

Abstract
The medical treatment of cancer with antineoplastic drugs is routine, but careful biomonitoring for these powerful drugs in individual healthcare worker exposure is necessary to ensure that engineering controls and exposure intervention measures are effective. This review describes published high performance liquid chromatography/mass spectrometry (HPLC-MS) methods for the determination of anticancer drugs in human urine as non-invasive tools for monitoring of healthcare worker exposure to antineoplastic and cytotoxic drugs. HPLC-MS is a sensitive and specific method for analysis of anticancer drugs and their metabolites in biological fluids. In this review, a tabular summary and overview of published HPLC-MS methods are presented, as well as future trends and limitations in this area of research.
Keywords: Liquid chromatography, mass spectrometry, urinary biomarker, anticancer drugs, antineoplastic drug, occupational biomonitoring
1. Introduction
Occupational exposure of healthcare workers to anticancer drugs has been a concern since the early 1980s [1, 2]. Workers may be exposed to such drugs throughout their working life. These workers include hospital shipping and receiving personnel, pharmacists and pharmacy technicians, nursing personnel, physicians, operating room personnel, environmental services personnel, research laboratory personnel, and workers in veterinary practices where hazardous drugs are used. The number of workers potentially exposed long term to all hazardous drugs or their toxic metabolites is estimated to be 11 million workers [3].
Although efforts to reduce exposures have been made, recent studies in the U.S. and several other countries show that workplace contamination with antineoplastic drugs is still occurring [4–17]. Contamination of drug preparation and administration areas can lead to exposure of healthcare workers to these drugs as evidenced by contamination of workers’ hands and measurement of the drugs in the urine of workers [11, 17].
The measurement of anticancer drugs in urine is key in detection of and characterizing occupational exposure in healthcare workers. Anticancer drug levels found in environmental monitoring of workplace surfaces and in the air in drug preparation areas, while reflecting the efficacy of measures to eliminate workplace contamination, these levels cannot be assumed to represent healthcare worker exposure which may include dermal exposure. Since the beginning of formal guidelines and their successful application to reduce exposure of healthcare works to anticancer drugs, the need for sensitive and accurate analytical methods to quantitate exposure are well met by the capability of HPLC-MS methodology. Most anticancer drugs are non-volatile, and thermolabile compounds making gas chromatographic separation and detection unsuitable [18]. Early liquid chromatography detection methods using ultraviolet, fluorescent and electrochemical detection, although sensitive, lacked specificity. Over time, liquid chromatographic separation with mass spectrometric detection has become the preferred method for detection and quantitation of anticancer drugs both in workplace area monitoring and healthcare worker biomonitoring [18].
The current review focuses on HPLC-MS determination of anticancer drugs or their metabolites in the urine of healthcare workers. Earlier analytical methods have been extensively reviewed [19,20] as well as those specifically using LC-MS methodology [18]. The majority of these procedures were developed for pre-clinical and clinical studies. Those for analysis of biological fluids have been developed for blood serum, and plasma or urine in clinical animal models or in patients given therapeutic doses of drug. In this review, HPLC-MS methods created for determination of anticancer drugs in urine of healthcare workers are summarized in tabular format and highlights of the sample preparation and chromatography techniques used in these methods are briefly described.
2. Tabular summaries of selected methods
Tables 1–4 summarize various HPLC-MS methods reported for the detection and quantification of various antineoplastic drugs in urine of exposed healthcare workers for use in occupational biomonitoring studies. The terminology and abbreviations appearing in these tables indicate sample preparation techniques, chromatographic conditions, and mass spectrometry detection modes reported for these methods, and are explained in more detail in the following sections of this review.
Table 1.
LC/MS determination of nitrogen mustard antineoplastic drugs in urine
