

Abstract
BACKGROUND
Exposures to ambient gaseous pollutants have been linked to cardiovascular diseases (CVDs), but the biological mechanisms remain uncertain.
OBJECTIVES
This study examined the changes in CVD marker levels resulting from elevated exposure to ambient gaseous pollutants in midlife women.
METHODS
Annual repeated measurements of several inflammatory, hemostatic and lipid makers were obtained from 2,306 midlife women enrolled in the longitudinal Study of Women’s Health Across the Nation (SWAN) between 1999 and 2004. Ambient carbon monoxide (CO), nitrogen dioxide (NO2), and sulfur dioxide (SO2) data were assigned to each woman based on proximity of the monitoring station to her residential address. Short- and long-term exposures were calculated, and their associations with markers were examined using linear mixed-effects regression models, adjusted for demographic, health and other factors.
RESULTS
Short-term CO exposure was associated with increased fibrinogen, i.e., every interquartile increase of average prior one-week exposure to CO was associated with 1.3% (95% CI: 0.6%, 2.0%) increase in fibrinogen. Long-term exposures to NO2 and SO2 were associated with reduced high-density lipoproteins and apolipoprotein A1, e.g., 4.0% (1.7%, 6.3%) and 4.7% (2.8%, 6.6%) decrease per interquartile increment in prior one-year average NO2 concentration, respectively. Fine particle (PM2.5) exposure confounded associations between CO/NO2 and inflammatory/hemostatic markers, while associations with lipoproteins were generally robust to PM2.5 adjustment.
CONCLUSIONS
Exposures to these gas pollutants at current ambient levels may increase thrombotic potential and disrupt cholesterol metabolism, contributing to greater risk of CVDs in midlife women. Caution should be exercised in evaluating the confounding by PM2.5 exposure.
Keywords: Cardiovascular, coagulation, fibrinolysis, carbon monoxide, nitrogen dioxide, sulfur dioxide, lipoprotein
Introduction
Cardiovascular diseases (CVDs) rank number one as the cause of death globally, accounting for about 30% of global deaths (World Health Organization, 2016). Many studies have observed associations between ambient gaseous pollutants and CVD morbidity. Carbon monoxide (CO) has been associated with hospital admissions and emergency department (ED) visits for ischemic heart disease (IHD), and congestive heart failure (CHF) (Lanki et al. 2006; Lee et al. 2007; Rosenlund et al. 2006; Szyszkowicz 2007). Consistent epidemiologic evidence has been reported for increases in incidence of myocardial infarction (MI) and IHD associated with exposure to nitrogen dioxide (NO2) (Beckerman et al. 2012; Cheng et al. 2009; Lipsett et al. 2011; Rosenlund et al. 2009). Positive associations have also been found for ambient sulfur dioxide (SO2) concentrations with ED visits and hospital admissions due to CVDs (Guo et al. 2010; Ito et al. 2011; Jalaludin et al. 2006), while some studies have observed negative associations (Chang et al. 2005; Llorca et al. 2005) or results were confounded by co-pollutants (Ballester et al. 2006). Based on an extensive literature review, the United States Environmental Protection Agency (USEPA) concluded that a likely or probable causal relationship existed between short-term exposure to these gases and CVD, but evidence was limited and inconclusive for long-term exposure (USEPA 2008,USEPA 2010,USEPA 2016).
While evidence for associations between gas exposure and CVDs has been increasing, the mechanisms behind the associations are not fully understood. Previous studies have proposed that CO and NO2 may cause oxidative stress and airway inflammation, which could migrate to other parts of the body, resulting in systemic inflammation and coagulation and further to development of CVDs (Chuang et al. 2007; Delfino et al. 2008; Riedl et al. 2012). Further, SO2 may cause oxidative stress to red blood cells and decrease blood viscosity (Baskurt et al. 1988). CO also enters the blood system and forms carboxyhemoglobin (COHb), which could trigger premature angina at high exposure levels (Allred et al. 1989). However, cardiovascular effects of gases at current ambient levels have not been confirmed (Channell et al. 2012; Li et al. 2011; Melin et al. 2005).
Researchers have identified a number of blood indices as CVD markers. Evaluation of these markers may help us better understand the relationship between gas exposures and CVD risks. Some studies have observed positive associations between short-term CO exposure and hs-CRP and fibrinogen (Delfino et al. 2009; Pekkanen et al. 2000), although the findings have been inconsistent across studies (Liao et al. 2005; Rückerl et al. 2007). Positive associations were also reported between short-term NO2 exposure and hs-CRP (Dadvand et al. 2014; Delfino et al. 2009), fibrinogen (Bind et al. 2012; Rich et al. 2012; Zhang et al. 2013), and thrombin (a coagulation marker) (Strak et al. 2013); however, the interpretation of these results has been uncertain due to potential confounding by co-pollutants, such as, CO and fine particles. A few studies have observed positive associations of short-term SO2 exposure with hs-CRP and fibrinogen (Khafaie et al. 2013; Zhang et al. 2013), but results overall have been insufficient and inconsistent, with inadequate control for co-pollutants. Meanwhile, a number of studies have reported no associations of gaseous air pollutants with inflammatory markers (Baccarelli et al. 2007; Langrish et al. 2010; Rückerl et al. 2007; Rudez et al. 2009), and others found a negative association (Rudez et al. 2009; Steinvil et al. 2008). Very few studies have reported an association between ambient gas exposure and lipoproteins (USEPA 2008,USEPA 2010,USEPA 2016). Evidence about the impact of long-term exposure to these gases on these markers is more scarce and inconsistent (Dadvand et al. 2014; Forbes et al. 2009; Huang et al. 2014; Panasevich et al. 2009). More research is needed to provide additional evidence about the relationship between ambient gas exposure and inflammatory, hemostatic and lipid markers to reconcile the available findings.
The present study was thus conducted using CVD marker data available from the Study of Women’s Health Across the Nation (SWAN), a longitudinal multi-site study designed to follow women through the menopausal transition (Sowers et al. 2000). Given the limited and inconsistent evidence mentioned above, we explored new evidence of the associations between exposure to ambient levels of three gaseous air pollutants, including CO, NO2 and SO2, and a number of inflammatory, hemostatic and lipid markers, to understand the mechanism and time frame of the adverse cardiovascular effects of each gas.
Methods
Study population
The study design and participant recruitment for SWAN has been previously described in detail (Sowers et al. 2000). The present study included data from six sites: Detroit, Michigan; Chicago, Illinois; Oakland, California; Los Angeles, California; Newark, New Jersey; and Pittsburgh, Pennsylvania. Between 1995 and 1997, SWAN recruited women who were 42 to 52 years of age, had an intact uterus and at least one ovary, were not using exogenous hormones, were not pregnant or lactating, and had at least one menstrual period in the previous three months. Multiple racial/ethnic groups were included, with Caucasians enrolled at every site and African Americans enrolled at three of these six sites and Hispanics, Chinese and Japanese recruited at one site each. Approximately 450 eligible women were recruited at each study site and have been followed up with clinical assessments and questionnaire interviews on a nearly annual basis. The SWAN protocols were approved by the Institutional Review Boards at all participating sites, and all participants provided written informed consent at baseline. The present analyses were based on participants with serum samples collected at SWAN visits 3 through 7 (1999–2004), when both PM2.5 measurements and the blood markers of interest were available.
Air pollutant data
Ambient CO, NO2 and SO2 data were obtained from the USEPA air monitoring network. The gases were monitored on an hourly basis. The data downloaded were maximum 8-hour average concentrations for CO and one-hour maximum concentrations for NO2 and SO2. Fine particles up to 2.5 micrometers in size (PM2.5) were a potential confounder of these analyses. PM2.5 was typically measured every three days, sometimes daily or every six days, and data were in the format of 24-hour average concentrations. Data for the entire U.S. were downloaded from USEPA’s Air Data website (https://www.epa.gov/airquality/airdata, accessed September 2010).
A residential history was maintained for each participant from the baseline visit to visit 7. The coordinate of each residence was geocoded and randomly moved up to 400 feet (about one block) to ensure confidentiality. We created 20 km circular buffer areas around each address using ArcGIS v10.0 (Environmental Systems Research Institute 1995–2016) and assigned exposures for participants within 20 km of monitors. If a participant moved (~13% of all women) during the year prior to her visit, exposure data from multiple addresses were weighted based on the time of move when assigning exposure, or evenly weighted if the move date was not available. More details about exposure assignment can be found in Green et al. (2016).
We calculated average exposure levels for one day, one week, one month, six months, and one year prior to each blood draw. Because PM2.5 measurements started in 1999, no matched one-year exposure data were available for some SWAN visits in 1999 and 2000. We, therefore, calculated the six-month average exposure to make use of more biomarker data, and expected similar associations observed for six-month and 1-year average exposures. Month was simplified to 30-day increments, six months to 180 days, and one year to 360 days. A minimum of three daily readings were required to qualify for the one-week exposures; at least nine daily readings were required for the one-month exposures; at least five months were required for the six-month averages; and at least ten months were required for the one-year averages. Otherwise, the specific exposure metric was considered missing. In over 95% of the cases, average exposures were calculated based on daily readings of >80% of exposure duration. We classified the exposure windows as short-term (the prior one-day, one-week, and one-month averages) and long-term (the prior six-month and one-year averages).
