAs and interact with each other regulate mRNA and translation (Sephton & Yu, 2015). Loss of synapses is one of the first events in neurodegeneration. Taken together, this shows the importance of TDP-43 in the maintenance of healthy synapses and how disruption of TDP-43 could lead to neurodegeneration.
Glutamate and neuroinflammation
Homeostasis of the excitatory neurotransmitter glutamate is important for maintenance of functional synapses (Zeng, et al., 2007). When glutamate is released by excitatory neurons, it binds to glutamate receptors on the postsynaptic cell. It is then converted in astrocytes to glutamine, which is then fed back to the presynaptic neuron for recycling into glutamate. Failure to maintain appropriate levels of glutamate can cause excitotoxicity. It is thought that excitotoxicity may contribute to motor neuron cell death in ALS by repeatedly activating postsynaptic glutamate receptors (Ilieva, Polymenidou, & Cleveland, 2009; Lee, et al., 2016). Under normal conditions, glutamate released into the synapse can be taken up by astrocytes via excitatory amino acid transporters (EAATs). It has been found that levels of EAAT2 are decreased in ALS (Rothstein, Martin, & Kuncl, 1992; Rothstein, Van Kammen, Levey, Martin, & Kuncl, 1995), suggesting that there is impaired glutamate uptake in ALS, leading to high levels of extracellular glutamate, which leads to excitotoxicity. One of the only two FDA-approved drugs for the treatment of ALS is Riluzole which acts on the glutamatergic system. Taken together, this indicates that glutamate homeostasis is likely disrupted in ALS and other neurodegenerative diseases, and restoration of neurotransmitter balance is important for treating disease.
ALS is a non-cell autonomous disease, affecting non-neuronal cells such as astrocytes and microglia (Lee, et al., 2016). Astrocyte dysfunction can disrupt their ability to support neurons, leading to motor and non-motor neuron cell death (Philips & Rothstein, 2014). Microglia are also involved in central nervous system (CNS) immunity and release pro-inflammatory and anti-inflammatory cytokines and chemokines in response to an insult and other cellular signals (Philips & Rothstein, 2014). Previous work with TDP-43 mouse models expressing mutant TDP-43A315T or TDP-43M337V shows increased reactive astrogliosis and microglial activation (Stallings, Puttaparthi, Luther, Burns, & Elliott, 2010; Wegorzewska, Bell, Cairns, Miller, & Baloh, 2010). This gliosis has also been observed in mouse models overexpressing human wild-type TDP-43 (Xu, et al., 2010). Further, TDP-43 pathology is observed not only in neurons of ALS/FTLD patients, but also in astrocytes and microglia (Kwong, Neumann, Sampathu, Lee, & Trojanowski, 2007). These findings suggest that cellular inflammation is another important aspect of TDP-43 pathology, and that multiple cell types in the CNS are affected in TDP-43 proteinopathies.
We found that overexpression of neuronal TDP-43 can affect astrocytes, as evidenced by a reduction in glial fibrillary acidic protein (GFAP) expression and alteration of glutamate, glutamine, and aspartate levels. (Heyburn, et al., 2016), suggesting either reduced efficiency or lack of involvement of astrocytes to detoxify glutamate. This change in astrocyte activity was independent of EAAT1/2 levels (Hebron, Chen, Miessau, Lonskaya, & Moussa, 2014; Heyburn, et al., 2016). This change in amino acid homeostasis was also associated with elevation of γ-amino butyric acid (GABA) neurotransmitter levels suggesting conversion of glutamate into GABA instead of glutamine, perhaps as an alternate cellular quality control mechanism to detoxify glutamate under conditions of astrocyte dysfunction (Heyburn, et al., 2016).
These alterations in synaptic glutamate and astrocyte function were not found to be associated with significant changes of brain inflammatory markers including microglial morphology or number (Heyburn, et al., 2016), suggesting that the suppression by TDP-43 of key synaptic proteins which mediate vesicular neurotransmitter release may sequester glutamate in vesicles and prevent its effects on neuroinflammation. Alternatively, neuronal overexpression of TDP-43 may lead to astrocytic dysfunction, attenuating production of inflammatory molecules that provoke microglial response to exacerbate neuroinflammation and lead to cell death.
Autophagy
RNA granules are assembled and disassembled as part of their homeostatic dynamics. However, when they persist or are unable to be disassembled, they must be degraded by some mechanism, or else they could become cytotoxic. These protein aggregates can be degraded by autophagy, a process of disposal of cellular components (Deter, Baudhuin, & De Duve, 1967). In autophagy, granules or aggregated proteins, for example, are tagged by ubiquitin, which signals to the autophagic machinery for degradation. A double membrane called an autophagosome forms around the ubiquitinated proteins. The autophagosome then fuses with the lysosome, forming an autophagolysosome, where the contents are degraded by acidic enzymes (Monahan, Shewmaker, & Pandey, 2016). Aggregated forms of TDP-43 are degraded via autophagy (Xia, et al., 2016).
Impaired protein degradation may be involved in neurodegenerative diseases, including in ALS and FTLD (Gotzl, Lang, Haass, & Capell, 2016). Autophagy has been shown to be dysfunctional in forms of ALS and caused by TARDBP mutations and it has been suggested that TDP-43 is involved in regulation of autophagy by affecting the biogenesis of autophagosomes and lysosomes (Filimonenko, et al., 2007; Bose, Huang, & Shen, 2011; Xia, et al., 2016; Ying, et al., 2016). Because of the importance of protein homeostasis in maintaining healthy neurons, a functional protein degradation system is highly important. This creates a possible target for treatment: activation of autophagic mechanisms to clear aberrant proteins. Indeed, studies have been done to investigate the effects of autophagic activation and inhibition on protein clearance. Activation of autophagy by rapamycin has been found to promote clearance of SGs and inhibition of autophagy led to the opposite (Buchan, Kolaitis, Taylor, & Parker, 2013). Scotter et al. found that treatment of cells with an autophagy inhibitor reduced degradation of TDP-43 (Scotter, et al., 2014). Treatment of TDP-43 transgenic mice with rapamycin decreases motor impairments (Wang, et al., 2012). Another pharmacological method of inducing autophagy is the use of tyrosine kinase inhibitors (TKIs). We have shown that TKIs induce autophagy and lead to protein degradation in mice and in vitro (Lonskaya, Hebron, Desforges, Franjie, & Moussa, 2013; Lonskaya, Hebron, Desforges, Schachter, & Moussa, 2014; Chen, et al., 2014). Tyrosine kinase inhibition has been shown to increase autophagy in other cell types and animal models (Bellodi, et al., 2009; Shaker, Ghani, Shiha, Ibrahim, & Mehal, 2013; Yu, et al., 2013; Mahul-Mellier, et al., 2014). TKIs, including the FDA-approved chronic myelogenous leukemia drugs Nilotinib and Bosutinib, reverse cell death in transgenic TDP-43 mice (Chen, et al., 2014) by differentially modifying TDP-43. Nilotinib reduces nuclear, soluble, cleaved, and insoluble TDP-43, while Bosutinib only reduces nuclear and soluble TDP-43 (Chen, et al., 2014). We also showed that Parkin ubiquitinates TDP-43 and trans-locates it from the nucleus to the cytosol, resulting in a prevention of cell death (Hebron, et al., 2013; Hebron, Chen, Miessau, Lonskaya, & Moussa, 2014). This translocation was also shown to be concurrent with autophagic and proteasomal clearance of TDP-43 (Hebron, et al., 2013; Chen, et al., 2014). Because both of these TKIs were brain protective, suggesting that insoluble TDP-43 may be a sequestration strategy (Chen, et al., 2014). TDP-43 overexpression models are associated with neuronal TDP-43 accumulation and suppression of pre-synaptic proteins as shown by a reduction of synapsin and synaptotagmin mRNAs and proteins (Heyburn, et al., 2016). This alteration in pre-synaptic proteins is associated with neuronal cell death and lack of glutamate detoxification by astrocytes (Heyburn, et al., 2016). Levels of pre-synaptic proteins and glutamate/glutamine were restored when TDP-43 levels were reduced and TDP-43 nucleocytoplasmic distribution was altered by treatment with TKIs (Chen, et al., 2014; Heyburn, et al., 2016). Together, these data show that reducing TDP-43 levels could be protective against neurodegeneration.