| Parent drug | Sample preparation | Chromatography | Interface/Detection | Target analyte | m/z of mass transition | Limit of Detection | Reference |
|---|---|---|---|---|---|---|---|
| cyclophosphamide (CP) | LLE ethylacetate |
RP C8/isocratic CH3CO2NH4/MeOH 4.6 × 150 mm, 5μm |
ESI/QQQ/MRM+ | CP IF |
261.2/140.2 261.2/92.0 |
0.05 μg/L | [21] |
| cyclophosphamide | LLE ethylacetate |
RP C18/gradient CH3COOH/MeOH 2.1 × 50 mm, 4μm |
ESI/QQQ/MRM+ | CP d6-CP |
263.1/142.1 267.1/140.3 |
0.01 μg/L | [22] |
| cyclophosphamide | SPE C18 ethylacetate |
RP C18/gradient HCO2NH4/ACN 3.0 × 150 mm, 3μm |
ESI+/QTrap | CP d4-CP |
261/140 265/140 |
0.05 μg/L | [23] |
| cyclophosphamide | SPE C18 ethylacetate/dichloromethane |
RP C18/isocratic CH3COOH/ACN 3.0 × 100 mm, 2.7μm |
ESI/QQQ/MRM+ | CP IF |
261/140 261/54 |
0.07 μg/L | [24] |
| cyclophosphamide ifosphamide (IF) |
salt-assisted LLE ethylacetate sodium borate |
RP C8/isocratic HCOOH/ACN 2.0 × 100 mm, 3μm |
ESI/QQQ/MRM+ | CP IF PCP |
261/154.1 261/140.1 249/164.1 |
0.1 μg/L 0.1 μg/L |
[25] |
| cyclophosphamide ifosphamide |
SPE C18 ethylacetate |
RP C8/gradient HCOOH/ACN/MeOH 4.6 × 100 mm, 5μm |
ESI/QQQ/SRM+ | CP IF TRP |
261.0/140.2 261.0/92.0 323.3/92.0 |
0.02 μg/L 0.04 μg/L |
[26] |
| cyclophosphamide ifosphamide |
SPE C18 MeOH |
RP C18/gradient HCOOH/ACN/MeOH 2.1 × 150 mm, 3μm |
ESI+/Ion Trap | CP IF PSL |
261/140 261/182 361/343 |
0.4 μg/L 0.4 μg/L |
[27] |
| cyclophosphamide ifosphamide |
LLE dichloromethane |
RP C18/gradient HCO2NH4/ACN 2.1 × 100 mm, 5 μm |
ESI/QQQ/MRM+ | CP d4-CP IF |
261/140 264/140 261/92 |
0.01 μg/L 0.01 μg/L |
[28] |
| cyclophosphamide 4-keto-CP ifosphamide |
LLE ethylacetate |
RP C18/gradient HCOOH/ACN 3.0 × 250 mm, 3.5μm |
ESI/QQQ/MRM+ | CP 4-keto-CP IF d6-CP |
261/140 267/140 275/106 261/154 |
0.1 μg/L 1.0 μg/L 0.05 μg/L |
[29] |
| cyclophosphamide 4-keto-CP carboxy-CP DCL-CP |
LLE MeOH |
RP C8/gradient HCOOH/MeOH 3.0 × 100 mm, 5μm |
ESI/QQQ/SRM+ | CP 4-keto-CP carboxy-CP DCL-CP d4-CP |
261/140 275/221 293/221 199/171 265/145 |
5 μg/L 5 μg/L 30 μg/L 1 μg/L |
[30] |
| bendamustine (BM) & phase I metabolites | SPE MeOH |
RP C18/gradient HCO2NH4/MeOH 2.0 × 150 mm, 4μm |
ESI/QQQ/MRM+ | BM BM-IS metabolite 3 metabolite 4 |
358/228 372/338 374/186 344/354 |
0.5 μg/L 0.5 μg/L 0.4 μg/L |
[31] |
| bendamustine phase I metabolite | SPE MeOH |
Polar RP/gradient HCO2NH4/MeOH 2.0 × 150 mm, 4μm |
ESI/QQQ/MRM+ | dihydroxy-BM α-DLA |
322/304 408/170 |
1 μg/L | [31] |
Legend of abbreviations used in Table 1. LC/MS determination of nitrogen mustard antineoplastic drugs in urine
ACN: acetonitrile, CH2Cl2, DCL-CP: N-dechloroethyl-cyclophosphamide, α-DLA: α-dansyl-L-arginine, ESI: electrospray ionization, LLE: liquid-liquid extraction, MeOH: methanol, MRM: multiple reaction monitoring, PCP: phencyclidine, PSL: prednisolone, QQQ: triple quadrupole, RP: reversed phase, SPE: solid phase extraction, SRM: single reaction monitoring, TRP: trophosphamide
Table 4.
LC/MS simultaneous determination of antineoplastic drugs in urine
| Parent drug | Sample preparation | Chromatography | Interface/Detection | Target analyte | m/z or mass transition | Limit of detection | Reference |
|---|---|---|---|---|---|---|---|
| multiple determination | ABN-SPE 96-well CHOOH/MeOH |
RP C12/gradient CH3CO2NH4/MeOH |
ESI/QQQ/MRM+ | [39] | |||
| 0.5 × 50 mm, 4μm | |||||||
| cyclophosphamide | CP TRP |
261/140 323/154 |
0.04 μg/L | ||||
| methotrexate | MTX | 455/308 | 0.2 μg/L | ||||
| multiple determination | SPE C18 CH2Cl2/2-propanol/MeOH |
RP C8, gradient CHOOH/ACN |
ESI/QQQ/MRM+ | [40] | |||
| 4.6 × 150 mm, 5μm | |||||||
| cyclophosphamide | CP TRP |
261/140 323/154 |
0.1 μg/L | ||||
| ifosphosphamide | IF | 261/92 | 0.1 μg/L | ||||
| doxorubicin | DXR | 528/321 | 0.2 μg/L | ||||
| epirubicin | EPI | 544/397 | 0.1 μg/L | ||||
| daunorubicin | DNR EDR |
528/321 528/321 |
0.1 μg/L | ||||
| multiple determination | dispersive SPE RP sorbent aqueous MeOH |
RP PFP/gradient CHOOH/ACN |
ESI/QTrap/SRM+ | [41] | |||
| 2.1 × 100 mm, 2.6 μm | |||||||
| cyclophosphamide | CP 2H4 -CP |
261/106 261/140 |
0.33 μg/L | ||||
| ifosphosphamide | IF 2H4 -IF |
261/154 261/92 |
0.13 μg/L | ||||
| doxorubicin | DXR 13C,2H3-DXR |
544/379 544/397 |
0.07 μg/L | ||||
| epirubicin | EPI 13C,2H3-EPI |
544/379 544/397 |
0.03 μg/L | ||||
| 5-fluorouracil | 5-FU 13C2,15N2-5-FU |
129/86 129/59 |
33.33 μg/L | ||||
| cytarabine | CYT 13C3-CYT |
244/95 244/112 |
0.33 μg/L | ||||
| gemcitabine | GCA 13C2,15N2- GEM |
264/95 264/112 |
0.67 μg/L | ||||
| dacarbazine | DAC 2H6 -DAC |
183/123 183/166 |
0.67 μg/L | ||||
| etoposide | ETOP 13C,2H3-ETOP |
589/425 589/229 |
0.17 μg/L | ||||
| methotrexate | MTX 2H3 -MTX |
455/134 455/308 |
0.01 μg/L | ||||
| vinblastine | VBL 2H3 -VBL |
