

Abstract
Heterotrophic marine bacterioplankton populations display substantive genomic diversity that is commonly explained to be the result of selective forces imposed by resource limitation or interactions with phage and predators. Here we use a mutation-accumulation experiment followed by whole-genome sequencing of mutation lines to determine an unbiased rate and molecular spectrum of spontaneous mutations for a model heterotrophic marine bacterium in the globally important Roseobacter clade, Ruegeria pomeroyi DSS-3. We find evidence for mutational bias towards deletions over insertions, and this process alone could account for a sizable portion of genome size diversity among roseobacters and also implies that lateral gene transfer and/or selection must also play a role in maintaining roseobacters with large genome sizes. We also find evidence for a mutational bias in favor of changes from A/T to G/C nucleobases, which explains widespread occurrences of G/C-enriched Roseobacter genomes. Using the calculated mutation rate of 1.39 × 10−10 per base per generation, we implement a ‘mutation-rate clock’ approach to date the evolution of roseobacters by assuming a constant mutation rate along their evolutionary history. This approach gives an estimated date of Roseobacter genome expansion in good agreement with an earlier fossil-based estimate of ~250 million years ago and is consistent with a hypothesis of a correlated evolutionary history between roseobacters and marine eukaryotic phytoplankton groups.
The ability of heterotrophic marine bacterioplankton lineages to drive critical transformations in global carbon and nutrient cycles is commonly attributed to their biochemical interactions with organic matter in dilute waters and transient micro-environments (Azam and Malfatti, 2007; Giovannoni, 2016). These adaptive strategies are backed up by substantive bacterioplankton genomic diversity manifested as large variations in metabolic and regulatory pathways, genome size and G+C content (Swan et al., 2013; Giovannoni et al., 2014; Luo and Moran, 2015). While it is often acknowledged that this diversity is the result of a long and complex evolutionary history through interactions between natural selection and mutation among other mechanisms, only selection has been subject to intensive discussions (Morris et al., 2012; Giovannoni et al., 2014; Luo and Moran, 2015). Mutation is often appreciated as a way to provide raw materials on which selection can work, but mutation itself may also respond to environmental changes such as acquisition of antibiotic resistance enhanced solely by increased mutation rate (Long et al., 2016). Despite their significant role in evolutionary dynamics and microbial adaptation, how the rate and spectrum of spontaneous mutations contribute to genetic diversity has not been assessed for any ecologically dominant marine bacterioplankton lineage.
Mutation-accumulation (MA) experiments followed by whole-genome sequencing (WGS) of derived MA lines are being used to determine spontaneous mutations of both prokaryotic and eukaryotic organisms (Sung et al., 2012; Dillon et al., 2016). This approach allows all but lethal mutations to accumulate and thus is considered an approximately unbiased method for mutation determination (Eyre-Walker and Keightley, 2007). Here we apply the MA/WGS strategy to characterize the genomic pattern of spontaneous mutations arising in the model heterotrophic marine bacterium Ruegeria pomeroyi DSS-3 (Moran et al., 2004), a member of the alphaproteobacterial Roseobacter clade. Roseobacters constitute a substantial proportion of marine bacterioplankton communities (5–20%), and are among the major drivers of global carbon and sulfur cycles (Luo and Moran, 2014; Voget et al., 2015). There is a considerable diversity among the clade members in genome size, genome content and base composition.
Our MA experiment allowed accumulation of mutations over 5,386 cell divisions in 80 independent MA lines initiated from a single founder colony of R. pomeroyi DSS-3 and passed through a single-cell bottleneck every two days. By sequencing genomes of 48 randomly selected lines at the end of the MA experiment and using a robust consensus method shown to achieve a low false-positive rate (Sung et al., 2012, 2015), we identified 161 base-substitution mutations over these sequenced lines (Figure 1 and Supplementary Table S1). The ratio of nonsynonymous to synonymous mutations did not significantly differ from the ratio of nonsynonymous to synonymous sites in the DSS-3 genome (χ2=1.66, P=0.20, df=1; Supplementary Table S2), which is evidence for minimal selective elimination of deleterious mutations during the MA process, confirming faithful representation of mutational pattern for DSS-3. Base-substitution mutation (BSM) was slightly biased toward intergenic regions compared to coding regions (χ2=11.73, P=0.0006, df=1; Supplementary Table S2), and the same trend was observed in other bacterial MA/WGS analyses (Lee et al., 2012; Sung et al., 2015; Dillon et al., 2016). One explanation is that mismatch repair may be more active in coding regions, though a minor role of selection in eliminating coding mutations cannot be excluded (Long et al., 2014; Dillon et al., 2016).