Animal Models
In order to understand the role of TDP-43 in disease pathogenesis, it is necessary to use animal models. Since the discovery of the involvement of TARDBP and TDP-43 protein in ALS-FTLD, researchers have aimed to create a model of TDP-43 pathology that recapitulates disease. Many animal models of TDP-43 pathology have been generated, with varying levels of reproduction of disease phenotype. Below are various animal models that have been created to study ALS-FTLD, with particular focus on TDP-43 models.
SOD1
The first gene found to be associated with ALS was superoxide dismutase 1 (SOD1) (Rosen, et al., 1993), and early animal models of ALS focused on SOD1 genetics (Philips & Rothstein, 2015). SOD1 knock-out mice appear to be normal, so SOD1 mutations are thought to lead to a toxic gain of function (Philips & Rothstein, 2015). Over 10 different rodent models overexpressing human mutant SOD1 have been created and characterized. Most of the mutant SOD1 lines develop adult onset progressive motor neuron deficits which resemble human ALS. They exhibit loss of spinal cord motor neurons, muscle wasting and atrophy leading to paralysis and death (Philips & Rothstein, 2015). These SOD1 models have been helpful for identifying disease-related mechanisms and for researching potential therapies. However, because SOD1 mutation is only responsible for a small percentage of all ALS cases and patients with SOD1 mutation do not exhibit TDP-43 pathology, these rodent models are not as useful for studying TDP-43 pathologies.
TARDBP/TDP-43
Animal models that exhibit TDP-43 pathology and recapitulate ALS-FTD symptoms are important for understanding how TDP-43 pathology contributes to disease. Total knock-out of TDP-43 is embryonically lethal (Wu, et al., 2009; Kraemer, et al., 2010; Sephton, et al., 2010). However, mice with one copy of TDP-43 knocked down (TARDBP+/−) have normal levels of TDP-43 and no neuropathology (Wu, et al., 2009; Kraemer, et al., 2010; Sephton, et al., 2010), suggesting that a single allele expression of TDP-43 may compensate for the cellular role of TDP-43. This is likely due to the fact that TDP-43 levels are very tightly regulated via TDP-43 auto-regulation. Because these knock-out and knock-down models proved to be insufficient to make a disease model, researchers have instead used human TDP-43 expression models which express the human form of the TDP-43 gene, leading to expression of human, rather than mouse, TDP-43 protein. These models vary in both the transgene as well as the promotor used to drive transgene expression.
Some groups have used the mouse prion promoter (PrP) in order to have ubiquitous transgene expression (Stallings, Puttaparthi, Luther, Burns, & Elliott, 2010; Wegorzewska, Bell, Cairns, Miller, & Baloh, 2010; Xu, et al., 2010; Xu, et al., 2011). Wegorzewska et al. created a mouse that expressed human TDP-43 with a A315T mutation. This line expresses about 3 time more endogenous mouse TDP-43 in the spinal cord (Wegorzewska, Bell, Cairns, Miller, & Baloh, 2010). The mice developed an abnormal gait and exhibited loss of cortical layer V neurons and a 20% loss of ventral horn motor neurons (Wegorzewska, Bell, Cairns, Miller, & Baloh, 2010). TDP-43 was found in the nucleus of most neurons and glia, but was expressed in the cytoplasm of layer V and ventral horn motor neurons, which also contained ubiquitinated aggregates (Wegorzewska, Bell, Cairns, Miller, & Baloh, 2010). Stallings et al. used the PrP to express human wild-type TDP-43 in one line and human A315T mutated TDP-43 in another (4x endogenous TDP-43 level). The TDP-43WT line did not exhibit any motor phenotype, but the mutant TDP-43 line exhibited loss of grip strength and death within 5 months (Stallings, Puttaparthi, Luther, Burns, & Elliott, 2010). Xu et al. also produced two PrP lines: one expressing human TDP-43WT (2.5x endogenous TDP-43) (Xu, et al., 2010) and one expressing human TDP-43M337V (2.7x endogenous TDP-43) (Xu, et al., 2011). Both lines had early death and increased phosphorylated TDP-43 in spinal cord motor neurons (Xu, et al., 2010; Xu, et al., 2011). In these PrP lines, increased expression of human TDP-43 led to motor deficits and early death, and the higher the expression, the more severe the phenotypes.