Blood measurement and analysis
Blood was drawn at each SWAN clinic visit and assayed for CVD markers as described previously (Green et al. 2016; Thurston et al. 2012). Inflammatory/hemostatic markers examined in this study included high-sensitivity C-reactive protein (hs-CRP), fibrinogen, factor VII coagulant (factor VIIc), tissue-type plasminogen activator antigen (tPA-ag), and plasminogen activator inhibitor Type 1 (PAI-1). When damage occurs to the endothelium of the blood vessels, fibrinogen and factor VIIc help in forming blood clots; tPA activates the fibrinolytic system to break down blood clots; PAI-1 is the major inhibitor of tPA by converting tPA into tPA-PAI-1 complex (Chandler et al. 1997).
We examined lipoproteins that have been linked to CVD risk (Burnett 2004; Kaptoge et al. 2012; Lowe et al. 2004; Smith et al. 2005). These included high-density lipoprotein cholesterol (HDL), low-density lipoprotein cholesterol (LDL), triglycerides, and total cholesterol, as well as lipoprotein(a) (Lp(a)) and lipoprotein A1 (LpA1). We also assessed apolipoprotein A1 (APOA1) and B (APOB). APOA1 is the primary protein component of HDL and APOB is the major protein component of all other lipoproteins. HDL-related lipoproteins, including HDL, LpA1 and APOA1, are considered to have protective effects on risk for CVDs (Navab et al. 2011), while elevated levels of LDL increases risk of CVD, as does Lp(a), which is an LDL-like particle that is involved in inhibition of fibrinolysis (Nordestgaard et al. 2010).
The majority of markers were measured at each visit, except fibrinogen, Factor VIIc, and LpA1 that were measured only at every other follow-up (Visits 3 and 5) in all women and in a subset of women at visit 7.
Statistical analysis
Summary statistics were calculated for each gas and CVD marker. Marker levels were log-transformed to better meet the normality assumption. Correlations between markers as well as between air pollutants were calculated based on visit 3 data, which has the largest sample size among the visits included.
To study the association between air pollutants and CVD markers, we applied linear mixed-effects regression models with marker concentration as a continuous dependent variable. Each gas metric was included in single-pollutant models along with the visit number. Site was included as a fixed effect, because participants were nested within site. As multiple longitudinal measurements collected from each woman may have been correlated, a random intercept was used to account for the covariance of measurements nested within the same participant. First-order ante-dependence structure was specified for the repeated measurements from each participant (Zimmerman and Núñez-Antón 1997).
In our statistical models, we evaluated a variety of factors for potential confounding, including study site, race/ethnicity, education (high school or less, some college, or college graduate), in addition to the following visit-specific time-varying variables: age at the blood draw (continuous), menopausal status (pre- / early peri- / late peri- / post- / unknown), body mass index (BMI), hormone use (yes/no), active smoking (yes/no) and alcohol consumption in the 24 hours before the blood draw (yes/no). We started with a full model including all covariates and performed a backward elimination of the variables based on statistical significance and Akaike Information Criterion (AIC) value, which allowed us to control for confounding without adding overfitted covariates.
Values of hs-CRP greater than 10 mg/L (9.9% of all observations) were excluded as they may indicate severe infection, major trauma, or chronic inflammatory diseases. For all other markers, values were excluded if they were outside the mean ± 3 times the standard deviations (0.02%-8.2% of observations, varied by markers). Eleven woman-visits conducted after major CVD events, including myocardial infarction, coronary heart failure, stroke, percutaneous coronary intervention and coronary artery bypass graft, were excluded from the analyses. We also censored the New Jersey data for visit 6 and 7, because only a few participants from that site had data for those two visits. Given the concern over the confounding by diabetes, we included women diagnosed with diabetes in the main analysis but excluded them in a sensitivity analysis.
Using the same data set, Green et al. (2016) reported that PM2.5 exposure was associated with levels of hs-CRP, fibrinogen, tPA-ag, and PAI-1. Therefore, we further estimated the associations between each gas exposure and CVD markers adjusting for PM2.5 exposure in the same time window (except for one-week window, one-week average PM2.5 exposure was not calculated given the PM2.5 measurement interval, one-month average PM2.5 exposure was used as a substitute). For CO and SO2, we used two-pollutant models incorporating each of these gases and PM2.5 concentrations simultaneously. For NO2, which was usually highly correlated with PM2.5 concentrations, we used a residual analysis to avoid potential collinearity (Bell et al. 2007). As illustrated by the equations below, we predicted PM2.5 concentrations by NO2 concentrations and calculated the residual of PM2.5 (rpm), in which rpm and NO2 concentrations were not correlated. The NO2 concentration and rpm were entered into the regression model simultaneously with other covariates to estimate marker levels. This procedure was repeated to calculate the residual of NO2 (rNO2).
If we found consistent results for NO2 concentrations and rNO2, we concluded NO2 results were robust even after controlling for PM2.5. Otherwise, the association with NO2 observed in the base model could have been confounded by PM2.5 exposure or other unknown factors. If the PM2.5 concentration and rpm were both associated with the outcome in each model, PM2.5 exposure was considered to have an association with that particular marker. Only visits 3–7 with matched PM2.5 concentrations were included in the analysis, so that the results of single-pollutant models can be compared with those adjusted for PM2.5 exposure.
As a sensitivity test, we also examined confounding between gases. Based on the correlation between individual gases, either a two-pollutant model or the residual model was used.
All analyses were conducted using SAS 9.4 (SAS Institute, Cary, NC). All tests were two-sided and p-values < 0.05 were considered statistically significant.
Results
Distribution of CVD marker levels
A total of 2,280 women had up to five annual visits between 1999 and 2004. Based on multiple-visit data, we observed that women with higher education levels, lower BMI, and who were premenopausal or early perimenopausal had lower levels of inflammatory and hemostatic markers as well as higher levels of HDL-related lipoproteins and lower levels of LDL-related lipoproteins and triglycerides (Table 1). Race/ethnicity, smoking, consuming alcohol, and having pre-existing conditions were all related to CVD marker levels.
Table 1.