811/751 8///224 |
0.83 μg/L | ||||
| vincristine | VCR 2H3 -VCR |
413/362 413/392 |
0.17 μg/L | ||||
| paclitaxel | TAX 2H5 -TAX |
854/569 854/286 |
0.33 μg/L |
Legend of abbreviations used in Table 4. LC/MS simultaneous determination of antineoplastic drugs in urine
ABN-SPE: Acid Base Neutral SPE, ACN: acetonitrile, CH2Cl2: methylene chloride, CHOOH: formic acid, EDR: epidaunorubicin, ESI: electrospray ionization, MeOH: methanol, MRM: multiple reaction monitoring, QQQ: triple quadrupole, RP: reversed phase, PFP: pentafluorophenyl, SPE: solid phase extraction, SRM: single reaction monitoring, TRP: trophosphamide
2.1. Sample preparation techniques
Successful determination of target analytes by HPLC-MS requires separation of target drugs and their metabolites from interfering components found in urine. These include proteins, numerous metabolites, salts and other components that make up the urinary sample matrix which interfere with the sensitive and specific detection of the target analytes. For example, salts can suppress the intensity of the analyte signal or similar metabolites may co-elute from the chromatographic column with the target drug or its metabolite. The necessary removal of these interferences make sample preparation as critical to success as any other part of the analysis. A variety of sample preparation techniques have been applied in the methods reviewed. The simplest clean up step is protein precipitation by acetonitrile and centrifugation prior to analysis [38]. Most methods use C18 solid phase extraction (SPE) for sample preparation and clean-up. In simple manual SPE techniques, urine is drawn into a syringe and manually pushed through a disk of chromatographic medium that is attached to the syringe after sample loading. Manual methods are used to extract small amounts of urine, generally 1–5 ml. For larger volumes, urine is applied to a bed of chromatographic medium in a syringe or cartridge, and is pulled through the medium under vacuum pressure. Target analytes are captured in the solid medium, and several volumes of wash solvent are used to remove sample matrix components. Concentrated and purified analytes then are washed free from the medium using solvent or solvent mixtures. Methods using SPE C18 sample preparation for nitrogen mustards (e.g. isofosamide) used either methanol or ethylacetate as eluent [23, 24, 26, 27, 30] while Sottani used methylene chloride/2-propanol mixtures for extraction of anthracyclines (e.g. doxorubicin) from C18 media [33, 40].
Sample preparation is often the labor intensive and rate-limiting step in most bioassay methods. SPE media in disk, cartridge and bed forms have been adapted to high-throughput popular 96-multi-well sample plate format when the speed of fully automated analysis is necessary. The convenience of 96- and other multi-well formats is also ideal for rapid development of sample extraction methods [42]. Rule et al., developed a 384-well plate sample extraction and sample handling technique for analysis of methotrexate and 7-hydroxy-methotrexate in human urine and plasma [37]. The authors describe optimization of sample extraction parameters, using spiked quality control samples to select between multiple C18 sorbent particle sizes and solvent elution volumes to allow direct injection into the LC/MS using an autosampler. The resulting peak areas were used to compare capture efficiency of analytes by sorbent particle size [37].
In addition to RP C18 phase, other SPE media are used to extract and concentrate target antineoplastic drugs from the urinary matrix. Ndaw et al. [34] and Sottani et al. [35] selected a HLB phase containing both hydrophilic and lipophilic monomers to extract antimetabolite pyrimidine drugs 5-fluorouracil (5-FU) [34] and gemcitabine (GCA) [35] from urine (Figure 1). To enhance analyte detection, Ndaw derivatized 5-FU with 2,4-dinitrofluorobenzene prior to HLB extraction. This combination of pre-column derivatization and SPE prior to hydrophilic interaction chromatography (HILIC) allowed a 1 μg/L LOD, a great improvement over 20 μg/L and 60 μg/L sensitivities of previous GC/MS methods [43,44]. Barbieri et al. [39]compared two polymer-based stationary phases for extraction of cyclophosphamide and methotrexate (Figure 1). These two most frequently used drugs in cancer therapy mixtures have very different chemical structures, and their simultaneous extraction from urine is not clear-cut. A co-polymer phase mixture (ABN) of two particle sizes, each with hydrophilic and hydrophobic groups was selected over a single particle highly cross-linked, non-polar phase (INV+) to retain both drugs for reproducible extraction [39].