Other groups have created lines where transgene expression is driven by the murine Thy1 promoter, which causes neuronal-specific expression. Shan et al. created a mouse expressing human TDP-43WT under the Thy1.2 promoter, a modified murine Thy1 promoter (Shan, Chiang, Price, & Wong, 2010). Males in this line have 4.6x endogenous TDP-43 and develop gait abnormalities, hindlimb clasp, and tremor (Shan, Chiang, Price, & Wong, 2010). They also have diffused nuclear TDP-43 staining in spinal cord neurons and intranuclear aggregates that colocalized with FUS (Shan, Chiang, Price, & Wong, 2010). Females of this line have 2.3x endogenous TDP-43 levels and have much less severe symptoms (Shan, Chiang, Price, & Wong, 2010). Wils et al. also made a line that expressed human TDP-43WT under the Thy1.2 promoter (Wils, et al., 2010). One homozygous line (TAR4/4) has 2x endogenous TDP-43, another (TAR6/6) has 1.2x endogenous levels, and the hemizygous (TAR4) line has 1x (Wils, et al., 2010). TAR4/4 have impaired motor function and spastic paralysis and die before 1 month (Wils, et al., 2010). TAR6/6 mice have motor deficits but live 6.7 months on average (Wils, et al., 2010). The TAR4 mice have impaired rotarod at 15 months (Wils, et al., 2010). TAR4/4 mice have nuclear and cytoplasmic protein aggregates in the spinal cord and layer V of the cortex which contain phosphorylated TDP-43. They have 30% loss of layer V neurons and 25% loss of spinal cord neurons (Wils, et al., 2010). These models all show both mutant and wild-type TDP-43 overexpression leads to motor deficits and pathology. Our lab has shown that the hemizygous overexpressing mice bred from the Wils et al. strain (Wils, et al., 2010) have increased TDP-43 expression in the brain, particularly in the hippocampus and forebrain (Chen, et al., 2014). These mice exhibit neuronal cell death in the cortex and decreased axonal myelination in the spinal cord, along with impairment on rotarod, Morris Water Maze, and novel object recognition tests (Chen, et al., 2014). Our laboratory demonstrated that overexpression of human wild type neuronal TDP-43 can mimic TDP-43 pathologies in a transgenic animal model that displays cognitive, behavioral, and motor symptoms (Chen, et al., 2014; Heyburn, et al., 2016). Homozygous TDP-43 overexpressing mice mimic MND pathology, displaying weakness, paralysis and hunch back; but hemizygous littermates exhibit symptoms that are closer to the FTLD-TDP phenotype, including anxiety and learning and memory deficits (Chen, et al., 2014; Heyburn, et al., 2016).
The Ca2+/calmodulin-dependent kinase II (CaMKII) promoter has also been used, to drive expression of mouse TDP-43 in the hippocampus and cortex, which led to behavioral and motor deficits and neuronal death in the cortex (Tsai, et al., 2010). This model has 2x endogenous murine TDP-43 and has impaired water maze performance and fear conditioning as well as impaired rotarod and abnormal clasping (Tsai, et al., 2010). Furthermore, Igaz et al. created a mouse line with an inducible CaMKII promoter that drives expression of human TDP-43WT, which is turned on at 28 days (Igaz, et al., 2009). This led to neuron loss in the hippocampus as well as limb clasping behavior (Igaz, et al., 2009). Swarup et al. used a full-length fragment from human bacterial artificial chromosome (BAC) to express human TDP-43WT, human TDP-43A315T, and human TDP-43G348C (Swarup, et al., 2011). All lines express 3x the endogenous level of TDP-43 and exhibit behavioral and motor deficits (Swarup, et al., 2011). A similar BAC method was used by Zhou et al. to express human TDP-43WT and human TDP-43M337V (Zhou, et al., 2010). The WT line did not exhibit paralysis, but the mutant line became paralyzed before 30 days (Zhou, et al., 2010). The same group also made an inducible rat that expresses human TDP-43M337V which exhibits paralysis and death within 2 months (Zhou, et al., 2010).
Because constitutive knock-out of TDP-43 proved to be lethal, (Wu, et al., 2009; Kraemer, et al., 2010; Sephton, et al., 2010) conditional knock-outs are necessary to study TARDBP depletion. Chiang et al. have developed a conditional knockout by flanking the 3rd exon of TARDBP by loxp with a neomycin resistance gene inserted into the 2nd intron (Chiang, et al., 2010). Floxed TARDBP mice crossed with CAG-Cre mice creates a heterozygous TARDBP knockout (+/−). These mice express a similar level of TDP-43 to wild-type mice and had a metabolic phenotype and premature death (Chiang, et al., 2010).
These TDP-43 animal models have mostly been evaluated as ALS models. Many of the lines have severe motor deficits, making them hard to evaluate as FTD models. However, in the studies that examined brain pathology, layer V of the cortex had TDP-43 pathology and loss of neurons (Wegorzewska, Bell, Cairns, Miller, & Baloh, 2010; Wils, et al., 2010). This reflects the vulnerability of layer V pyramidal neurons observed in FTLD (Roberson, 2012). Protein pathology similar to that in ALS and FTLD was observed in most of the discussed TDP-43 mouse models. There was phenotypic variation in the lines, which exhibited ALS-like symptoms of motor deficits and weakness as well as FTD-like symptoms such as memory and social deficits.
In addition to these many rodent models of TDP-43 pathology, there are also many models created in other species, including zebrafish, C. elegans, Drosophila, and yeast. The yeast Saccharomyces cerevisiae has been used to study TDP-43 aggregation and toxicity and the effects of mutated TDP-43 (Johnson, McCaffery, Lindquist, & Gitler, 2008; Johnson, et al., 2009). The fruit fly Drosophila melanogaster has been used to express wild-type and mutant human and Drosophila TDP-43, and various fly models exhibit cell death, reduced lifespan, and ALS-associated phenotypes (Li, et al., 2010; Feiguin, et al., 2009; Zhan, Hanson, Kim, Tare, & Tibbetts, 2013). The roundworm Caenorhabditis elegans has a TDP-43 ortholog called tdp-1. Various C. elegans models have been made which have motor phenotypes, protein aggregation, and impaired synaptic function (Ash, et al., 2010; Liachko, Guthrie, & Kraemer, 2010; Zhang, Hwang, Hao, Talbot, & Wang, 2012; Vaccaro, Tauffenberger, Ash, Carlomagno, & Parker, 2012). The zebrafish Danio rerio has been used to model TDP-43 pathology either by introducing exogenous TDP-43 or by alteration of endogenous zebrafish TDP-43. These models exhibit motor deficits, motor axonopathy, and muscle degeneration (Schmid, et al., 2013; Kabashi, et al., 2010; Laird, et al., 2010). In addition to these models, there have also been recent studies using induced pluripotent stem cells (iPSCs) from patients which can be induced to become motor neurons, which exhibit decreased survival, TDP-43 mislocalization, and increased TDP-43 insolubility (Bilican, et al., 2012).