Distribution of inflammatory, hemostatic and lipid biomarkers by demographic factors for SWAN cohort, 1999–2004a
| Variable | Nb | hs-CRP | Fibrinog en | Factor VIIc | TPA-ag | PAI-1 | HDL | LDL | Triglyceri des | Total Choleste rol | Lp(a) | LpA1 | APOA1 | APOB | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| (unit) | mg/L | mg/dl | % | ng/ml | ng/ml | mg/dl | mg/dl | mg/dl | mg/dl | mg/dl | mg/dl | mg/dl | mg/dl | ||
|
| |||||||||||||||
| N of samples | 7951 | 4039 | 3930 | 8382 | 8309 | 8902 | 8549 | 8665 | 8900 | 8386 | 4432 | 8761 | 8683 | ||
|
| |||||||||||||||
| All participants | 2280 | 1.4 (3.0) | 267 (70) | 119 (35) | 7.0 (4.3) | 14 (19) | 57 (21) | 115 (42) | 105 (75) | 199 (48) | 16 (36) | 52 (19) | 167 (38) | 108 (37) | |
|
| |||||||||||||||
| Race/Ethnicity | African American | 26% | 2.5 (4.1) | 292 (78) | 117 (37) | 7.8 (4.6) | 17 (20) | 54 (18) | 116 (44) | 99 (62) | 193 (48) | 38 (54) | 50 (17) | 160 (36) | 107 (36) |
| Chinese | 10% | 0.7 (1.1) | 262 (58) | 118 (31) | 6.2 (3.8) | 12 (17) | 61 (20) | 112 (38) | 106 (73) | 201 (46) | 9 (19) | 57 (19) | 173 (35) | 107 (35) | |
| Hispanic | 11% | 2.4 (3.8) | 279 (74) | 117 (35) | 8.7 (4.1) | 21 (22) | 49 (17) | 117 (41) | 122 (84) | 198 (48) | 16 (32) | 46 (17) | 155 (36) | 115 (39) | |
| Japanese | 7% | 0.5 (1.1) | 242 (61) | 118 (34) | 6.3 (3.6) | 10 (16) | 62 (20) | 117 (42) | 109 (77) | 205 (45) | 12 (20) | 55 (18) | 175 (34) | 111 (35) | |
| White | 46% | 1.6 (3.0) | 267 (62) | 122 (37) | 6.8 (4.1) | 13 (20) | 57 (21) | 115 (43) | 108 (82) | 199 (48) | 11 (29) | 52 (18) | 167 (38) | 108 (37) | |
|
| |||||||||||||||
| Education | <12 yrs | 24% | 1.7 (3.4) | 279 (87) | 121 (36) | 7.4 (4.6) | 16 (21) | 54 (18) | 116 (42) | 113 (84) | 198 (47) | 20 (42) | 50 (19) | 163 (36) | 110 (37) |
| some college | 33% | 1.7 (3.2) | 267 (70) | 121 (34) | 7.3 (4.3) | 15 (20) | 56 (20) | 117 (42) | 108 (75) | 200 (48) | 18 (41) | 51 (19) | 166 (40) | 111 (38) | |
| college graduated/post grad | 43% | 1.2 (2.5) | 262 (66) | 119 (37) | 6.5 (4.1) | 12 (18) | 59 (21) | 113 (42) | 100 (70) | 198 (47) | 13 (31) | 53 (18) | 169 (38) | 106 (35) | |
|
| |||||||||||||||
| Menopausal status | Pre | 510 | 1.4 (2.6) | 262 (66) | 115 (37) | 6.8 (4.3) | 15 (20) | 53 (21) | 111 (38) | 99 (68) | 189 (38) | 12 (30) | 50 (18) | 161 (37) | 106 (36) |
| Early peri | 3419 | 1.2 (2.5) | 267 (66) | 115 (31) | 6.9 (3.9) | 14 (19) | 56 (19) | 111 (39) | 98 (68) | 192 (44) | 16 (36) | 52 (18) | 163 (35) | 105 (35) | |
| Late peri | 917 | 1.4 (3.1) | 273 (74) | 123 (38) | 7.4 (4.5) | 17 (23) | 57 (21) | 122 (44) | 109 (85) | 206 (51) | 19 (43) | 51 (18) | 167 (38) | 112 (39) | |
| Post | 3022 | 1.5 (3.4) | 273 (74) | 125 (38) | 7.1 (4.7) | 14 (19) | 58 (21) | 121 (45) | 113 (77) | 206 (49) | 17 (36) | 52 (19) | 170 (40) | 112 (38) | |
| Unknown | 1050 | 1.8 (3.5) | 262 (62) | 123 (39) | 6.5 (4.0) | 11 (16) | 59 (22) | 112 (42) | 112 (80) | 199 (47) | 12 (33) | 52 (19) | 173 (41) | 109 (37) | |
|
| |||||||||||||||
| BMI (kg/m2) | <25, normal/under | 3294 | 0.6 (1.0) | 251 (57) | 112 (31) | 5.3 (3.2) | 8.1 (10) | 65 (21) | 111 (40) | 90 (50) | 197 (46) | 12 (25) | 57 (19) | 177 (36) | 101 (33) |
| 25–29.9, overweight | 2414 | 1.6 (2.4) | 267 (62) | 124 (37) | 7.0 (3.6) | 14 (17) | 56 (19) | 118 (42) | 112 (81) | 201 (47) | 15 (35) | 51 (18) | 166 (36) | 111 (38) | |
| ≥30, obese | 2998 | 3.7 (4.2) | 292 (72) | 125 (37) | 8.8 (4.1) | 23 (24) | 50 (16) | 118 (44) | 124 (92) | 198 (49) | 22 (47) | 48 (16) | 157 (33) | 113 (38) | |
|
| |||||||||||||||
| Current smoker | Yes | 1126 | 2.0 (3.5) | 292 (83) | 118 (34) | 7.9 (4.4) | 18 (23) | 52 (19) | 117 (45) | 117 (85) | 197 (50) | 21 (45) | 48 (18) | 161 (40) | 113 (41) |
| No | 7563 | 1.3 (2.9) | 267 (66) | 120 (36) | 6.8 (4.2) | 13 (18) | 58 (21) | 115 (42) | 104 (74) | 199 (47) | 15 (34) | 52 (19) | 167 (38) | 107 (36) | |
|
| |||||||||||||||
| Alcohol in last 24 hours | Yes | 1285 | 1.2 (2.2) | 251 (64) | 117 (36) | 6.8 (4.3) | 13 (17) | 65 (22) | 113 (43) | 94 (63) | 200 (47) | 13 (37) | 56 (18) | 180 (38) | 106 (36) |
| No | 7520 | 1.5 (3.1) | 273 (70) | 120 (34) | 7.0 (4.3) | 14 (19) | 56 (19) | 116 (42) | 107 (76) | 198 (47) | 16 (36) | 51 (19) | 165 (37) | 109 (37) | |
|
| |||||||||||||||
| Hormone use since last visit | Yes | 1719 | 1.9 (3.6) | 262 (58) | 123 (41) | 6.2 (4.0) | 11 (14) | 60 (22) | 112 (40) | 115 (80) | 200 (45) | 12 (30) | 54 (19) | 177 (40) | 108 (36) |
| No | 7201 | 1.3 (2.8) | 267 (70) | 119 (34) | 7.1 (4.4) | 15 (21) | 56 (20) | 116 (42) | 103 (74) | 198 (48) | 17 (38) | 51 (18) | 164 (37) | 108 (37) | |
|
| |||||||||||||||
| diagnosed diabetes | Yes | 704 | 4.2 (4.7) | 300 (89) | 131 (35) | 9.1 (4.4) | 26 (29) | 46 (15) | 112 (44) | 159 (127) | 194 (52) | 23 (50) | 48 (15) | 155 (34) | 116 (43) |
| No | 8229 | 1.3 (2.8) | 267 (66) | 119 (35) | 6.8 (4.2) | 13 (18) | 58 (21) | 116 (42) | 103 (70) | 199 (47) | 15 (35) | 52 (19) | 168 (38) | 108 (35) | |
|
| |||||||||||||||
| Any cardio-vascular event | Yes | 104 | 2.8 (3.6) | 308 (72) | 135 (30) | 8.6 (5.1) | 20 (32) | 50 (19) | 121 (51) | 155 (125) | 200 (64) | 24 (38) | 51 (14) | 156 (33) | 115 (47) |
| No | 8832 | 1.4 (2.9) | 267 (70) | 119 (35) | 6.9 (4.3) | 14 (19) | 57 (21) | 115 (42) | 105 (74) | 199 (48) | 16 (36) | 52 (19) | 167 (38) | 108 (36) | |
Data shown in each grid is median (interquartile range).
For ethnicity/education, the % show in this column are the percentage of participants in each category among all participants. For the visit-specific variables, N is the number of observations, not women; each participant could have data from multiple visits, and could be in different categories at different visits. Sample size varied by biomarkers. Visits without any blood data (N=69) or any matched exposure data (N=5147) were excluded. Visits 6 and 7 in New Jersey site (N=55) were censored due to small sample size. Visits happened after any CVD events (N=11 from 4 participants) were excluded. Marker values out of reasonable ranges (0.02–8.2% varied by markers) were excluded. For hs-CRP, values greater than 10 mg/L (N=863) were not included due to the concern of possible severe inflammation.
As expected, some CVD markers were correlated (Table S1). Two inflammatory markers, hs-CRP and fibrinogen, were moderately correlated (Pearson correlation coefficient (r) = 0.46). PAI-1 was correlated with tPA-ag (r = 0.54), and both were positively correlated with triglycerides but negatively correlated with HDL. Lipoproteins were correlated with each other, with highly positive correlations between LDL, APOB and total cholesterol (r = 0.80–0.88) as well as between HDL, LpA1 and APOA1 (r = 0.71–0.80).
Distribution of air pollutant levels
The gas concentrations were similar for different exposure windows; thus, only the prior-day and one-year average gas concentrations, which represented short- and long-term exposures, respectively, are shown stratified by study site (Table 2). For most of the locations, over 75% of the serum samples had matched gas exposure data. Both CO and NO2 concentrations were higher in Los Angeles and Newark than the other four sites. SO2 concentrations were remarkably lower in the two California sites than non-California sites, with the highest levels in Detroit.
Table 2.