Figure 1.
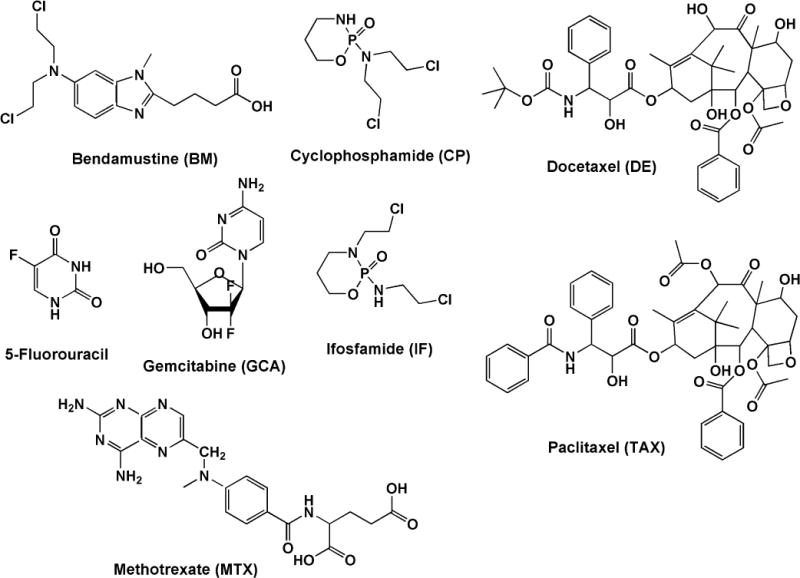
Chemical structures of various anticancer drugs.
A second extraction technique liquid-liquid extraction (LLE) was used in several nitrogen mustard methods [21,22, 25, 28, 29,30] and in the methods for taxanes (e.g. docetaxel) [23, 32]. In this technique, an immiscible solvent is added to an aqueous sample, and the two phases are mixed thoroughly. Then the phases are allowed to separate, extracting analytes from the urinary matrix through selective partitioning of analytes and contaminants between the two phases.
The target antineoplastic drugs in the methods reviewed here represent a variety of structural classes and have a wide range of polarities, pKa and LogP (hydrophilicity) values. A single technique to extract and concentrate these dissimilar analytes from urine is not straightforeward, and no single systematic approach for simultaneous extraction can be recommended. Two methods aimed at their simultaneous determination in a single chromatographic run, use either C18 or other RP SPE medium with a mixture of organic solvents: acetic acid/methanol [39] or methylene chloride/2-propanol/methanol [40] for analyte elution (Table 4). A third multi-analyte method by Fabrizi et al. [41] used a customized dispersive SPE technique (dSPE) to extract 13 drugs from urine. In this technique, diluted urine was acidified and two SPE powdered sorbents, a C18 phase and a hydrophilic-lipophilic balanced (HLB) phase, were added and thoroughly mixed to absorb the target analytes from urine. The mixture was centrifuged, and the aqueous methanol supernatant was discarded. To release the adsorbed analytes, the sorbents were washed with methanol, mixed as before, and centrifuged. Then the methanol supernatant was collected and dried down for later analysis [41].
2.2. Liquid Chromatography
2.2.1. Reversed-phase chromatography
With one exception, the methods reviewed here use reversed-phase (RP) columns containing either non-polar stationary phase consisting of alkane chains (C18, C12 or C8) or polar ether-linked pentafluorophenyl functional groups (PFP). Commonly used mobile phases for tandem MS analysis contain volatile acids or buffers such as formic acid (HCO2H), acetic acid (CH3CO2H), ammonium formate (HCO2NH4), or ammonium acetate (CH3CO2NH4). Organic modifiers such as methanol (MeOH) and acetonitrile (ACN) are typically used with either isocratic or gradient conditions for analyte elution.
2.2.2. Hydrophilic interaction chromatography
World-wide, 5-Fluorouracil (5-FU) is the most commonly used anticancer drug [45]. An antimetabolite drug, 5-FU is anabolized to a cytotoxic pyrimidine analog, or it is catabolized to an unnatural amino acid, α-fluoro-β-alanine (FBAL). FBAL has a low molecular weight and is similar in structure to other endogenous pyrimidine metabolites found in urine. To enhance chromatographic separation of FBAL from urinary matrix components, Ndaw et al. [34] used 2,4-dinitrofluorobenzene (DNFB) to derivatized both FBAL and an internal standard, β-alanine in urinary matrix prior to sample extraction (Table 3). For chromatographic separation, three different C18 reversed-phase columns using water and acetonitrile as a mobile phase were evaluated. Of those evaluated, either both analytes would be unretained, or they formed broad tailing peaks. To resolve these problems, Ndaw used hydrophilic interaction chromatography (HILIC). In HILIC separations, retention increases with hydrophilicity and polarity of the analyte. Analytes partition into a water-enriched layer formed over a polar HILIC stationary phase. The layer forms initially when water in a 5–15% aqueous/polar organic solvent mobile phase containing volatile ammonium formate or ammonium acetate associates with the polar stationary phase. Analyte elution is driven when more water is introduced to play the role of a stronger eluting solvent phase containing high organic content, usually acetonitrile or alternatively methanol. Typically HILIC mobile phases use high organic content (> 80%), which are ideal for ESI-MS analysis, and may enhance ES-MS response [46]. The HILIC mechanism has been discussed in great detail [47]. The application of HILIC in quantitative bioanalysis of other compounds of pharmaceutical interest has been described [48].