C9ORF72
The link between C9ORF2 and TDP-43 has been studied (Zufiria, et al., 2016). The presence of anti-sense foci for the HRE is correlated with mislocalization and accumulation of TDP-43 in the cerebellum neurons in ALS patients (Zufiria, et al., 2016). Knock-out of C9ORF72 does not cause motor neuron death or decreased survival (Koppers, et al., 2015). An Adeno-associated virus (AAV) model with 66 repeats showed phosphorylated TDP-43 inclusions, motor deficits, and anxiety and social dysfunction (Chew, et al., 2015). Jiang et al. created a BAC mouse model that has 450 repeats that have RNA foci and age-dependent cognitive deficits (Jiang, et al., 2016). Liu et al. created a BAC model with 500 repeats which have TDP-43 inclusions as well as muscle denervation, motor neuron death, anxiety, and paralysis (Liu, et al., 2016). This area of model creation is still relatively new and further development of C9ORF72 models will likely provide insight into disease pathogenesis as well as the relationship between C9ORF72 and TDP-43.
Conclusion
Figure 1 shows the role of TDP-43 in synaptic function and maintenance as well as how alterations in TDP-43 expression affects the synapse. TDP-43 pathology is associated with neurodegenerative diseases, particularly ALS and FTLD. Its extensive role as an RNA-binding protein underlines the importance of impaired RNA metabolism in disease pathogenesis. Alteration of TDP-43 protein homeostasis can lead to many downstream deficits, each of which can contribute to development of neurodegenerative processes. Understanding the process of protein imbalance as well as the countless molecular and cellular effects is necessary for developing targets for intervention. treating TDP-43 pathologies via modulation of its role in RNA metabolism or via autophagic clearance represent an important area of progress, as there is currently no cure for ALS or FTLD. Elucidation of disease mechanisms will provide the missing link necessary for creating effective disease treatments.
Figure 1. TDP-43 at the synapse.
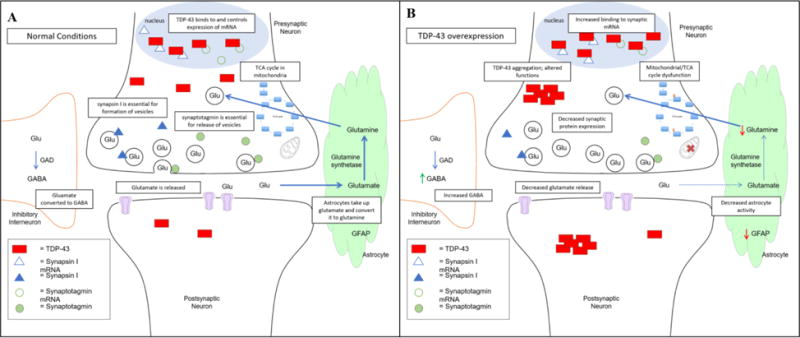
A) Under normal conditions, TDP-43 controls the expression of synaptic proteins such as synaptotagmin and synapsin I. It is present in postsynaptic dendrites, where it is involved in local protein translation. Normal TDP-43 expression is necessary for synaptic maintenance, neurotransmitter balance, and mitochondrial function. B) When TDP-43 is overexpressed, there is aberrant binding to its mRNA targets, leading to alterations in synaptic function. TDP-43 accumulates in the cytoplasm, forming protein aggregates. This aggregation can lead to loss of function or a toxic gain of function, which contribute to disease. TDP-43 overexpression is also associated with neurotransmitter imbalance, astrocyte dysfunction, and mitochondrial impairment. Maintenance of TDP-43 levels is crucial for normal synaptic maintenance and function.
Highlights.
TDP-43 is an RNA-binding protein implicated in neurodegeneration
This review summarizes existing knowledge and new contributions to the TDP-43 field
TDP-43 disruption leads to synaptic dysfunction and neurotransmitter balance
Acknowledgments
Funding
This work was supported by Georgetown University funding to Charbel Moussa and NINDS grant [5T32NS041218] to Lanier Heyburn
Abbreviations
- AAV
adeno-associated virus
- ALS
amyotrophic lateral sclerosis
- BAC
bacterial artificial chromosome
- bvFTD
behavioral variant FTD
- C9ORF72
chromosome 9 open reading frame 72
- CaMKII
Calcium/calmodulin-dependent kinase II
- CFTR
cystic fibrosis transmembrane conductance regulator
- EAAT
excitatory amino acid transporter
- FTD
frontotemporal dementia
- FTLD
frontotemporal lobar degeneration
- FUS
fused in sarcoma
- GABA
γ-amino butyric acid
- GFAP
glial fibrillary acidic protein
- GRN
granulin
- GU
guanine, uracil
- hnRNP
heterogenous nuclear ribonucleoprotein
- HRE
hexanucleotide repeat expansion
- IBMPFD
Paget’s disease of bone and frontotemporal dementia
- IL-6
interleukin 6
- miRNA
micro RNA
- MND
motor neuron disease
- NCI
neuronal cytoplasmic inclusions
- NES
nuclear export signal
- NLS
nuclear localization sequence
- OPTN
optineurin
- P-body
processing body
- PFN1
profilin 1
- PNFA
progressive nonfluent aphasia
- PrP
prion protein promoter
- PSD-95
post-synaptic density 95
- RRM
RNA recognition motif
- SD
semantic dementia
- SG
stress granule
- SOD1
superoxide dismutase 1
- SQSTM1
sequestosome
- TDP-43
transactive response DNA binding protein 43
- TIA-1
T-cell intracellular antigen 1
- TKI
tyrosine kinase inhibitor
- tRNP
transport ribonucleoprotein
- UBQLN2
ubiquilin 2
- VCP
Valosin-containing protein
- VEGFA
vascular endothelial growth factor A
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure: Charbel Moussa is listed as an inventor of a patent to use tyrosine kinase inhibitors as a treatment for neurodegenerative diseases.