Distribution of prior one-day and one-year average CO, NO2, and SO2 concentrations by SWAN site, 1999–2004
| 1-day average concentration | 1-year average concentration | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| site | N | % a | Mean | SD | Median | IQR | N | % a | Mean | SD | Median | IQR |
|
| ||||||||||||
| CO (ppm) | ||||||||||||
|
| ||||||||||||
| Detroit, MI | 758 | 45 | 0.58 | 0.39 | 0.50 | 0.40 | 791 | 47 | 0.61 | 0.16 | 0.56 | 0.10 |
| Chicago, IL | 1096 | 77 | 0.95 | 0.48 | 0.90 | 0.60 | 1141 | 80 | 0.98 | 0.21 | 0.96 | 0.40 |
| Oakland, CA | 1670 | 95 | 0.90 | 0.54 | 0.80 | 0.70 | 1674 | 95 | 0.95 | 0.19 | 0.92 | 0.31 |
| Los Angeles, CA | 1908 | 95 | 1.19 | 1.07 | 0.80 | 1.00 | 1957 | 97 | 1.55 | 0.40 | 1.43 | 0.54 |
| Newark, NJ | 366 | 69 | 1.72 | 0.86 | 1.60 | 1.00 | 394 | 75 | 1.82 | 0.50 | 1.79 | 0.59 |
| Pittsburgh, PA | 1333 | 87 | 0.99 | 0.58 | 0.90 | 0.80 | 1462 | 95 | 1.02 | 0.34 | 1.01 | 0.64 |
|
| ||||||||||||
| NO2 (ppb) | ||||||||||||
|
| ||||||||||||
| Detroit, MI | 647 | 39 | 39.6 | 17.2 | 39.0 | 16.0 | 607 | 36 | 39.1 | 2.6 | 39.6 | 3.2 |
| Chicago, IL | 1063 | 74 | 40.8 | 13.0 | 40.0 | 18.0 | 1083 | 76 | 39.8 | 2.1 | 39.1 | 2.8 |
| Oakland, CA | 1506 | 86 | 31.9 | 13.7 | 32.0 | 19.0 | 1524 | 87 | 31.2 | 2.7 | 31.4 | 3.4 |
| Los Angeles, CA | 1913 | 95 | 44.7 | 18.5 | 43.0 | 19.0 | 1962 | 98 | 48.1 | 4.3 | 47.8 | 5.4 |
| Newark, NJ | 374 | 71 | 50.7 | 14.6 | 50.0 | 18.0 | 402 | 76 | 49.4 | 3.9 | 49.2 | 6.0 |
| Pittsburgh, PA | 1448 | 94 | 37.4 | 12.5 | 37.0 | 18.0 | 1484 | 97 | 37.4 | 3.8 | 37.6 | 3.0 |
|
| ||||||||||||
| SO2 (ppb) | ||||||||||||
|
| ||||||||||||
| Detroit, MI | 721 | 43 | 24.6 | 26.5 | 15.0 | 33.0 | 745 | 44 | 24.7 | 4.2 | 24.6 | 5.6 |
| Chicago, IL | 1295 | 91 | 17.6 | 18.2 | 12.0 | 16.0 | 1360 | 95 | 18.0 | 7.8 | 15.3 | 15.5 |
| Oakland, CA | 1055 | 60 | 4.5 | 4.1 | 3.0 | 4.0 | 1046 | 60 | 4.5 | 0.9 | 4.3 | 0.9 |
| Los Angeles, CA | 1851 | 92 | 7.5 | 7.8 | 6.0 | 7.0 | 1910 | 95 | 7.5 | 2.5 | 8.2 | 3.2 |
| Newark, NJ | 440 | 83 | 20.0 | 13.2 | 17.0 | 15.0 | 448 | 85 | 20.1 | 3.9 | 21.2 | 6.9 |
| Pittsburgh, PA | 1241 | 81 | 22.9 | 17.3 | 20.0 | 17.0 | 1409 | 92 | 23.6 | 5.1 | 22.0 | 4.5 |
Percentage of blood samples with geographically matched gas concentrations from monitors within 20 km buffer from participants’ residences.
The CO and NO2 concentrations matched by exposure window were moderately to highly correlated (r = 0.61–0.78) (Table S2). NO2 concentrations were also correlated with PM2.5 concentrations for the same exposure window (r = 0.39–0.70), while CO was less correlated with PM2.5 (r = 0.25–0.41). The SO2 concentrations were weakly correlated with other gases or PM2.5 (r = −0.26–0.36).
Associations between air pollutants and CVD markers
Final regression models between gases and CVD markers were adjusted for study site, race/ethnicity and education, and time-varying visit number, menopausal status, BMI, hormone use, active smoking, and alcohol consumption in the 24 hours prior to blood draw. The associations were expressed as the percent change in marker levels per interquartile increase in the concentration of a gas pollutant, which was 0.68 ppm for CO, 13 ppb for NO2, and 15 ppb for SO2. The associations observed for the previous 1-day, 1-week and 1-month exposure windows followed the same trend, as did the previous 6-month and 1-year exposure windows; therefore, results of 1-week and 1-year exposure windows were presented as examples of short- and long-term effects, respectively (Table 3, full results can be found in Table S3–S5).
Table 3.
Adjusteda associations between gas exposure and cardiovascular disease markers for SWAN cohort, 1999–2004
| Marker | Short-term (1-week) | Long-term (1-year) | ||||
|---|---|---|---|---|---|---|
| CO | NO2 | SO2 | CO | NO2 | SO2 | |
| hs-CRP | 1.2 (−1.4, 3.8) | 0.7 (−1.7, 3.2) | −2.2 (−5.1, 0.7) | 1.2 (−6.2, 9.2) | 3.9 (−8.4, 17.8) | 3.8 (−3.9, 12) |
| Fibrinogen | 1.3 (0.6, 2.0)** | 0.7 (−0.1, 1.4) | −0.5 (−1.5, 0.4) | 0.05 (−2.1, 2.2) | 0.2 (−3.4, 3.9) | −2.0 (−4.6, 0.6) |
| Factor VIIc | 0.9 (0.1, 1.7)* | 1.0 (0.1, 1.8)* | −0.6 (−1.8, 0.5) | −1.0 (−3.5, 1.7) | 2.1 (−2.3, 6.7) | −1.6 (−4.6, 1.5) |
| tPA-ag | −1.4 (−2.6, −0.2)* | −1.5 (−2.7, −0.3)* | −0.2 (−1.6, 1.2) | 3.3 (−0.3, 7.1) | −1.6 (−7.2, 4.5) | 3.1 (−0.6, 6.9) |
| PAI-1 | 0.1 (−2.2, 2.6) | 0.9 (−1.4, 3.4) | 0.4 (−2.4, 3.3) | 6.1 (−1.3, 14.1) | 12.4 (−0.1, 26.6) | −3.4 (−10.3, 4.0) |
| HDL | 0.9 (0.4, 1.4)** | 0.2 (−0.2, 0.7) | −0.6 (−1.1, 0)* | −1.2 (−2.6, 0.3) | −4.0 (−6.3, −1.7)** | −1.3 (−2.7, 0.1) |
| LDL | −0.02 (−0.7, 0.6) | −0.03 (−0.7, 0.6) | 0.2 (−0.5, 1.0) | 1.9 (−0.3, 4.0) | 0.7 (−2.6, 4.2) | 0.3 (−1.7, 2.4) |
| Triglycerides | −1.4 (−2.5, −0.4)* | −1.6 (−2.6, −0.5)** | −0.6 (−1.8, 0.7) | 1.2 (−2.2, 4.7) | 3.1 (−2.6, 9.1) | −0.7 (−4.0, 2.7) |
| Total cholesterol | 0.1 (−0.3, 0.6) | −0.2 (−0.6, 0.3) | −0.1 (−0.6, 0.4) | 0.7 (−0.6, 2.0) | −0.6 (−2.7, 1.6) | −0.3 (−1.6, 1.0) |
| APOA1 | 0.3 (−0.1, 0.7) | −0.3 (−0.7, 0.1) | −0.7 (−1.2, −0.3)** | −0.8 (−2.0, 0.4) | −4.7 (−6.6, −2.8)** | −2.1 (−3.3, −0.9)** |
| APOB | 0.1 (−0.4, 0.7) | −0.2 (−0.7, 0.3) | −0.05 (−0.7, 0.6) | 1.5 (−0.2, 3.3) | −1.2 (−3.9, 1.6) | 1.6 (−0.1, 3.3) |
| Lp(a) | −0.6 (−2.3, 1.1) | −0.5 (−2.1, 1.1) | 1.4 (−0.5, 3.4) | −0.9 (−6.4, 5.0) | −3.4 (−12.3, 6.5) | 5.7 (0.2, 11.5)* |
| LpA1 | 0.7 (−0.1, 1.5) | −0.1 (−1.0, 0.7) | −0.1 (−1.2, 1.1) | −0.8 (−3.3, 1.8) | −0.7 (−5.0, 3.7) | −0.1 (−3.3, 3.3) |
Note: Results shown are percent of change in biomarker level per an interquartile increase of exposure, which is 0.68 ppm for CO, 13 ppb for NO2, and 15 ppb for SO2.
Analyses were based on log-transformed biomarker levels, adjusted for study site, visit number, race/ethnicity, education, menopause status, BMI, active smoking status, hormone use since last visit, alcohol consumption in the past 24 hours before blood draw.
p<0.05;
p<0.01
Short-term exposure to CO and NO2 were positively associated with fibrinogen and factor VIIc, but negatively associated with tPA-ag (Table 3). For example, an interquartile increase in the one-month average concentrations of CO and NO2 was associated with 1.3% (95% confidence interval: 0.5%, 2.1%) and 1.4% (0.4%, 2.3%) increases in fibrinogen levels, respectively. After adjusting for PM2.5, associations with CO became weaker and the residuals of NO2 were not statistically significant (Figure 1a). Also, the negative associations of tPA-ag with short-term exposure of CO and NO2 were confounded by PM2.5. PM2.5 had associations with fibrinogen, factor VIIc, and tPA-ag in the same direction as NO2 and with similar magnitude, but the residuals of neither NO2 nor PM2.5 met statistical significance in most of the cases, making it difficult to differentiate the associations of NO2 and PM2.5 exposures with the markers. In a few cases, PM2.5 exposures showed stronger associations than NO2. Long-term exposure to NO2 was positively associated with PAI-1, with a 12.4% (-0.1%, 26.6%) increase of PAI-1 levels per an interquartile increase in average prior one-year NO2 concentration, but this association was also confounded by PM2.5 (Figure 1b). Exposure to SO2 was generally not statistically associated with inflammatory or hemostatic markers we examined, except for a negative association between six-month SO2 exposure and fibrinogen.