Table 3.
LC/MS determination of antimetabolite drugs in urine
| Parent drug | Sample preparation | Chromatography | Interface/Detection | Target analyte | m/z of mass transition | Limit of Detection | Reference |
|---|---|---|---|---|---|---|---|
| 5-fluorouracil | DNFB derivatization & SPE HLB MeOH/ACN |
ZIC-HILIC CHCOONH4/ACN 2.1 × 100mm, 5μm |
ESI/QQQ/MRM- | α-fluoro-β-alanine d4-β-alanine |
272/182 258/182 |
1 μg/L | [34] |
| gemcitabine & metabolite | SPE MeOH |
RP C8/gradient CH3COOH/ACN 4.6 × 150 mm, 5μm |
ESI/QQQ/MRM+ | dFdC 2dFdU 2deC |
264/112 265/113 228/112 |
0.05 μg/L 0.3 μg/L |
[35] |
| methotrexate (MTX) |
SPE C18 ethylacetate |
RP C18/isocratic CH3COONH4/MeOH 4.6 × 150 mm, 5μm |
ESI/QQQ/MRM+ | MTX 7-OH-MTX |
455/308 471/324 |
0.2 μg/L | [36] |
| methotrexate | SPE-HAX methanol/acetic acid |
RP C18/gradient CHCOONH4/ACN 2.1 × 100 mm, 5μm |
ESI/QQQ/MRM+ | MTX d3-MTX |
455/308 458/311 |
0.01 μg/L | [28] |
| methotrexate 7-hydroxy-methotrexate |
SPE C18 384-well aqueous CHOOH/MeOH |
RP C8/gradient CHCOOH/ACN 2.0 × 10 mm, 5μm |
ESI/QQQ/SRM+ | MTX d3-MTX 7-OH-MTX d3 7-OH-MTX |
445/308 458/311 471/191 474/191 |
1 μg/L 50 μg/L |
[37] |
| methotrexate 7-hydroxy-methotrexate |
protein precipitation |
RP C18/gradient CHCOOH/ACN 2.1 × 100 mm, 1.9μm |
ESI/QQQ/SRM+ | MTX d3-MTX 7-OH-MTX |
455/308 458/311 471/324 |
1 μg/L 5 μg/L |
[38] |
Legend of abbreviations used in Table 3. LC/MS determination of antimetabolite drugs in urine.
ACN: acetonitrile, DNFB: 2,4 dinitrofluorobenzene, ESI: electrospray ionization, dFdC: 2′,2′-difluorodeoxycytidine, 2dFdU: 2′,2′-difluorodeoxyuridine, 2deC: 2′-deoxycytidine, MeOH: methanol, MRM: multiple reaction monitoring, QQQ: triple quadrupole, RP: reversed phase, SPE: solid phase extraction, SPE-HAX: SPE non-polar and strong anion exchange, SPE HLB: SPE hydrophilic-lipophilic balance medium, SRM: selective reaction monitoring, ZIC-HILIC: zwitterionic-hydrophilic interaction chromatography
2.3. Detection modes by MS/MS
In tandem HPLC-MS/MS analysis, after target analytes are separated chromatographically, they are introduced into the mass spectrometer for analysis. This is done at the HPLC-MS interface where the chromatographic eluate is vaporized and the analyte molecules are ionized for mass selection and detection in the mass analyzer. The ion source used in the methods reviewed here is electrospray ionization (ESI). Atmospheric chemical ionization (APCI) was not used in the methods reviewed here. However, both these ionization techniques allow easy and robust interfacing of HPLC to tandem mass spectrometry [49].
The basic function of a mass spectrometer is to measure the mass-to-charge ratios (m/z) of analyte ions. Mass spectrometers have various designs which have been reviewed elsewhere in the literature [50]. Although mass spectrometers are used in qualitative identification of compounds, the monitoring of specific ions for quantitative determination is the focus of this discussion. While three determinations use either Q-Trap or Ion-Trap design [23, 27, 41] the majority of the methods found in this review use tandem transmission quadrupole instruments (MS/MS). These analyses use a triple quadruple configuration (QQQ) where precursor ions are selected in the first quadrupole, and allowed to pass into a second quadrupole collision chamber for collision-induced dissociation fragmentation into product ions. Transmitted from the collision chamber, fragmentation product ions will be separated by the third quadrupole for detection. The majority of the methods described here use multiple reaction mode (MRM) detection that allows for analyte identification and quantification by both its molecular ion and a typical fragment. Thus tandem mass spectrometry used with MRM detection provides the greatest level of sensitivity and specificity for the analysis method. This allows for detection of analytes even in the presence of biological sample matrix components that would otherwise interfere with ultraviolet or fluorescence detection [51]. For this reason, tandem MS detection is considered the method of choice for quantitation of drugs and their metabolites in biological fluids [52]. The high sensitivity of the MS/MS detection is of particular importance in investigating low-level exposure in healthcare workers in whom urinary antineoplastic drugs may be present in μg/L or ng/L levels.