References
- Alami N, Smith R, Carrasco M, Williams L, Winborn C, Han S, Chandran S. Axonal transport of TDP-43 mRNA granules in neurons is impaired by ALS-causing mutations. Neuron. 2014;81(3):536–543. doi: 10.1016/j.neuron.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash P, Zhang YJ, Roberts C, Saldi T, Hutter H, Buratti E, Link C. Neurotoxic effects of TDP-43 overexpression in C. elegans. Human Molecular Genetics. 2010;19(16):3206–3218. doi: 10.1093/hmg/ddq230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala Y, Pantano S, D’Ambrogio A, Buratti E, Brindisi A, Marchetti C, Baralle FE. Human, Drosophila, and C.elegans TDP43: nucleic acid binding properties and splicing regulatory function. Journal of Molecular Biology. 2005;348(3):575–588. doi: 10.1016/j.jmb.2005.02.038. [DOI] [PubMed] [Google Scholar]
- Ayala Y, De Conti L, Avendano-Vazquez SE, Dhir A, Romano M, D’Ambrogio A, Baralle FE. TDP-43 regulates its mRNA levels through a negative feedback loop. The EMBO Journal. 2010;30(2):277–288. doi: 10.1038/emboj.2010.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baralle M, Buratti E, Baralle FE. The role of TDP-43 in the pathogenesis of ALS and FTLD. Biochemical Society Transactions. 2013;41(6):1536–1540. doi: 10.1042/BST20130186. [DOI] [PubMed] [Google Scholar]
- Barbee SA, Estes PS, Cziko AM, Hillebrand J, Luedeman RA, Coller JM, Nakamura, et al. Staufen- and FMRP-Containing Neuronal RNPs Are Structurally and Functionally Related to Somatic P Bodies. Neuron. 2006;52(6):997–1009. doi: 10.1016/j.neuron.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bellodi C, Lidonnici M, Hamilton A, Helgason G, Soliera A, Ronchetti M, et al. Targeting autophagy potentiates tyrosine kinase inhibitor–induced cell death in Philadelphia chromosome–positive cells, including primary CML stem cells. Journal of Clinical Investigation. 2009;119(5):1109–1123. doi: 10.1172/JCI35660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, Golfier V. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Annals of Neurology. 2009;65(4):470–473. doi: 10.1002/ana.21612. [DOI] [PubMed] [Google Scholar]
- Bilican B, Serio A, Barmada S, Nishimura A, Sullivan G, Carrasco M, Hardingham G. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. PNAS. 2012;109(15):5803–5808. doi: 10.1073/pnas.1202922109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroni B, Bonvicini C, Alberici A, Buratti E, Agosti C, Archetti S, Padovani A. Mutation within TARDBP leads to Frontotemporal Dementia without motor neuron disease. Human Mutation. 2009;30(11):E974–E983. doi: 10.1002/humu.21100. [DOI] [PubMed] [Google Scholar]
- Borroni B, Archetti S, Del Bo R, Papetti A, Buratti E, Bonvicini C, Turla M. TARDBP Mutations in Frontotemporal Lobar Degeneration: Frequency, Clinical Features, and Disease Course. Rejuvenation Research. 2010;13(5):509–517. doi: 10.1089/rej.2010.1017. [DOI] [PubMed] [Google Scholar]
- Bose JK, Huang CC, Shen CKJ. Regulation of Autophagy by Neuropathological Protein TDP-43. Journal of Biological Chemistry. 2011;286:44441–44448. doi: 10.1074/jbc.M111.237115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan RJ, Kolaitis RM, Taylor JP, Parker R. Eukaryotic Stress Granules Are Cleared by Autophagy and Cdc48/VCP Function. Cell. 2013;153:1461–1474. doi: 10.1016/j.cell.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. Characterization and Functional Implications of the RNA Binding Properties of Nuclear Factor TDP-43, a Novel Splicing Regulator ofCFTR Exon 9. Journal of Biological Chemistry. 2001;276:36337–36343. doi: 10.1074/jbc.M104236200. [DOI] [PubMed] [Google Scholar]
- Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala Y, Baralle FE. TDP-43 Binds Heterogeneous Nuclear Ribonucleoprotein A/B through Its C-terminal Tail. Journal of Biological Chemistry. 2005;280:37572–37584. doi: 10.1074/jbc.M505557200. [DOI] [PubMed] [Google Scholar]
- Cardarelli R, Kertesz A, Knebl J. Frontotemporal dementia: a review for primary care physicians. American Family Physician. 2010;82(11):1372–1377. [PubMed] [Google Scholar]
- Chaudhury A, Chander P, Howe PH. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: Focus on hnRNP E1’s multifunctional regulatory roles. RNA. 2010;16(8):1449–1462. doi: 10.1261/rna.2254110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, Lin R, Hsieh E, Tu PH, Chen R, Liao TY, Huang J. Induction of Amyloid Fibrils by the C-Terminal Fragments of TDP-43 in Amyotrophic Lateral Sclerosis. Journal of the American Chemical Society. 2010;132(4):1186–1187. doi: 10.1021/ja9066207. [DOI] [PubMed] [Google Scholar]
- Chen W, Lonskaya I, Hebron M, Ibrahim Z, Olszewski R, Neale J, Moussa C. Parkin-mediated reduction of nuclear and soluble TDP-43 reverses behavioral decline in symptomatic mice. Human Molecular Genetics. 2014;23(18):4960–4969. doi: 10.1093/hmg/ddu211. [DOI] [PubMed] [Google Scholar]
- Chen-Plotkin A, Lee V, Trojanowski J. TAR DNA-binding protein 43 in neurodegenerative disease. Nature Reviews Neurology. 2010;6(4):211–220. doi: 10.1038/nrneurol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew J, Gendron T, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Stankowski J. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015;348(6239):1151–1154. doi: 10.1126/science.aaa9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang PM, Ling J, Jeong YH, Price D, Aja S, Wong P. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. PNAS. 2010;107(37):16320–16324. doi: 10.1073/pnas.1002176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio A, Calvo A, Moglia C, Restagno G, Ossola I, Brunetti M, Borghero G. Amyotrophic Lateral Sclerosis–Frontotemporal Lobar Dementia in 3 Families With p.Ala382Thr TARDBP Mutations. Archives of Neurology. 2010;67(8):1002–1009. doi: 10.1001/archneurol.2010.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen TJ, Lee VM, Trojanowski JQ. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol Med. 2011;17(11):659–667. doi: 10.1016/j.molmed.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, Buratti E, Ratti A. TDP-43 is recruited to stress granules in conditions of oxidative insult. Journal of Neurochemistry. 2009;111:1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x. [DOI] [PubMed] [Google Scholar]