Figure 1.
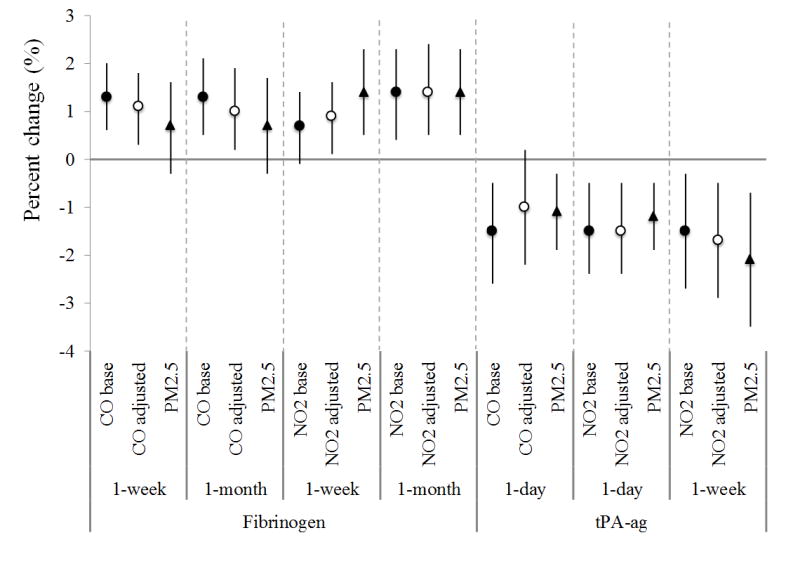
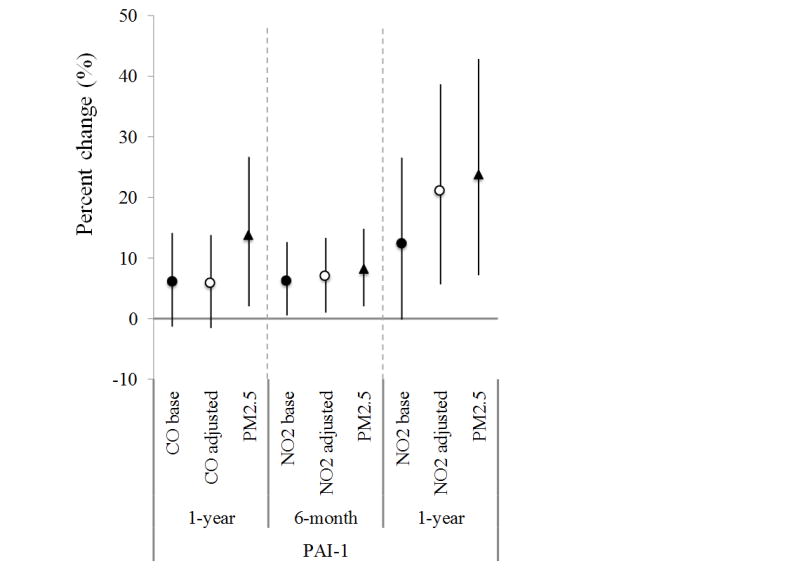
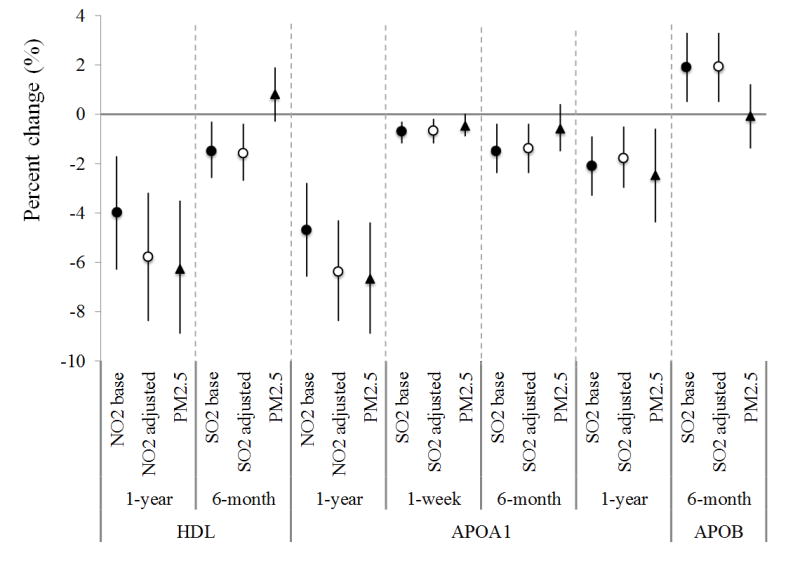
The association between gas exposure and selected CVD markers in selected exposure windows, before and after adjustment of PM2.5 exposure for SWAN cohort, 1999–2004. (Dots/circles/triangles are percent of change in the levels of markers per interquartile increase of exposure, and error bars are 95th% of confidence intervals. Dots - CO / NO2 / SO2 base – the change of the gas in the single-pollutant model with one gas only; circles - CO / NO2 / SO2 adj by PM – the change of the gas in the two-pollutant model with one gas and PM2.5; triangles - PM2.5 – the change of PM2.5 in two-pollutant model)
For lipoproteins, long-term exposures to NO2 and SO2 were strongly associated with reduced HDL-related lipoproteins (Figure 1c). An interquartile increment of the one-year average NO2 concentration was associated with an average 4.0% (1.7%, 6.3%) decrease of HDL and 4.7% (2.8%, 6.6%) decrease of APOA1. These negative associations were also observed for six-month and one-week average concentrations of SO2. Meanwhile, every interquartile increase of the six-month average SO2 concentration was associated with a 1.9% (0.5%, 3.3%) increase of APOB. SO2 exposure was also associated with increases in LDL and Lp(a) (Table S5). Short-term CO and NO2 exposures were associated with increased HDL and decreased triglycerides (Table 3). Results on lipoproteins remained robust or became stronger after adjusting for PM2.5.
The sensitivity analysis excluding women diagnosed with diabetes obtained basically same results with stronger statistical significance. Another sensitivity test on the confounding between gases suggested consistent associations with those observed in the single-pollutant models.
Discussion
The multi-site SWAN cohort provided valuable data on a large sample of midlife women to explore the associations between ambient gases and a number of CVD markers while adjusting for covariates. We observed increased levels of fibrinogen associated with short-term CO exposure. Short-term exposure to NO2 was positively associated with fibrinogen and factor VIIc but negatively associated with tPA-ag, although some associations were difficult to differentiate from the associations with PM2.5. These findings suggested increased inflammation and coagulation but suppressed fibrinolysis in association with NO2 and/or CO. Occurring simultaneously, these gases may increase the risk of thrombosis. Altered vascular reactivity and reduced blood flow may eventually lead to myocardial ischemia and other CVDs (Davies and Thomas 1984). In addition, long-term gas exposures have the potential to lower the “good cholesterol” and increase the “bad cholesterol”. Together, these results suggest ambient gas exposures increase the potential of inflammation and thrombosis and disrupt cholesterol balance, which may further increase the risk of myocardial infarction or other CVDs.
As mentioned earlier, previous findings regarding the associations between short-term gas exposure and inflammatory/hemostatic markers have been inconsistent. For example, Delfino et al. (2009) studied elderly individuals in Los Angeles and found that daily exposure to CO and NO2 were positively associated with hs-CRP. Bind et al. (2012) found that an IQR increase in one-day exposure to NO2 was associated with 1.7% (95% CIs: 0.2, 3.3) increase in fibrinogen but not with hs-CRP, also among elderly men. Based on a large midlife cohort from four centers in the U.S., Liao et al. (2005) did not observe statistically significant association between short-term exposure (1–3 days prior) to CO, NO2 and SO2 and fibrinogen. In our study, no statistically signficant association was observed between gas exposure in any window and hs-CRP levels. However, fibrinogen was associated with one-week and one-month exposure to CO and NO2, which provides evidence that short-term exposure to CO and NO2 increases the tendency of inflammation.