3. Overview of the methods
3.1. Nitrogen mustard drugs
The Oxazaphosphoramine drugs cyclophosphamide (CP) and ifosphamide (IF) are chiral isomers (Figure 1) and are administered as a racemic mixture of the 2 enantiomers. In four of the methods for their quantitation (Table 1), CP is the single target using either IF [21,24] or deuterated-CP [22,23] as an internal standard (IS). The remainder target both CP and IF using structural analogs phencyclidine, trophosphamide, and prednisolone as ISs [25–27], while three target CP, IF and one or more stable metabolites using a deuterated-CP IS [28–30]. The majority use sample preparation with either LLE or SPE C18 with ethylacetate to remove matrix interferences, while one method uses methanol to remove protein while capturing polar CP metabolites [30]. To maintain positive ions, all use acidic mobile phases containing either acetic or formic acid and some use a combination of these with a small amount of ammonium hydroxide. At the ionization interface, all use a positive ESI; the majority detecting with MRM+ in tandem, while two methods use either Q-Trap [23] or Ion-Trap [27] detection.
Most of the methods summarized in Table 1 were developed specifically for occupational biomonitoring and several methods were applied to specimens from occupationally exposed workers. However, three methods were developed using clinical specimens from cancer patents [22, 30, 31]. Hedmer et al. [22] tested the validity of occupational biomonitoring using urinary CP concentration versus blood plasma CP concentration with clinical specimens. By using pharmacokinetic methods to study CP renal clearance from high and low plasma concentrations in 16 cancer patients, CP renal clearance was found to be independent of plasma concentration. However, patients given the same dose of CP had highly variable plasma and urine CP concentrations. Further, excretion of CP in urine was dependent on urine flow. The authors concluded that while urinary CP concentration would be a valid indicator of occupational exposure, the high individual variation of CP excretion into urine, and its dependence on urine flow indicate that urinary CP concentration does not reflect internal dose. The authors caution that urinary excretion of CP could either over or under estimate the risk of CP exposure compared to concomitant measurements of plasma CP concentration. Kasel et al. [30] also used clinical urine specimens for method development. Taking advantage of the higher levels of drug and metabolites to be found in 24 hour urine collections from cancer patients, a method for determination of CP, and 3 relatively stable metabolites including 4-keto-CP was developed and validated using FDA and ICH guidance. B’Hymer and Cheever [29] also applied FDA guidance to develop a method with improved sensitivity sufficient to detect CP, IF and 4-keto-CP at the trace levels expected in biomonitoring. During analyte recovery studies, two different Agilent Zorbax C18 columns packed with 3.5 μm particles were used to obtain consistent and reproducible results. This demonstrated robustness for the chromatographic conditions of the method using columns from this manufacturer.
3.2. Taxanes
The majority of LC-MS methods for taxanes have been developed for support of pre-clinical and clinical studies and are optimized for tissues, plasma, urine or other biological matrices containing high levels of drugs [53]. Of two methods validated for human urine that are included here (Table 2) only one was developed specifically for occupational biomonitoring. One, for both paclitaxel and docetaxel (Figure 1), was optimized for processing and analysis of both extracted urine and homogenized fecal samples and has LODs for both drugs of 0.5 μg/L [32]. The other for paclitaxel only, was developed specifically for low or trace level occupational drug biomonitoring and has a greater sensitivity of 0.05 μg/L [23]. Both make use of LLE with tert-butylmethylether widely used for extraction of taxanes to achieve high recoveries, and reduce or minimize endogenous matrix effects [53]. Further, both methods use deuterated isotopic IS to further counter matrix effects. Both use C18 columns and their mobile phases include ammonium-containing additives to reduce adduct formation. Addition of ammonium hydroxide or ammonium formate also creates an alkaline pH which can increase the ionization response of taxanes [53].
Table 2.
LC/MS determination of taxane and anthracycline drugs in urine
| Parent drug | Sample preparation | Chromatography | Interface/Detection | Target analyte | m/z of mass transition | Limit of Detection | Reference |
|---|---|---|---|---|---|---|---|
| paclitaxel (TAX) | LLE methyl-tert-butyl ether/ACN |
RP C18/gradient HCOONH4/ACN 4.6 × 50 mm, 1.8μm |
ESI+/QTrap | TAX d5-PA |
854/105 859/105 |
0.05 μg/L | [23] |
| paclitaxel docetaxel (DE) |
LLE methyl-tert-butyl ether/aqueous MeOH |
RP C18/isocratic MeOH/NH4OH 2.1 × 150 mm, 5μm |
ESI/QQQ/MRM+ | TAX 13C6-PA DE d9-DE |
854/509 860/515 808/527 817/527 |
0.5 μg/L 0.5 μg/L |
[32] |
| anthracyclines epirubicin doxorubicin daunoribicin idarubicin |
SPE C18 CH2Cl2/2-propanol |
RP C8/gradient HCOOH/ACN/MeOH 4.6 × 150 mm, 5μm |
ESI/QQQ/MRM+ | EPI DXR DNR IDA epi-IDA |
544/321 544/397 528/321 498/291 528/321 |
0.04 μg/L 0.04 μg/L 0.01 μg/L 0.01 μg/L |
[33] |
Legend of abbreviations used in Table 2. LC/MS determination of taxane and anthracycline drugs in urine.