Figure 1.

The genomic locations of the base-substitution mutations and insertion/deletion mutations in the 48 independent mutation accumulation (MA) lines of Ruegeria pomeroyi DSS-3. From outer to inner rings scaled to genome size: (1)–(3) The three outermost rings represent gene density (gray), G/C content (orange) and A/T content (green), respectively, calculated in non-overlapping 1 kb blocks. If the value of the 1 kb block is above the genome-wide mean, the 1 kb block is colored; (4) locus tag of genes with mutations; (5) position of each base-substitution mutation (A/T→G/C (red), G/C→A/T (blue) and A↔T or G↔C (grey)), as well as insertion/deletions (green) in MA lines, with each ring representing the genome of an individual MA line. The chromosome (a) and megaplasmid (b) of DSS-3 are not plotted in proportion to their number of nucleotides in order to make the features of the latter visible.
The data derived from this MA/WGS procedure have important implications for understanding marine Roseobacter evolution. First, the base changes gave an average mutation rate (μ) of 1.39 × 10−10 per base per generation. Direct, unbiased estimates of natural mutation rates have been determined for only a handful of bacteria thus far, and this is among the first determinations for a marine bacterial species (see also Dillon et al., 2016). Previous studies have measured rates that vary from 1.28 × 10−10 per base per generation (for Bacillus anthracis) to 9.78 × 10−9 (for Mesoplasma florum) (Sung et al., 2012). Our estimate for DSS-3 is at the lower end of this range. On the basis of this mutation rate, a genome size of 4 601 048 bp, and an average growth rate of 45 generations per year inferred based on a well-articulated linear relationship between growth rate and rRNA/rDNA ratio in a laboratory culture of DSS-3 and measures of this ratio in field populations (Lankiewicz et al., 2016), it takes ~35 years for a Roseobacter cell in the surface ocean to gain one base-substitution mutation. If the DSS-3 natural population has an effective population size of 3.0 × 108 (Supplementary Methods; Supplementary Table S3), an average of 8.7 × 106 mutations are expected to arise in the population each year. The frequency of selectively advantageous mutations from this pool of spontaneous mutations is likely to be extremely low (Eyre-Walker and Keightley, 2007) but may make an important contribution to the evolution of this lineage.
A second implication of the MA/WGS data emerges from evidence of a nucleobase bias in spontaneous mutations. The 161 base-substitution mutations included 61 from G/C to A/T and 73 from A/T to G/C. Correcting for the genomic base composition (A/T:G/C=0.56:1), a significantly higher rate of mutations from A/T to G/C was verified using a bionomial test for comparison of proportions with continuity correction (prop.test function of R, P<0.001, df=1). The most abundant surface ocean bacterioplankton lineages such as the alphaproteobacterial SAR11 clade, the gammaproteobacterial SAR86 clade and the cyanobacterial Prochlorococcus clade that is adapted to a high light environment, have genomic G+C contents of only 29–32% (Giovannoni et al., 2014). Roseobacters are unusual in this respect because many members have genomic G+C content above 50% (Zhang et al., 2016), including R. pomeroyi DSS-3 with 64.1%. Reduced G+C content in many major bacterioplankton lineages has been explained as an adaptive strategy to cope with N-limited surface waters (since one AT pair uses one less nitrogen than one GC pair) (Swan et al., 2013; Giovannoni et al., 2014; Luo et al., 2015). It was thus perplexing to find a prevalence of roseobacters with higher G+C content. Our MA/WGS data provide a proximal mechanism for this contrasting pattern of G+C content between roseobacters and other successful lineages. In fact, a mutational bias from A/T to G/C was recently shown through the MA/WGS data of a soil bacterium Burkholderia cenocepacia HI2424 (Dillon and Cooper, 2016), which also has high genomic G+C content (66.7%).