Only a few studies were available regarding the associations between long-term exposure to the gases and inflammatory/hemostatic markers, with mixed results. Some studies found CO and carboxyhemoglobin (COHb) (a CO product) were positively associated with hs-CRP (Davutoglu et al. 2009; Huang et al. 2014). Panasevich et al. (2009) observed positive but not significant associations of hs-CRP with long-term exposures to CO, NO2 and SO2 up to 30 years, but not for fibrinogen or PAI-1. In contrast, two studies found no association between annual average NO2 or SO2 and CRP or fibrinogen (Dadvand et al. 2014; Forbes et al. 2009). Our study supports null associations between long-term gas exposure and inflammation/hemostatic markers, except the unexplained negative association between six-month SO2 exposure and fibrinogen.
Information from prior studies on the relations of ambient gases with lipid markers were even more scarce. Among an elderly population in Taiwan, Chuang et al. (2011) found one-year average NO2 concentrations were associated with elevated total cholesterol but confounded by PM2.5, and no changes were due to exposure to SO2. Similarly, no stable association with total cholesterol was observed in our study. Instead, we found negative associations between HDL and long-term NO2 and SO2 exposure, as well as increased APOB with six-month SO2 exposure. Both indicate greater risks of CVDs. Furthermore, we observed negative associations of short-term exposures to all gases with triglycerides, which could be confounded by recent diet, although fasting blood samples were used. We note that levels of triglycerides increased as the women entered menopause (Derby et al. 2009), while the gas levels decreased over time during our follow-up period. This may have led to elevated levels of triglycerides at lower gas exposures.
One strength of our study is that multiple exposure windows were examined, shedding light on the time frame required for the body’s responses to ambient gas exposure. Previous studies mostly focused on short follow-up times, with lags from hours to several days. USEPA (2010) summarized that CVD outcomes due to CO exposure usually occurred within three days after exposure. Dadvand et al. (2014) found that the associations between NO2 and inflammatory markers were strongest at lags of 4–5 days but not at lags of 0–2 days. Bind et al. (2012) studied exposure window from hours to a moving average of 28-day lag and found a positive association between CO/NO2 and fibrinogen from a 1- to a 28-day lag. In the present study, we covered the exposure windows from day and year scales. Given the short half-life of CO in the environment, the associations with markers we examined were mostly observed in short-term exposure windows up to one-month. NO2 and SO2 influence human health not only in gas form but also by forming secondary particles, which are temporally more stable, and the associations with CVD markers were observed in both short- and long-term exposure windows. From the perspective of markers, the inflammatory and hemostatic markers, such as fibrinogen and tPA-ag, responded to gas exposures in all short-term windows we examined from prior one-day to one-month. Repeated inflammation and thrombosis raise the risk of developing chronic cardiovascular outcomes, such as, atherosclerosis (Pearson et al. 2003). In contrast, for lipoproteins, adverse associations were observed for long-term exposure and thus demonstate the long-term impact of ambient gas exposure. Greater variations were observed for apolipoproteins, APOA1 and APOB, than the traditional lipid profile, indicating that they were more sensitive to gas exposure.
We also considered the confounding by PM2.5 while evaluating the health impacts of gaseous pollutants. Previous studies showed that the associations of CO with health outcomes generally remained robust in co-pollutant models (USEPA 2010), but for NO2 and SO2, such associations of each gas have not been clearly defined with co-pollutants controlled due to potential collinearity issue. Across our analyses with all three gaseous pollutants, PM2.5 appeared consistently associated with reduced tPA-ag level in short-term exposure windows as well as increased PAI-1 in long-term exposure windows, indicating risks of thrombosis. We used a residual analysis for NO2 to avoid collinearity, and found that PM2.5 confounded the associations between CO/NO2 and several inflammatory/hemostatic markers (Table S3–S4). The 1-year average PM2.5 exposure was associated with decreased HDL and APOA1; however, the associations between gases and lipoproteins were generally robust and were not confounded by PM2.5 exposure.
This study had several limitations to be considered when interpreting the results. First, using the regulatory monitoring network with a 20 km buffer may have introduced exposure misclassification, and thus the ambient gas exposures we obtained were only approximations of personal exposure. The 20 km buffer was selected by taking into consideration the impacts of sample size, exposure misclassification, and population characteristics. By using a larger buffer, we obtained similar exposure estimates to those from the 10 km buffer but a 2–3 times increased sample sizes. A bigger buffer distance also enabled us to keep a diverse population, because areas close to monitors where sources are usually located tend to be low socioeconomic communities (Ebisu et al. 2014). Furthermore, we tracked the changes of participants’ home addresses, which maximized accuracy of the exposure estimation.
Secondly, some known cardiovascular risk factors were not considered in this study, such as traffic noise, ultrafine particles, and ozone. As a result of incomplete combustion from traffic, CO may serve as a marker of freshly emitted ultrafine particles, which are considered more toxic than PM2.5 (Araujo and Nel 2009). Both CO and NO2 are involved in ozone formation, and ozone has been shown to affect cardiovascular health (Hoffmann et al. 2012). Future studies with consideration of these factors are warranted. Third, although the SWAN data included many covariates, some potential confounding factors were not included due to the incompleteness of data, e.g., dietary intake and physical activity, which might have resulted in residual confounding. Finally, in this study, participants were recruited with a series of selection criteria and not randomly selected; therefore, the study population may not be representative of the general US female midlife population.
Conclusions
In summary, this study provides valuable additions to the current literature on the associations between ambient gaseous pollutants and CVD markers. Our results suggest that short-term ambient exposures to CO and NO2 may increase the potential for thrombosis and exposure to NO2 and SO2 may affect cholesterol metabolism, suggesting that ambient gas at normally observed level could pose additional CVD risks. One should also be cautious of the confounding of PM2.5 exposure on the associations between CO/NO2 and inflammatory/hemostatic markers, while the associations between gases and lipoproteins were generally robust to PM2.5 exposure adjustment.
This study was conducted among midlife women who normally have not been considered as a sensitive population. The disrupted cholesterol levels and increasing vascular inflammation due to menopause make this population more vulnerable to environmental factors that may affect CVD risks. The air pollutant levels below the NAAQS could cause inflammation and coagulation as well as changes in their lipoprotein levels, posing additional cardiovascular burden beyond the impact of menopause.
Supplementary Material
Highlights.
We examined the associations between CVD markers and ambient gases.
Short-term CO exposure was associated with increased levels of coagulation markers.
Long-term exposures to NO2 and SO2 were associated with reduced HDL and APOA1.
PM2.5 exposure confounded associations between CO/NO2 and hemostatic markers.
Exposure to ambient gases at current NAAQS may increase CVD risks in midlife women.
Acknowledgments
The opinions expressed in this paper are solely those of the authors and do not represent the policy or position of the State of California or the California Environmental Protection Agency.
The Study of Women’s Health Across the Nation (SWAN) has grant support from the National Institutes of Health (NIH), DHHS, through the National Institute on Aging (NIA), the National Institute of Nursing Research (NINR) and the NIH Office of Research on Women’s Health (ORWH) (Grants U01NR004061; U01AG012505, U01AG012535, U01AG012531, U01AG012539, U01AG012546, U01AG012553, U01AG012554, U01AG012495). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIA, NINR, ORWH or the NIH.
Clinical Centers: University of Michigan, Ann Arbor – Siobán Harlow, PI 2011 – present, MaryFran Sowers, PI 1994–2011; Massachusetts General Hospital, Boston, MA – Joel Finkelstein, PI 1999 – present; Robert Neer, PI 1994 – 1999; Rush University, Rush University Medical Center, Chicago, IL – Howard Kravitz, PI 2009 – present; Lynda Powell, PI 1994 – 2009; University of California, Davis/Kaiser – Ellen Gold, PI; University of California, Los Angeles – Gail Greendale, PI; Albert Einstein College of Medicine, Bronx, NY – Carol Derby, PI 2011 – present, Rachel Wildman, PI 2010 – 2011; Nanette Santoro, PI 2004 – 2010; University of Medicine and Dentistry – New Jersey Medical School, Newark – Gerson Weiss, PI 1994 – 2004; and the University of Pittsburgh, Pittsburgh, PA – Karen Matthews, PI.
NIH Program Office: National Institute on Aging, Bethesda, MD – Winifred Rossi 2012 - present; Sherry Sherman 1994 – 2012; Marcia Ory 1994 – 2001; National Institute of Nursing Research, Bethesda, MD – Program Officers.
Central Laboratory: University of Michigan, Ann Arbor – Daniel McConnell (Central Ligand Assay Satellite Services).
Coordinating Center: University of Pittsburgh, Pittsburgh, PA – Maria Mori Brooks, PI 2012 - present; Kim Sutton-Tyrrell, PI 2001 – 2012; New England Research Institutes, Watertown, MA - Sonja McKinlay, PI 1995 – 2001.
Steering Committee: Susan Johnson, Current Chair, Chris Gallagher, Former Chair
We thank the study staff at each site and all the women who participated in SWAN.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare they have no actual or potential competing financial interests.