ACN: acetonitrile, CH2Cl2: methylene chloride, ESI: electrospray ionization, MeOH: methanol, LLE: liquid-liquid extraction, MRM: multiple reaction monitoring, QQQ: triple quadrupole, RP: reversed phase, SPE: solid phase extraction, SRM: single reaction monitoring
3.3. Methotrexate
As is the case with detection and quantification of taxanes in biological matrices, the majority of published methods for methotrexate (MTX) were developed for pre-clinical and clinical studies (Figure 1) [18, 36]. The methods summarized in Table 3 have similar chromatographic and detection conditions using acidic mobile phases to promote positive ion formation for MRM+ [28, 36] or SRM+ detection [37, 38] and all use similar mass transitions for detection and quantification. Rule et al. [37], uses a microbore column (2mm) and focuses on a high-throughput determination of MTX and its metabolite 7-OH-MTX, The method features a multi-well plate SPE C18 extraction, 0.15 ml/minute flow rate and a 2.2 minute run-time [37]. To monitor low-dose adherence in rheumatoid arthritis patients, Bluett et al. [38] also determined MTX and the 7-OH metabolite using a small-bore column (2.1mm) and a flow rate of 0.3 ml/min. Bluett reports a lower LOD than that of Rule for 7-OH-MTX, 5 μg/L vs 50 μg/L, but the former has an 11 minute run-time. Oddly, neither method includes the amount of urine that was extracted to achieve the reported LOD values. In contrast, Turci et al. [36] and Canal-Raffin et al. [28] developed determinations specifically for biomonitoring of healthcare workers. Turci et al., [36] used manual SPE and a conventional RP analytical column. To achieve the sensitivity required for urinary biomonitoring, Turci and collaborators first optimized sample preparation. Several extraction parameters were investigated; analyte retention efficiency on eight sorbent types; the effect of initial sample volume and pH on analyte retention, and the effect of elution volume on analyte recovery and concentration. In the final extraction, 5 ml of urine, diluted 1:1 with buffer was manually applied to SPE C18 medium, and the medium washed with ethylacetate. Analytes were eluted from sorbent medium in three 1-ml aliquots of methanol and dried under nitrogen. The final dry extract was reconstituted in 200 μl of mobile phase. A 10 μl injection provided for an LOD of 0.2 μg/L for MTX as single the analyte [36]. In the more recent method, Canal-Raffin et al., [28] also used SPE with HAX medium that combines non-polar (C8) interactions and strong anion exchange for analyte concentration. To remove matrix components from 5 ml of urine, samples were diluted with an equal volume of formate buffer. After the application of diluted sample, cartridge matrix was extensively washed using a vacuum manifold; first with 5 ml of ammonium acetate buffer, followed by 5 ml equal parts methanol/water, and finally 5 ml of acetic acid buffer as the final washing step. MTX was eluted twice with 2.5 ml methanol/acetic acid. After drying and resuspension in mobile phase, a 20 μl sample injection yielded a reported 0.01 μg/L LOD [28]
3.3. Simultaneous determinations
The majority of the methods described here determine a single drug, drug isomers or their metabolites that have similar polarities, pKa values, LogP (hydrophilicity) or other properties. Structurally related drugs have similar extraction properties and undergo similar interactions with chromatographic media. Anticancer drugs, however, are a heterogenous group of compounds having very different chemical properties and chromatographic behaviors. Ongoing improvements in sample preparation, chromatographic media, column technology and in tandem mass spectrometers used with HPLC analysis have led to increased speed and efficiency in analyte separation. Subsequently, methods capable of determining chemically dissimilar analytes in a single chromatographic run have become more popular.
Two simultaneous determinations were developed by Sottani and collaborators [39, 40]. In the first Barbieri et al. [39] combined chromatographic and tandem MS detection parameters used in two earlier methods for the nitrogen mustard, cyclophosphamide [21] and the more polar anti-metabolite drug, methotrexate [36]. Sample preparation in this combined method used Acid Base Neutral (ABN) SPE with formic acid and methanol to capture and purify both analytes in place of LLE or C18 SPE with ethylacetate used in the earlier single determinations (Table 4). Modifications to chromatographic parameters featured a linear elution gradient and use of a 50 mm length capillary column with a microflow rate of 10 μl/min [39]. A second simultaneous method developed by the same group determines CP, IF and three anthracyclines, doxorubicin (DXR), epirubicin (EPI), and daunoribicin (DNR) (Figure 2) [40]. The sample preparation used previously for anthracycline extraction [33] was modified to a mixture of methylene chloride, 2-propanol and methanol for elution of analytes from C18 SPE medium. The RP chromatographic conditions used by Sottani in CP and IF determination [26], and in determination of 3 anthracyclines [33] were used again with minor modification of the mobile phase. Similar transitions for MS/MS detection reported in the earlier single determinations were used (Table 4).
Figure 2.