- Colombrita C, Onesto E, Megiorni F, Pizzuti A, Baralle F, Buratti E, Ratti A. TDP-43 and FUS RNA-binding Proteins Bind Distinct Sets of Cytoplasmic Messenger RNAs and Differently Regulate Their Post-transcriptional Fate in Motoneuron-like Cells. Journal of Biological Chemistry. 2012;287(19):15635–15647. doi: 10.1074/jbc.M111.333450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conicella A, Zerze G, Mittal J, Fawzi N. ALS Mutations Disrupt Phase Separation Mediated by α-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure. 2016;24(9):1537–1549. doi: 10.1016/j.str.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Current Opinions in Neurobiology. 2011;21(6):904–919. doi: 10.1016/j.conb.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie I, Boeve B, Boxer A, Baker M, Rutherford N, Hsiung GY. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deter R, Baudhuin P, De Duve C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. Journal of Cell Biology. 1967;35(2) doi: 10.1083/jcb.35.2.C11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey CM, Cenik B, Sephton CF, Johnson BA, Herz J, Yu G. TDP-43 Aggregation In Neurodegeneration: Are Stress Granules The Key? Brain Research. 2012;1462:16–25. doi: 10.1016/j.brainres.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C, Bassell G, Rossoll W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Human Molecular Genetics. 2012;21(16):3703–3718. doi: 10.1093/hmg/dds205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiguin Fabian, Godena V, Romano G, D’Ambrogio A, Klima R, Baralle F. Depletion of TDP-43 affects drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Letters. 2009;583(10):1586–1592. doi: 10.1016/j.febslet.2009.04.019. [DOI] [PubMed] [Google Scholar]
- Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerod L, Fisher EM, Simonsen A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. Journal of Cell Biology. 2007;179(3):485–500. doi: 10.1083/jcb.200702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilks N, Kedersha N, Ayodele M, Shen L, Stoeklin G, Dember LM, Anderson P. Stress Granule Assembly Is Mediated by Prion-like Aggregation of TIA-1. Molecular Biology of the Cell. 2004;15(12):5383–5398. doi: 10.1091/mbc.E04-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godena VK, Romano G, Romano M, Appocher C, Klima R, Buratti E, Feiguin F. TDP-43 regulates Drosophila neuromuscular junctions growth by modulating Futsch/MAP1Blevels and synaptic microtubules organization. PLoS One. 2011;6:e17808. doi: 10.1371/journal.pone.0017808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotzl JK, Lang CM, Haass C, Capell A. Impaired protein degradation in FTLD and related disorders. Ageing Research Reviews. 2016;32:122–139. doi: 10.1016/j.arr.2016.04.008. [DOI] [PubMed] [Google Scholar]
- Hardy J, Rogaeva E. Motor neuron disease and frontotemporal dementia: sometimes related, sometimes not. Experimental Neurology. 2014;262:75–83. doi: 10.1016/j.expneurol.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Hebron M, Lonskaya I, Sharpe K, Weerasinghe PP, Algarzae N, Shekoyan A, Moussa C. Parkin Ubiquitinates Tar-DNA Binding Protein-43 (TDP-43) and Promotes Its Cytosolic Accumulation via Interaction with Histone Deacetylase 6 (HDAC6) Journal of Biological Chemistry. 2013;288(6):4103–4115. doi: 10.1074/jbc.M112.419945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebron M, Chen W, Miessau M, Lonskaya I, Moussa C. Parkin reverses TDP-43-induced cell death and failure of amino acid homeostasis. Journal of Neurochemistry. 2014;129(2):350–361. doi: 10.1111/jnc.12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyburn L, Hebron M, Smith J, Winston C, Bechara J, Li Z, Moussa C. Tyrosine kinase inhibition reverses TDP-43 effects on synaptic protein expression, astrocytic function and amino acid dis-homeostasis. Journal of Neurochemistry. 2016;139(4):610–623. doi: 10.1111/jnc.13763. [DOI] [PubMed] [Google Scholar]
- Honda D, Ishigaki S, Iguchi Y, Fujioka Y, Udagawa T, Masuda A, Sobue G. The ALS/FTLD-relatedRNA-binding proteins TDP-43 and FUS have common downstreamRNA targets in cortical neurons. FEBS Open Bio. 2014;4:1–10. doi: 10.1016/j.fob.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaz L, Kwong L, Chen-Plotkin A, Winton M, Unger T, Xu Y, Lee V. Expression of TDP-43 C-terminal Fragments in Vitro Recapitulates Pathological Features of TDP-43 Proteinopathies. Journal of Biological Chemistry. 2009;284(13):8516–8524. doi: 10.1074/jbc.M809462200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland D. Non–cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. Journal of Cell Biology. 2009;187(6):761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens J, Van Broeckhoven C. Pathological mechanisms underlying TDP-43 driven neurodegeneration in FTLD–ALS spectrum disorders. Human Molecular Genetics. 2013;22(R1):R77–R87. doi: 10.1093/hmg/ddt349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Zhu Q, Gendron T, Saberi S, McAlonis-Downes M, Seelman A, Engelhardt J. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron. 2016;90(3):535–550. doi: 10.1016/j.neuron.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B, McCaffery JM, Lindquist S, Gitler A. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. PNAS. 2008;105(17):6439–6444. doi: 10.1073/pnas.0802082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B, Snead D, Lee J, McCaffery JM, Shorter J, Gitler A. TDP-43 Is Intrinsically Aggregation-prone, and Amyotrophic Lateral Sclerosis-linked Mutations Accelerate Aggregation and Increase Toxicity. J Biol Chem. 2009;284(30):20329–20339. doi: 10.1074/jbc.M109.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs K, Hodges J, Snowden J, Mackenzie I, Neumann M, Mann D, Dickson D. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathologica. 2011;122:137–153. doi: 10.1007/s00401-011-0839-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Lin L, Tradewell M, Dion P, Bercier V, Bourgouin P, Drapeau P. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Human Molecular Genetics. 2010;19(4):671–683. doi: 10.1093/hmg/ddp534. [DOI] [PubMed] [Google Scholar]
- Koppers M, Blokhuis A, Westeneng HJ, Terpstra M, Zundel C, de Sa R, Pasterkamp RJ. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Annals of Neurology. 2015;78(3):426–438. doi: 10.1002/ana.24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs G, Murrell J, Horvath S, Haraszti L, Majtenyi K, Molnar M, Spina S. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Movement Disorders. 2009;24(12):1843–1847. doi: 10.1002/mds.22697. [DOI] [PubMed] [Google Scholar]
- Kraemer B, Schuck T, Wheeler J, Robinson L, Trojanowski JQ, Lee V, Schellenberg G. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathologica. 2010;119(4):409–419. doi: 10.1007/s00401-010-0659-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong L, Neumann M, Sampathu D, Lee V, Trojanowski JQ. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathologica. 2007;144(1):63–70. doi: 10.1007/s00401-007-0226-5. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Hutt KR, Viu A, Baughn M, Huelga S, Cleveland D. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Naure Neuroscience. 2012;15(11):1488–1497. doi: 10.1038/nn.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird A, Van Hoecke AD, Timmers M, Van Den Bosch L, Van Damme P, Robberecht W. Progranulin is Neurotrophic In Vivo and Protects against a Mutant TDP-43 Induced Axonopathy. PLoS ONE. 2010;5(10):e13368. doi: 10.1371/journal.pone.0013368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CW, Peng HB. The Function of Mitochondria in Presynaptic Development at the Neuromuscular Junction. Molecular Biology of the Cell. 2008;19(1):150–158. doi: 10.1091/mbc.E07-05-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee T, Lee E, Kang S, Park A, Kim SW, Park B. Identification of a subnuclear body involved in sequence-specific cytokine RNA processing. Nature Communications. 2015;6 doi: 10.1038/ncomms6791. [DOI] [PubMed] [Google Scholar]