References
- Allred EN, Bleecker ER, Chaitman BR, Dahms TE, Gottlieb SO, Hackney JD, et al. Short-term effects of carbon monoxide exposure on the exercise performance of subjects with coronary artery disease. N Engl J Med. 1989;321:1426–1432. doi: 10.1056/NEJM198911233212102. [DOI] [PubMed] [Google Scholar]
- Araujo JA, Nel AE. Particulate matter and atherosclerosis: role of particle size, composition and oxidative stress. Part Fibre Toxicol. 2009;6:24. doi: 10.1186/1743-8977-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarelli A, Zanobetti A, Martinelli I, Grillo P, Hou L, Giacomini S, et al. Effects of exposure to air pollution on blood coagulation. J Thromb Haemost. 2007;5:252–260. doi: 10.1111/j.1538-7836.2007.02300.x. [DOI] [PubMed] [Google Scholar]
- Ballester F, Rodriguez P, Iniguez C, Saez M, Daponte A, Galan I, et al. Air pollution and cardiovascular admissions association in spain: Results within the emecas project. J Epidemiol Community Health. 2006;60:328–336. doi: 10.1136/jech.2005.037978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskurt OK. Acute hematologic and hemorheologic effects of sulfur dioxide inhalation. Arch Environ Health. 1988;43:344–348. doi: 10.1080/00039896.1988.9934946. [DOI] [PubMed] [Google Scholar]
- Beckerman BS, Jerrett M, Finkelstein M, Kanaroglou P, Brook JR, Arain MA, et al. The association between chronic exposure to traffic-related air pollution and ischemic heart disease. J Toxicol Environ Health A. 2012;75:402–411. doi: 10.1080/15287394.2012.670899. [DOI] [PubMed] [Google Scholar]
- Bell ML, Dominici F, Ebisu K, Zeger SL, Samet JM. Spatial and temporal variation in pm(2.5) chemical composition in the united states for health effects studies. Environ Health Perspect. 2007;115:989–995. doi: 10.1289/ehp.9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bind MA, Baccarelli A, Zanobetti A, Tarantini L, Suh H, Vokonas P, et al. Air pollution and markers of coagulation, inflammation, and endothelial function: Associations and epigene-environment interactions in an elderly cohort. Epidemiology. 2012;23:332–340. doi: 10.1097/EDE.0b013e31824523f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett JR. Lipids, lipoproteins, atherosclerosis and cardiovascular disease. Clin Biochem Rev. 2004;25:2. [PMC free article] [PubMed] [Google Scholar]
- Chandler WL, Alessi MC, Aillaud MF, Henderson P, Vague P, Juhan-Vague I. Clearance of tissue plasminogen activator (tpa) and tpa/plasminogen activator inhibitor type 1 (pai-1) complex: Relationship to elevated tpa antigen in patients with high pai-1 activity levels. Circulation. 1997;96:761–768. doi: 10.1161/01.cir.96.3.761. [DOI] [PubMed] [Google Scholar]
- Chang CC, Tsai SS, Ho SC, Yang CY. Air pollution and hospital admissions for cardiovascular disease in taipei, taiwan. Environ Res. 2005;98:114–119. doi: 10.1016/j.envres.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Channell MM, Paffett ML, Devlin RB, Madden MC, Campen MJ. Circulating factors induce coronary endothelial cell activation following exposure to inhaled diesel exhaust and nitrogen dioxide in humans: Evidence from a novel translational in vitro model. Toxicol Sci. 2012;127:179–186. doi: 10.1093/toxsci/kfs084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MF, Tsai SS, Yang CY. Air pollution and hospital admissions for myocardial infarction in a tropical city: Kaohsiung, taiwan. J Toxicol Environ Health A. 2009;72:1135–1140. doi: 10.1080/15287390903091756. [DOI] [PubMed] [Google Scholar]
- Chuang KJ, Chan CC, Su TC, Lee CT, Tang CS. The effect of urban air pollution on inflammation, oxidative stress, coagulation, and autonomic dysfunction in young adults. Am J Respir Crit Care Med. 2007;176:370–376. doi: 10.1164/rccm.200611-1627OC. [DOI] [PubMed] [Google Scholar]
- Chuang KJ, Yan YH, Chiu SY, Cheng TJ. Long-term air pollution exposure and risk factors for cardiovascular diseases among the elderly in taiwan. Occup Environ Med. 2011;68:64–68. doi: 10.1136/oem.2009.052704. [DOI] [PubMed] [Google Scholar]
- Dadvand P, Nieuwenhuijsen MJ, Agusti A, de Batlle J, Benet M, Beelen R, et al. Air pollution and biomarkers of systemic inflammation and tissue repair in copd patients. Eur Respir J. 2014;44:603–613. doi: 10.1183/09031936.00168813. [DOI] [PubMed] [Google Scholar]
- Davies MJ, Thomas A. Thrombosis and acute coronary-artery lesions in sudden cardiac ischemic death. N Engl J Med. 1984;310:1137–1140. doi: 10.1056/NEJM198405033101801. [DOI] [PubMed] [Google Scholar]
- Davutoglu V, Zengin S, Sari I, Yildirim C, Al B, Yuce M, et al. Chronic carbon monoxide exposure is associated with the increases in carotid intima-media thickness and c-reactive protein level. Tohoku J Exp Med. 2009;219:201–206. doi: 10.1620/tjem.219.201. [DOI] [PubMed] [Google Scholar]
- Delfino RJ, Staimer N, Tjoa T, Polidori A, Arhami M, Gillen DL, et al. Circulating biomarkers of inflammation, antioxidant activity, and platelet activation are associated with primary combustion aerosols in subjects with coronary artery disease. Environ Health Perspect. 2008;116:898–906. doi: 10.1289/ehp.11189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delfino RJ, Staimer N, Tjoa T, Gillen DL, Polidori A, Arhami M, et al. Air pollution exposures and circulating biomarkers of effect in a susceptible population: Clues to potential causal component mixtures and mechanisms. Environ Health Perspect. 2009;117:1232–1238. doi: 10.1289/ehp.0800194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derby CA, Crawford SL, Pasternak RC, Sowers M, Sternfeld B, Matthews KA. Lipid changes during the menopause transition in relation to age and weight: The study of women’s health across the nation. Am J Epidemiol. 2009;169:1352–1361. doi: 10.1093/aje/kwp043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebisu K, Belanger K, Bell ML. The association between airborne pm2.5 chemical constituents and birth weight-implication of buffer exposure assignment. Environ Res Lett. 2014;9 doi: 10.1088/1748-9326/9/8/084007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes LJ, Patel MD, Rudnicka AR, Cook DG, Bush T, Stedman JR, et al. Chronic exposure to outdoor air pollution and markers of systemic inflammation. Epidemiology. 2009;20:245–253. doi: 10.1097/EDE.0b013e318190ea3f. [DOI] [PubMed] [Google Scholar]
- Green R, Broadwin R, Malig B, Basu R, Gold EB, Qi L, et al. Long- and short-term exposure to air pollution and inflammatory/hemostatic markers in midlife women. Epidemiology. 2016;27:211–220. doi: 10.1097/EDE.0000000000000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Tong S, Li S, Barnett AG, Yu W, Zhang Y, et al. Gaseous air pollution and emergency hospital visits for hypertension in beijing, china: A time-stratified case-crossover study. Environ Health. 2010;9:57. doi: 10.1186/1476-069X-9-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann B, Luttmann-Gibson H, Cohen A, Zanobetti A, de Souza C, Foley C, et al. Opposing effects of particle pollution, ozone, and ambient temperature on arterial blood pressure. Environ Health Perspect. 2012;120:241–246. doi: 10.1289/ehp.1103647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WH, Yen TH, Chan MJ, Su YJ. Environmental carbon monoxide level is associated with the level of high-sensitivity c-reactive protein in peritoneal dialysis patients. Medicine (Baltimore) 2014;93:e181. doi: 10.1097/MD.0000000000000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Mathes R, Ross Z, Nadas A, Thurston G, Matte T. Fine particulate matter constituents associated with cardiovascular hospitalizations and mortality in new york city. Environ Health Perspect. 2011;119:467–473. doi: 10.1289/ehp.1002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalaludin B, Morgan G, Lincoln D, Sheppeard V, Simpson R, Corbett S. Associations between ambient air pollution and daily emergency department attendances for cardiovascular disease in the elderly (65+ years), sydney, australia. J Expo Sci Environ Epidemiol. 2006;16:225–237. doi: 10.1038/sj.jea.7500451. [DOI] [PubMed] [Google Scholar]