A third implication of the study is that it provides a new type of data for molecular dating of Roseobacter lineages. Like most free-living heterotrophic bacterial lineages, roseobacters do not have lineage-specific fossil records, and thus studying their evolutionary history relies on geochemical fossil evidence from cyanobacteria (Luo et al., 2013). Using this approach, a previous analysis predicted a major genome expansion event of roseobacters, coincident with the start of marine dinoflagellate and coccolithophore radiation around 250 million years ago (Mya) (Falkowski et al., 2004). This was consistent with the present-day ecological associations between these microbial groups (Gonzalez et al., 2000; Jasti et al., 2005) and subsequent hypotheses regarding reciprocal exchanges of metabolites between phytoplankton and roseobacters such as organic matter and vitamins (Luo et al., 2013; Luo and Moran, 2014; Durham et al., 2015). However, the substantial evolutionary distance between roseobacters and cyanobacteria suggests that this estimate may have large uncertainties (Nei et al., 2001; Smith and Peterson, 2002; Bromham and Penny, 2003). Furthermore, new evidence from ultraclean Archaean samples argues against the validity of the previous cyanobacterial fossils used in various analyses (French et al., 2015), including the Roseobacter dating study.
For these reasons, it is useful to have an approach that does not rely on fossils and instead makes use of an unbiased measure of the DSS-3 mutation rate. Here the molecular sequence divergence time (T) was derived from mutation rate (μ; per base per generation) of R. pomeroyi DSS-3 and growth rate of oceanic samples of roseobacters (γ; number of generations per year), based on 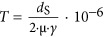 (Myr ago), where dS is the number of synonymous substitutions per synonymous site and meaningful only for closely related sequences (here, dS⩽0.74 (±0.42); Supplementary Figures S1B, S2). This principle is based on the assumption that mutations at silent sites are largely invisible to natural selection and thus accumulate freely, though there is evidence for selection on these sites and that the strength and nature of this selection varies among lineages (Hildebrand et al., 2010; Raghavan et al., 2012; Luo et al., 2015). With the estimated T, an iterative linear regression approach (Supplementary Methods) was used to identify Roseobacter gene families showing evidence of a local molecular clock (Supplementary Figure S2), manifested as a linear correlation between T and genetic distance of these closely related proteins (
(Myr ago), where dS is the number of synonymous substitutions per synonymous site and meaningful only for closely related sequences (here, dS⩽0.74 (±0.42); Supplementary Figures S1B, S2). This principle is based on the assumption that mutations at silent sites are largely invisible to natural selection and thus accumulate freely, though there is evidence for selection on these sites and that the strength and nature of this selection varies among lineages (Hildebrand et al., 2010; Raghavan et al., 2012; Luo et al., 2015). With the estimated T, an iterative linear regression approach (Supplementary Methods) was used to identify Roseobacter gene families showing evidence of a local molecular clock (Supplementary Figure S2), manifested as a linear correlation between T and genetic distance of these closely related proteins (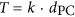 , where k is the slope and unique for each clock-like family and dPC is the Poisson-corrected distances of protein sequences).
, where k is the slope and unique for each clock-like family and dPC is the Poisson-corrected distances of protein sequences).
A local clock derived from closely related sequences does not guarantee that there exists a global clock in which sequence evolutionary rate remains constant throughout the evolutionary history of the Roseobacter clade. To test whether these families showing evidence of a local clock also fit the global clock model, the likelihood of each gene phylogeny was calculated using PAML (Yang, 2007) with and without a global clock model, and the Bayesian Information Criterion (BIC) was used to assess whether the global clock model was superior. Subsequently, the linear models for gene families showing evidence of a global clock were used to calculate separation time of more divergent sequences (T′), according to 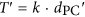 , where dPC′ is the Poisson-corrected distances of divergent protein sequences. This procedure generated T′ for all possible pairs of sequences in each clock-like family and construction of a chronogram for each using the UPGMA (Michener and Sokal, 1957) method. By averaging T′ from these clock-like families, we were able to estimate the divergence time of any two Roseobacter lineages (Figure 2a; Supplementary Methods).
, where dPC′ is the Poisson-corrected distances of divergent protein sequences. This procedure generated T′ for all possible pairs of sequences in each clock-like family and construction of a chronogram for each using the UPGMA (Michener and Sokal, 1957) method. By averaging T′ from these clock-like families, we were able to estimate the divergence time of any two Roseobacter lineages (Figure 2a; Supplementary Methods).