- Lee J, Hyeon SJ, Im H, Ryu H, Kim Y, Ryu H. Astrocytes and Microglia as Non-cell Autonomous Players in the Pathogenesis of ALS. Experimental Neurobiology. 2016;25(5):233–240. doi: 10.5607/en.2016.25.5.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Ray P, Rao E, Shi C, Guo W, Chen X, Wu J. A Drosophila model for TDP-43 proteinopathy. PNAS. 2010;107(7):3169–3174. doi: 10.1073/pnas.0913602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liachko N, Guthrie C, Kraemer B. Phosphorylation Promotes Neurotoxicity in a Caenorhabditis elegans Model of TDP-43 Proteinopathy. Journal of Neuroscience. 2010;30(48):16208–16219. doi: 10.1523/JNEUROSCI.2911-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MJ, Cheng CW, Shen CKJ. Neuronal Function and Dysfunction of Drosophila dTDP. PLoS One. 2011;6(6) doi: 10.1371/journal.pone.0020371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt D, Ranum L. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron. 2016;90(3):521–534. doi: 10.1016/j.neuron.2016.04.005. [DOI] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderwyde T, Citro A, Mehta T, Wolozin B. Tar DNA Binding Protein-43 (TDP-43) Associates with Stress Granules: Analysis of Cultured Cells and Pathological Brain Tissue. PLoS One. 2010;5(10) doi: 10.1371/journal.pone.0013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonskaya I, Hebron M, Desforges N, Franjie A, Moussa C. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Molecular Medicine. 2013;5(8):1247–1262. doi: 10.1002/emmm.201302771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonskaya I, Hebron M, Desforges N, Schachter J, Moussa C. Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. Journal of Molecular Medicine. 2014;92(4):373–386. doi: 10.1007/s00109-013-1112-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie I, Bigio E, Ince P, Geser F, Neumann M, Cairns N, Kirby J. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Annals of Neurology. 2007;61(5):427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- Mackenzie I, Neumann M. Reappraisal of TDP-43 pathology in FTLD-U subtypes. Acta Neuropathologica. 2017;134(1):79–96. doi: 10.1007/s00401-017-1716-8. [DOI] [PubMed] [Google Scholar]
- Mahul-Mellier AL, Fauvet B, Gysbers A, Dikiy I, Oueslati A, Georgeon S, Lashuel H. c-Abl phosphorylates α-synuclein and regulates its degradation: implication for α-synuclein clearance and contribution to the pathogenesis of Parkinson’s disease. Human Molecular Genetics. 2014;23(11):2858–2879. doi: 10.1093/hmg/ddt674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazroui R, Di Marco S, Kaufman RK, Gallouzi IE. Inhibition of the ubiquitin-proteasome system induces stress granule formation. Molecular Biology of the Cell. 2007;18:2603–2618. doi: 10.1091/mbc.E06-12-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan Z, Shewmaker F, Pandey UB. Stress granules at the intersection of autophagy and ALS. Brain Research. 2016;1649:189–200. doi: 10.1016/j.brainres.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima-Yasuda H, Uryu K, Robinson J, Xie S, Hurtig H, Duda J, Trojanowski J. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathologica. 2007;114(3):221–229. doi: 10.1007/s00401-007-0261-2. [DOI] [PubMed] [Google Scholar]
- Neary D, Snowden J, Gustafson L, Passant U, Stuss D, Black S, Benson D. Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–1554. doi: 10.1212/WNL.51.6.1546. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Lee V. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Onyike C, Diehl-Schmid J. The Epidemiology of Frontotemporal Dementia. International Review of Psychiatry. 2013;25(2):130–137. doi: 10.3109/09540261.2013.776523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou S, Wu F, Harrich D, Garcia-Martinez L, Gaynor R. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. Journal of Virology. 1995;69(6):3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesiridis GS, Lee V, Trojanowski JQ. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Human Molecular Genetics. 2009;18(R2):R156–R162. doi: 10.1093/hmg/ddp303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips T, Rothstein J. Glial cells in Amyotrophic Lateral Sclerosis. Experimental Neurology. 2014;262PB:111–120. doi: 10.1016/j.expneurol.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips T, Rothstein J. Rodent Models of Amyotrophic Lateral Sclerosis. Current Protocols in Pharmacology. 2015;69 doi: 10.1002/0471141755.ph0567s69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TLC, Shiue L. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nature Neuroscience. 2011;14(4):459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prut L, Belzung C. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. European Journal of Pharmacology. 2003;463(1-3):3–33. doi: 10.1016/S0014-2999(03)01272-X. [DOI] [PubMed] [Google Scholar]
- Ratti A, Buratti E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. Journal of Neurochemistry. 2016;138(Suppl. 1):95–111. doi: 10.1111/jnc.13625. [DOI] [PubMed] [Google Scholar]
- Reineke LC, Lloyd RE. Diversion of stress granules and P-bodies during viral infection. Virology. 2013;436:255–267. doi: 10.1016/j.virol.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton A, Chio A, Traynor B. State of play in amyotrophic lateral sclerosis genetics. Nature Neuroscience. 2014;17(1):17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton A, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick J. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson E. Mouse Models of Frontotemporal Dementia. Neurological Progress. 2012;72(6):837–849. doi: 10.1002/ana.23722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano G, Klima R, Buratti E, Verstreken P, Baralle F, Feiguin F. Chronological requirements of TDP-43 function in synaptic organization and locomotive control. Neurobiology of Disease. 2014;71:95–109. doi: 10.1016/j.nbd.2014.07.007. [DOI] [PubMed] [Google Scholar]
- Rosen D, Siddique T, Patterson D, Figlewicz D, Sapp P, Hentati A, Deng HX. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;51(1):59–62. doi: 10.1002/ajmg.1320510114. [DOI] [PubMed] [Google Scholar]
- Rothstein J, Martin L, Kuncl R. Decreased Glutamate Transport by the Brain and Spinal Cord in Amyotrophic Lateral Sclerosis. New England Journal of Medicine. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- Rothstein J, Van Kammen M, Levey A, Martin L, Kuncl R. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Annals of Neurology. 1995;38(1):73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- Saini A, Chauhan VS. Delineation of the Core Aggregation Sequences of TDP-43 C-Terminal Fragment. ChemBioChem. 2011;12(16):2495–2501. doi: 10.1002/cbic.201100427. [DOI] [PubMed] [Google Scholar]