- Kaptoge S, Di Angelantonio E, Pennells L, Wood AM, White IR, Gao P, et al. Emerging risk factors collaboration: C-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med. 2012;367:1310–1320. doi: 10.1056/NEJMoa1107477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khafaie MA, Salvi SS, Ojha A, Khafaie B, Gore SS, Yajnik CS. Systemic inflammation (c-reactive protein) in type 2 diabetic patients is associated with ambient air pollution in pune city, india. Diabetes Care. 2013;36:625–630. doi: 10.2337/dc12-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish JP, Lundback M, Barath S, Soderberg S, Mills NL, Newby DE, et al. Exposure to nitrogen dioxide is not associated with vascular dysfunction in man. Inhal Toxicol. 2010;22:192–198. doi: 10.3109/08958370903144105. [DOI] [PubMed] [Google Scholar]
- Lanki T, Pekkanen J, Aalto P, Elosua R, Berglind N, D’Ippoliti D, et al. Associations of traffic related air pollutants with hospitalisation for first acute myocardial infarction: The heapss study. Occup Environ Med. 2006;63:844–851. doi: 10.1136/oem.2005.023911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IM, Tsai SS, Ho CK, Chiu HF, Yang CY. Air pollution and hospital admissions for congestive heart failure in a tropical city: Kaohsiung, taiwan. Inhal Toxicol. 2007;19:899–904. doi: 10.1080/08958370701479406. [DOI] [PubMed] [Google Scholar]
- Li H, Han M, Guo L, Li G, Sang N. Oxidative stress, endothelial dysfunction and inflammatory response in rat heart to no(2) inhalation exposure. Chemosphere. 2011;82:1589–1596. doi: 10.1016/j.chemosphere.2010.11.055. [DOI] [PubMed] [Google Scholar]
- Liao D, Heiss G, Chinchilli VM, Duan Y, Folsom AR, Lin HM, et al. Association of criteria pollutants with plasma hemostatic/inflammatory markers: A population-based study. J Expo Anal Environ Epidemiol. 2005;15:319–328. doi: 10.1038/sj.jea.7500408. [DOI] [PubMed] [Google Scholar]
- Lipsett MJ, Ostro BD, Reynolds P, Goldberg D, Hertz A, Jerrett M, et al. Long-term exposure to air pollution and cardiorespiratory disease in the california teachers study cohort. Am J Respir Crit Care Med. 2011;184:828–835. doi: 10.1164/rccm.201012-2082OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorca J, Salas A, Prieto-Salceda D, Chinchon-Bengoechea V, Delgado-Rodriguez M. Nitrogen dioxide increases cardiorespiratory admissions in torrelavega (spain) J Environ Health. 2005;68:30–35. [PubMed] [Google Scholar]
- Lowe GD, Danesh J, Lewington S, Walker M, Lennon L, Thomson A, et al. Tissue plasminogen activator antigen and coronary heart disease. Prospective study and meta-analysis. Eur Heart J. 2004;25:252–259. doi: 10.1016/j.ehj.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Melin A, Bonnet P, Eder V, Antier D, Obert P, Fauchier L. Direct implication of carbon monoxide in the development of heart failure in rats with cardiac hypertrophy subjected to air pollution. Cardiovasc Toxicol. 2005;5:311–320. doi: 10.1385/ct:5:3:311. [DOI] [PubMed] [Google Scholar]
- Navab M, Reddy ST, Van Lenten BJ, Fogelman AM. Hdl and cardiovascular disease: Atherogenic and atheroprotective mechanisms. Nat Rev Cardiol. 2011;8:222–232. doi: 10.1038/nrcardio.2010.222. [DOI] [PubMed] [Google Scholar]
- Nordestgaard BG, Chapman MJ, Ray K, Boren J, Andreotti F, Watts GF, et al. Lipoprotein(a) as a cardiovascular risk factor: Current status. Eur Heart J. 2010;31:2844–2853. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panasevich S, Leander K, Rosenlund M, Ljungman P, Bellander T, de Faire U, et al. Associations of long- and short-term air pollution exposure with markers of inflammation and coagulation in a population sample. Occup Environ Med. 2009;66:747–753. doi: 10.1136/oem.2008.043471. [DOI] [PubMed] [Google Scholar]
- Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO, 3rd, Criqui M, et al. Markers of inflammation and cardiovascular disease: Application to clinical and public health practice: A statement for healthcare professionals from the centers for disease control and prevention and the american heart association. Circulation. 2003;107:499–511. doi: 10.1161/01.cir.0000052939.59093.45. [DOI] [PubMed] [Google Scholar]
- Pekkanen J, Brunner EJ, Anderson HR, Tiittanen P, Atkinson RW. Daily concentrations of air pollution and plasma fibrinogen in london. Occup Environ Med. 2000;57:818–822. doi: 10.1136/oem.57.12.818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich DQ, Kipen HM, Huang W, Wang G, Wang Y, Zhu P, et al. Association between changes in air pollution levels during the beijing olympics and biomarkers of inflammation and thrombosis in healthy young adults. JAMA. 2012;307:2068–2078. doi: 10.1001/jama.2012.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl MA, Diaz-Sanchez D, Linn WS, Gong H, Jr, Clark KW, Effros RM, et al. Allergic inflammation in the human lower respiratory tract affected by exposure to diesel exhaust. Res Rep Health Eff Inst. 2012:5–43. discussion 45–64. [PubMed] [Google Scholar]
- Rosenlund M, Berglind N, Pershagen G, Hallqvist J, Jonson T, Bellander T. Long-term exposure to urban air pollution and myocardial infarction. Epidemiology. 2006;17:383–390. doi: 10.1097/01.ede.0000219722.25569.0f. [DOI] [PubMed] [Google Scholar]
- Rosenlund M, Bellander T, Nordquist T, Alfredsson L. Traffic-generated air pollution and myocardial infarction. Epidemiology. 2009;20:265–271. doi: 10.1097/EDE.0b013e318190ea68. [DOI] [PubMed] [Google Scholar]
- Rückerl R, Greven S, Ljungman P, Aalto P, Antoniades C, Bellander T, et al. Air pollution and inflammation (interleukin-6, c-reactive protein, fibrinogen) in myocardial infarction survivors. Environ Health Perspect. 2007;115:1072–1080. doi: 10.1289/ehp.10021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudez G, Janssen NA, Kilinc E, Leebeek FW, Gerlofs-Nijland ME, Spronk HM, et al. Effects of ambient air pollution on hemostasis and inflammation. Environ Health Perspect. 2009;117:995–1001. doi: 10.1289/ehp.0800437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A, Patterson C, Yarnell J, Rumley A, Ben-Shlomo Y, Lowe G. Which hemostatic markers add to the predictive value of conventional risk factors for coronary heart disease and ischemic stroke? The caerphilly study. Circulation. 2005;112:3080–3087. doi: 10.1161/CIRCULATIONAHA.105.557132. [DOI] [PubMed] [Google Scholar]
- Sowers M, Crawford S, Sternfeld B, Morganstein D, Gold E, Greendale G, et al. Swan: A multicenter, multiethnic, community-based cohort study of women and the menopausal transition. In: Lobo JK RA, Marcus R, editors. Menopause: Biology and pathobiology. San Diego: Academic Press; 2000. pp. 175–188. [Google Scholar]
- Steinvil A, Kordova-Biezuner L, Shapira I, Berliner S, Rogowski O. Short-term exposure to air pollution and inflammation-sensitive biomarkers. Environ Res. 2008;106:51–61. doi: 10.1016/j.envres.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Strak M, Hoek G, Steenhof M, Kilinc E, Godri KJ, Gosens I, et al. Components of ambient air pollution affect thrombin generation in healthy humans: The raptes project. Occup Environ Med. 2013;70:332–340. doi: 10.1136/oemed-2012-100992. [DOI] [PubMed] [Google Scholar]
- Szyszkowicz M. Air pollution and emergency department visits for ischemic heart disease in montreal, canada. Int J Occup Med Environ Health. 2007;20:167–173. doi: 10.2478/v10001-007-0015-3. [DOI] [PubMed] [Google Scholar]
- Thurston RC, El Khoudary SR, Sutton-Tyrrell K, Crandall CJ, Sternfeld B, Joffe H, et al. Vasomotor symptoms and insulin resistance in the study of women’s health across the nation. J Clin Endocrinol Metab. 2012;97:3487–3494. doi: 10.1210/jc.2012-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. EPA. Integrated science assessment for sulfur oxides - health criteria. Research Triangle Park NC: 2008. (EPA/600/R-08/047F). [Google Scholar]
- U.S. EPA. Integrated science assessment for carbon monoxide. Research Triangle Park, NC: 2010. (EPA/600/R-09/019F). [Google Scholar]
- U.S. EPA. Integrated science assessment for oxides of nitrogen - health criteria. Research Triangle Park, NC: 2016. (EPA/600/R-15/068). [Google Scholar]
- World Health Organization. Cardiovascular diseases (cvds) fact sheet. 2016 Available: http://www.who.int/mediacentre/factsheets/fs317/en/ [accessed September 27 2016]
- Zhang J, Zhu T, Kipen H, Wang G, Huang W, Rich D, et al. Cardiorespiratory biomarker responses in healthy young adults to drastic air quality changes surrounding the 2008 beijing olympics. Res Rep Health Eff Inst. 2013:5–174. [PMC free article] [PubMed] [Google Scholar]
- Zimmerman DL, Núñez-Antón V. Modelling longitudinal and spatially correlated data. Springer; New York: 1997. Structured antedependence models for longitudinal data; pp. 63–76. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.