Figure 2.

The molecular dating pipeline developed in this study, and the lognormal distribution of time estimates for the most ancient Roseobacter ancestors. (a) Of the three main inputs required (growth rate, mutation rate and the sequence data of orthologous gene families), the growth rate (γ: number of generations per year) of oceanic roseobacters was obtained from recent literature (Lankiewicz et al., 2016); the mutation rate (μ; per base per generation) was determined using a mutation accumulation experiment followed by whole-genome sequencing of derived mutation lines; and the sequence data were downloaded from NCBI. For the molecular dating calculation, 456 shared single-copy orthologous genes were used to determine the number of synonymous substitutions per synonymous site (dS) and the Poisson-corrected distances (dPC) of protein sequences. Note that dS is meaningful only for closely related sequences (here dS⩽0.74±0.42). The time since separation (T) was calculated by 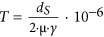 (Myr ago). An iterative linear regression analysis was performed using T against dPC, along with the likelihood test of a global molecular clock, which led to the identification of 232 clock-like families (Figure 2; Supplementary Figure S1A). Next, slope (k) was obtained from the regression model and used to calculate the divergence time (T′) of more divergent sequences using
(Myr ago). An iterative linear regression analysis was performed using T against dPC, along with the likelihood test of a global molecular clock, which led to the identification of 232 clock-like families (Figure 2; Supplementary Figure S1A). Next, slope (k) was obtained from the regression model and used to calculate the divergence time (T′) of more divergent sequences using 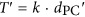 . This procedure estimated divergence time for all possible pairs of sequences in each of the clock-like families, which allowed for construction of a chronogram for each of these families using UPGMA, and a distribution was obtained for the timing of a given ancestral node by pooling chronograms at this node. Among the 232 clock-like families, 189 correctly placed HTCC2255 as the basal lineage of the Roseobacter clade (Supplementary Figure S1A). (b) A Roseobacter phylogenomic tree illustrates the ancestral nodes R38 and R37. The notation of these two most ancient nodes was used in a previous publication (Luo et al., 2013). (c) The consensus divergence times of R38 and R37 estimated with a growth rate of 45 generations per year calculated based on recent data (Lankiewicz et al., 2016). From top to bottom, each panel represents: the lower boundary of the 95% prediction interval derived from the linear regression models (R38: MODE=296 Myr ago; R37: MODE=240 Myr ago); the estimated T′ calculated directly from the linear equations
. This procedure estimated divergence time for all possible pairs of sequences in each of the clock-like families, which allowed for construction of a chronogram for each of these families using UPGMA, and a distribution was obtained for the timing of a given ancestral node by pooling chronograms at this node. Among the 232 clock-like families, 189 correctly placed HTCC2255 as the basal lineage of the Roseobacter clade (Supplementary Figure S1A). (b) A Roseobacter phylogenomic tree illustrates the ancestral nodes R38 and R37. The notation of these two most ancient nodes was used in a previous publication (Luo et al., 2013). (c) The consensus divergence times of R38 and R37 estimated with a growth rate of 45 generations per year calculated based on recent data (Lankiewicz et al., 2016). From top to bottom, each panel represents: the lower boundary of the 95% prediction interval derived from the linear regression models (R38: MODE=296 Myr ago; R37: MODE=240 Myr ago); the estimated T′ calculated directly from the linear equations 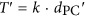 (R38: MODE=390 Myr ago; R37: MODE=317 Myr ago); and the upper boundary of the 95% prediction interval derived from the linear regression models (R38: MODE=474 Myr ago; R37: MODE=387 Myr ago). The lower and upper boundaries of the 95% prediction interval represent the uncertainty associated with the estimates of T′. The dotted vertical lines in all three panels point to the mode values derived from the fitted models for the divergence times obtained from the 189 clock-like families. The horizontal bars correspond to the 95% confident interval for the Half-Range Modes (HRM) calculated by 100 bootstrap replicates. The dot on each bar displays the mean of the bootstrapped mode estimates (HRM-BME) (Hedges and Shah, 2003).