- Schmid B, Hruscha A, Hogl S, Banzhaf-Strathmann J, Strecker K, van der Zee J, Haass C. Loss of ALS-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth. PNAS. 2013;110(13):4986–4991. doi: 10.1073/pnas.1218311110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotter EL, Vance C, Nishimura AL, Lee YB, Chen HJ, Urwin H, Shaw CE. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. Journal of Cell Science. 2014;127:1263–1278. doi: 10.1242/jcs.140087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton C, Good S, Atkin S, Dewey C, Mayer Paul, Yu G. TDP-43 Is a Developmentally Regulated Protein Essential for Early Embryonic Development. Journal of Biological Chemistry. 2010;285(9):6826–6834. doi: 10.1074/jbc.M109.061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton C, Cenik C, Kucukural A, Dammer E, Cenik B, Han Y, Yu G. Identification of Neuronal RNA Targets of TDP-43-containing Ribonucleoprotein Complexes. Journal of Biological Chemistry. 2011;286(2):1204–1215. doi: 10.1074/jbc.M110.190884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, Yu G. The function of RNA-binding proteins at the synapse: implications for neurodegeneration. Cellular and Molecular Life Sciences. 2015;72:3621–3635. doi: 10.1007/s00018-015-1943-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaker M, Ghani A, Shiha G, Ibrahim T, Mehal W. Nilotinib induces apoptosis and autophagic cell death of activated hepatic stellate cells via inhibition of histone deacetylases. Biochimica et Biophysica Acta – Molecular Cell Research. 2013;1833(8):1992–2003. doi: 10.1016/j.bbamcr.2013.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan X, Chiang PM, Price DL, Wong PC. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. PNAS. 2010;107(37):16325–16330. doi: 10.1073/pnas.1003459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U, Parker R. Decapping and Decay of Messenger RNA Occur in Cytoplasmic Processing Bodies. Science. 2003;300(5620):805–808. doi: 10.1126/science.1082320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair I, Tripathi V, Hu X, Vance C, Rogelj B, Shaw C. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallings N, Puttaparthi K, Luther C, Burns D, Elliott J. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiology of Disease. 2010;40:404–414. doi: 10.1016/j.nbd.2010.06.017. [DOI] [PubMed] [Google Scholar]
- Sun Y, Chakrabartty A. Phase to Phase with TDP-43. Biochemistry. 2017;56:809–823. doi: 10.1021/acs.biochem.6b01088. [DOI] [PubMed] [Google Scholar]
- Swarup V, Phaneuf D, Bareil C, Robertson J, Rouleu G, Kriz J, Julien JP. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain. 2011;134(9):2610–2626. doi: 10.1093/brain/awr159. [DOI] [PubMed] [Google Scholar]
- Tsai KJ, Yang CH, Fang YH, Cho KH, Chien WL, Wang WT, Shen CKJ. Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. Journal of Experimental Medicine. 2010;207(8):1661–1673. doi: 10.1084/jem.20092164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccaro A, Tauffenberger A, Ash P, Carlomagno YP, Parker JA. TDP-1/TDP-43 Regulates Stress Signaling and Age-Dependent Proteotoxicity in Caenorhabditis elegans. PLoS Genetics. 2012;8(7):e1002806. doi: 10.1371/journal.pgen.1002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Wu LS, Chang HY, Shen CKJ. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. Journal of Neurochemistry. 2008;105(3):797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]
- Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ, Shen CKJ. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. PNAS. 2012;109(37):15024–15029. doi: 10.1073/pnas.1206362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegorzewska I, Bell S, Cairns N, Miller T, Baloh R. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. PNAS. 2010;106(44):18809–18814. doi: 10.1073/pnas.0908767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cujit I, Kumar-Singh S. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. PNAS. 2010;107(8):3858–3863. doi: 10.1073/pnas.0912417107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Dugger B, Dickson D, Wang DS. TDP-43 in aging and Alzheimer’s disease – a review. Int J Clin Exp Pathol. 2011;4(2):147–155. [PMC free article] [PubMed] [Google Scholar]
- Wu LS, Cheng WC, Hou SC, Yan YT, Jiang ST, Shen CJ. TDP-43, a neuropathosignature factor, is essential for early mouse embryogenesis. Genesis. 2009;48(1):56–62. doi: 10.1002/dvg.20584. [DOI] [PubMed] [Google Scholar]
- Xia Q, Wang H, Hao Z, Fu C, Hu Q, Gao F, Wang G. TDP‐ 43 loss of function increases TFEB activity and blocks autophagosome–lysosome fusion. The EMBO Journal. 2016;35:121–142. doi: 10.15252/embj.201591998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YF, Gendron T, Zhang WL, D’Alton S, Sheng H, Castanedes M, Petrucelli L. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits and early mortality in transgenic mice. Journal of Neuroscience. 2010;30(32):10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YF, Zhang YJ, Lin WL, Cao X, Stetler C, Dickson DW, Petrucelli L. Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Molecular Neurodegeneration. 2011;6(73) doi: 10.1186/1750-1326-6-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Z, Xia Q, Hao Z, Xu D, Wang M, Wang H. TARDBP/TDP-43 regulates autophagy in both MTORC1-dependent and MTORC1-independent manners. Autophagy. 2016;12:707–708. doi: 10.1080/15548627.2016.1151596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HC, Lin CS, Tai WT, Liu CY, Shiau CW, Chen KF. Nilotinib Induces Autophagy in Hepatocellular Carcinoma through AMPK Activation. Journal of Biological Chemistry. 2013;288:18249–18259. doi: 10.1074/jbc.M112.446385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Ouyang Y, Gazit V, Cirrito J, Jansen L, Ess K, Wong M. Abnormal glutamate homeostasis and impaired synaptic plasticity and learning in a mouse model of tuberous sclerosis complex. Neurobiology of Disease. 2007;28(2):184–196. doi: 10.1016/j.nbd.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan L, Hanson K, Kim SH, Tare A, Tibbetts R. Identification of genetic modifiers of TDP-43 neurotoxicity in drosophila. PLoS ONE. 2013;8(2):e57214. doi: 10.1371/journal.pone.0057214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Hwang HY, Hao H, Talbot C, Wang J. Caenorhabditis elegans RNA-processing Protein TDP-1 Regulates Protein Homeostasis and Life Span. Journal of Biological Chemistry. 2012;287:8371–8382. doi: 10.1074/jbc.M111.311977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Huang C, Chen H, Wang D, Landel C, Xia PY, Xia XG. Transgenic Rat Model of Neurodegeneration Caused by Mutation in the TDP Gene. PLoS Genetics. 2010;6(3) doi: 10.1371/journal.pgen.1000887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufiria M, Gil-Bea F, Fernandez-Torron R, Poza JJ, Munoz-Blanco JL, Rojas-Garcia R, Lopez de Munain A. ALS: A bucket of genes, environment, metabolism and unknown ingredients. Progress in Neurobiology. 2016;142:104–129. doi: 10.1016/j.pneurobio.2016.05.004. [DOI] [PubMed] [Google Scholar]