(R38: MODE=390 Myr ago; R37: MODE=317 Myr ago); and the upper boundary of the 95% prediction interval derived from the linear regression models (R38: MODE=474 Myr ago; R37: MODE=387 Myr ago). The lower and upper boundaries of the 95% prediction interval represent the uncertainty associated with the estimates of T′. The dotted vertical lines in all three panels point to the mode values derived from the fitted models for the divergence times obtained from the 189 clock-like families. The horizontal bars correspond to the 95% confident interval for the Half-Range Modes (HRM) calculated by 100 bootstrap replicates. The dot on each bar displays the mean of the bootstrapped mode estimates (HRM-BME) (Hedges and Shah, 2003).
From chronograms of 189 protein families (Supplementary Figure S1; Supplementary Table S4) among 77 Roseobacter genomes (Supplementary Table S5) that show evidence of a global clock and that correctly place the phylogenetically basal lineage of the Roseobacter clade at strain HTCC2255 (Newton et al., 2010; Luo et al., 2014) (Supplementary Figure S3), a lognormal distribution was generated for each of the two most ancient ancestral nodes of the clade according to a model selection procedure (Supplementary Figure S4); these nodes correspond to R38 and R37 (Figure 2b) in a previous study (Luo et al., 2013). Using the mode of the distribution as the divergence time estimate (Figure 2c) (Kumar and Hedges, 1998; Hedges and Shah, 2003; Hedges et al., 2015) and taking a growth rate of 45 cell divisions per year corresponding to the upper limit of the measures in the Delaware estuary Roseobacter populations (Lankiewicz et al., 2016), the model predicted that the R38 and R37 nodes occurred at 296 and 240 Myr ago, respectively, taking the values corresponding to the lower boundary of the 95% prediction intervals (Figure 2c; upper panel). This is consistent with the fossil-based prediction (Luo et al., 2013).
One caveat of this analysis is that it assumes a constant mutation rate through the evolutionary history of the clade (that is, a ‘mutation-rate clock’). A future analysis should determine the mutation rate for other representative strains across the phylogeny and use the derived timing information for each major lineage through the above approach to calibrate the phylogeny of the Roseobacter clade using r8s and other conventional dating programs, which also allows varying evolutionary rate among lineages of the clade (Supplementary Figure S5). Another caveat is that it does not consider growth rate variation among lineages. If a lower growth rate of 26 generations per year derived from the average of Lankiewicz’s measures (Lankiewicz et al., 2016) was used instead, for instance, the mutation-rate clock model predicts a more ancient origin of the roseobacters (513 Myr ago and 416 Myr ago for R38 and R37, respectively). A more accurate estimate of the Roseobacter diversification time will thus require improved measures of growth rates for each major Roseobacter lineage and incorporating this variation into the mutation-rate clock model. In general, this mutation-rate clock method is useful to date more recently evolved lineages and thus is complementary to the traditional estimates based on the cyanobacterial fossils.
A fourth implication of the MA/WGS study comes from the evidence that DSS-3 displays a mutational bias toward deletion over insertion, shown by both the number of deletion versus insertion events (18 versus 12) and the number of deleted versus inserted nucleobases (67 versus 23) over the 48 sequenced MA lines (Supplementary Table S6). This observation is consistent with previous findings that deletion bias is a near universal trend (Mira et al., 2001; Kuo and Ochman, 2009). Assuming 45 generations per year for Roseobacters in the ocean (Lankiewicz et al., 2016) and a 296 million years’ evolutionary history for the lineage, the measured net loss rate translates to a deletion of 2.27 Mb. In other words, if mutation would have been the only evolutionary force giving rise to genome size diversity, the ~300 million years’ evolution would have transformed a typical Roseobacter (that is, DSS-3) to a streamlined genome (2.33 Mb) that has a similar size as the basal strain HTCC2255 (2.43 Mb). Therefore, mutation alone could account for the occurrences of several streamlined Roseobacter lineages in today’s ocean. On the other hand, most sequenced roseobacters have a large genome size. For instance, a CheckM (Parks et al., 2015) analysis of the 122 Roseobacter genomes (Supplementary Table S7) deposited in the NCBI RefSeq database showed that the mean genome size of roseobacters is 4.33 (±0.72) Mb. The prevalence of roseobacters with large genomes despite deletion bias suggests that other evolutionary mechanisms such as lateral gene transfer and/or natural selection act to increase genome size in this bacterial clade.
The MA/WGS experiments, while performed on rich media under laboratory conditions, are valuable for estimating the rate and spectrum of spontaneous mutations in bacteria, with implications for understanding bacterial ecological diversification and evolutionary history. Comparing this mutation accumulation study for a marine heterotrophic bacterium with similar analyses of other major bacterial groups in the ocean will improve understanding of the evolutionary history of marine microbial plankton.
Acknowledgments
We thank Samuel F Miller, Christa Smith and Yongjie Huang for their assistance. This research was funded by a grant from the Hong Kong RGC Early Career Scheme (24101015); the NSFC grant (41576141); the Direct Grant (4930062, 4053105) from the Chinese University of Hong Kong; the Gordon and Betty Moore Foundation (GBMF538.01).
Footnotes
Supplementary Information accompanies this paper on The ISME Journal website(http://www.nature.com/ismej)
The authors declare no conflict of interest.
Supplementary Material
References
- Azam F, Malfatti F. (2007). Microbial structuring of marine ecosystems. Nat Rev Microbiol 5: 782–791. [DOI] [PubMed] [Google Scholar]
- Bromham L, Penny D. (2003). The modern molecular clock. Nat Rev Genet 4: 216–224. [DOI] [PubMed] [Google Scholar]
- Dillon MM, Cooper VS. (2016). The fitness effects of spontaneous mutations nearly unseen by selection in a bacterium with multiple chromosomes. Genetics 204: 1225–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon MM, Sung W, Sebra R, Lynch M, Cooper VS. (2016). Genome-wide biases in the rate and molecular spectrum of spontaneous mutations in Vibrio cholerae and Vibrio fischeri. Mol Biol Evol 34: 93–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham BP, Sharma S, Luo HW, Smith CB, Amin SA, Bender SJ et al. (2015). Cryptic carbon and sulfur cycling between surface ocean plankton. Proc Natl Acad Sci USA 112: 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley PD. (2007). The distribution of fitness effects of new mutations. Nat Rev Genet 8: 610–618. [DOI] [PubMed] [Google Scholar]
- Falkowski PG, Katz ME, Knoll AH, Quigg A, Raven JA, Schofield O et al. (2004). The evolution of modern eukaryotic phytoplankton. Science 305: 354–360. [DOI] [PubMed] [Google Scholar]
- French KL, Hallmann C, Hope JM, Schoon PL, Zumberge JA, Hoshino Y et al. (2015). Reappraisal of hydrocarbon biomarkers in Archean rocks. Proc Natl Acad Sci USA 112: 5915–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni SJ, Cameron Thrash J, Temperton B. (2014). Implications of streamlining theory for microbial ecology. ISME J 8: 1553–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni SJ. (2016). SAR11 bacteria: the most abundant plankton in the oceans. Ann Rev Mar Sci 9: 231–255. [DOI] [PubMed] [Google Scholar]
- Gonzalez JM, Simo R, Massana R, Covert JS, Casamayor EO, Pedros-Alio C et al. (2000). Bacterial community structure associated with a dimethylsulfoniopropionate-producing North Atlantic algal bloom. Appl Environ Microbiol 66: 4237–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges SB, Shah P. (2003). Comparison of mode estimation methods and application in molecular clock analysis. BMC Bioinformatics 4: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges SB, Marin J, Suleski M, Paymer M, Kumar S. (2015). Tree of life reveals clock-like speciation and diversification. Mol Biol Evol 32: 835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand F, Meyer A, Eyre-Walker A. (2010). Evidence of selection upon genomic GC-content in bacteria. PloS Genetics 6: e1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasti S, Sieracki ME, Poulton NJ, Giewat MW, Rooney-Varga JN. (2005). Phylogenetic diversity and specificity of bacteria closely associated with Alexandrium spp. and other phytoplankton. Appl Environ Microbiol 71: 3483–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Hedges SB. (1998). A molecular timescale for vertebrate evolution. Nature 392: 917–920. [DOI] [PubMed] [Google Scholar]
- Kuo CH, Ochman H. (2009). Inferring clocks when lacking rocks: the variable rates of molecular evolution in bacteria. Biology Direct 4: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lankiewicz TS, Cottrell MT, Kirchman DL. (2016). Growth rates and rRNA content of four marine bacteria in pure cultures and in the Delaware estuary. ISME J 10: 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Popodi E, Tang HX, Foster PL. (2012). Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci USA 109: E2774–E2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, Sung W, Miller SF, Ackerman MS, Doak TG, Lynch M. (2014). Mutation rate, spectrum, topology, and context-dependency in the DNA mismatch repair-deficient Pseudomonas fluorescens ATCC948. Genome Biol Evol 7: 262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, Miller SF, Strauss C, Zhao C, Cheng L, Ye Z et al. (2016). Antibiotic treatment enhances the genome-wide mutation rate of target cells. Proc Natl Acad Sci USA 113: E2498–E2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Csuros M, Hughes AL, Moran MA. (2013). Evolution of divergent life history strategies in marine alphaproteobacteria. mBio 4: e00373–e00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Moran MA. (2014). Evolutionary ecology of the marine Roseobacter clade. Microbiol Mol Biol Rev 78: 573–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Swan BK, Stepanauskas R, Hughes AL, Moran MA. (2014). Evolutionary analysis of a streamlined lineage of surface ocean Roseobacters. ISME J 8: 1428–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Moran MA. (2015). How do divergent ecological strategies emerge among marine bacterioplankton lineages? Trends Microbiol 23: 577–584. [DOI] [PubMed] [Google Scholar]
- Luo H, Thompson LR, Stingl U, Hughes AL. (2015). Selection maintains low genomic GC content in marine SAR11 lineages. Mol Biol Evol 32: 2738–2748. [DOI] [PubMed] [Google Scholar]
- Michener CD, Sokal RR. (1957). A quantitative approach to a problem in classification. Evolution 11: 130–162. [Google Scholar]
- Mira A, Ochman H, Moran NA. (2001). Deletional bias and the evolution of bacterial genomes. Trends Genet 17: 589–596. [DOI] [PubMed] [Google Scholar]
- Moran MA, Buchan A, Gonzalez JM, Heidelberg JF, Whitman WB, Kiene RP et al. (2004). Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment. Nature 432: 910–913. [DOI] [PubMed] [Google Scholar]
- Morris JJ, Lenski RE, Zinser ER. (2012). The black queen hypothesis: evolution of dependencies through adaptive gene loss. MBio 3: e00036–00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Xu P, Glazko G. (2001). Estimation of divergence times from multiprotein sequences for a few mammalian species and several distantly related organisms. PNAS 98: 2497–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton RJ, Griffin LE, Bowles KM, Meile C, Gifford S, Givens CE et al. (2010). Genome characteristics of a generalist marine bacterial lineage. ISME J 4: 784–798. [DOI] [PubMed] [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25: 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan R, Kelkar YD, Ochman H. (2012). A selective force favoring increased G+C content in bacterial genes. Proc Natl Acad Sci USA 109: 14504–14507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AB, Peterson KJ. (2002). Dating the time of origin of major clades: molecular clocks and the fossil record. Annu Rev Earth Planet Sci 30: 65–88. [Google Scholar]
- Sung W, Ackerman MS, Miller SF, Doak TG, Lynch M. (2012). Drift-barrier hypothesis and mutation-rate evolution. PNAS 109: 18488–18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, Ackerman MS, Gout JF, Miller SF, Williams E, Foster PL et al. (2015). Asymmetric context-dependent mutation patterns revealed through mutation-accumulation experiments. Mol Biol Evol 32: 1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan BK, Tupper B, Sczyrba A, Lauro FM, Martinez-Garcia M, González JM et al. (2013). Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. PNAS 110: 11463–11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voget S, Wemheuer B, Brinkhoff T, Vollmers J, Dietrich S, Giebel HA et al. (2015). Adaptation of an abundant Roseobacter RCA organism to pelagic systems revealed by genomic and transcriptomic analyses. ISME J 9: 371–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24: 1586–1591. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Sun Y, Jiao N, Stepanauskas R, Luo H. (2016). Ecological genomics of the uncultivated marine Roseobacter lineage CHAB-I-5. Appl Environ Microbiol 82: 2100–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.