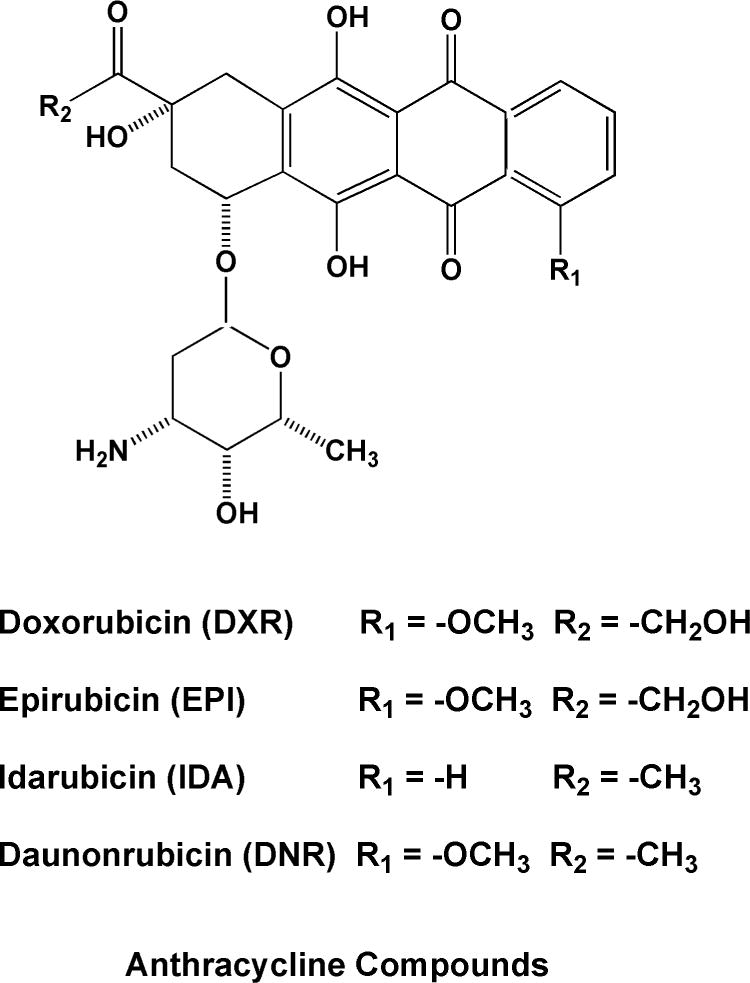
Chemical structures of anthracycline anticancer drugs.
Fabrizi et al., [41] developed a multi-analyte method using a small-bore 2.1 mm column filled with reduced size (2.6 μm) core-shell pentafluorophenyl functional group medium. Reduced particle size and smaller column diameters generally decrease analyte dispersion, enhance peak resolution and allow for decreased sample loading. These improvements can be expected to increase the number of theoretical plates [54]. This in turn may increase sensitivity by increasing the signal-to-noise ratio of the detector based on use of the tandem mass spectrometer. These improvements may in some cases match or exceed the sensitivity of standard chromatographic systems.
3.4. Comments on method validation
In order to reliably determine trace urinary concentrations of analytes, the critical elements of method validation or figures of merit, that is the precision within and between determination series, the limit of detection (LOD), the lower limit of quantitation (LLOQ), linearity and specificity must be established [55]. The majority of methods reviewed here reported only the minimal level of detail on precision, including intra- and inter-day values; accuracy; method range, linearity of the calibration curve; limits of detection and recovery data. Only a few papers failed to report these minimal validation parameters. However, a few did provide full validation parameters including analyte carry-over, analyte and reagent stability studies, or supportive evidence regarding method ruggedness or robustness [56]. Equally, few publications provide results or cite reports detailing the application of the method to actual biomonitoring samples. Some of the papers reviewed were a demonstration of the methodology or supported field work, while a few others focused on a through method validation with field applications to follow or reported elsewhere. Roughly half of the cited works reviewed did refer to the guidelines for method development and validation. These references included the use of US Food and Drug Administration Guidelines (FDA)[57] which was cited by seven studies and those of various European agencies which generally mirror the guidelines of the US FDA [58, 59]. Although these guidelines, published in the US and in Europe, may not be fully detailed or developed, they do represent efforts to bring standardization to the validation of bioanalytical methods and greater consistency in analytical results for biomonitoring studies.
In biomonitoring studies, reliable determination of trace urinary concentrations of drugs that have been absorbed at very low levels is required. Drug absorption, metabolism and target analyte excretion rate must be considered to choose an optimal specimen collection time. For analytes with a long biological retention time, or half-life, 24 hour specimen collections are generally representative. For detection of rapidly excreted agents like CP, a collection at the end of the work shift is more appropriate [55]. Following collection, specimens must be processed promptly to assure analyte preservation until the time of specimen analysis.
4.0 Future Directions
Ongoing improvements in chromatographic separation media with smaller particle diameters have allowed for increased theoretical plate counts and improved peak resolution. These improvements led to shorter columns, faster analysis times, and shorter equilibration periods [54]. As described earlier, Fabrizi et al., used improved separation media and a shorter column to create a multi-analyte method [41]. These improvements have been combined in ultra-high performance liquid chromatography (UHPLC) that utilizes shorter columns, 3–5 cm long, and reduced particle sizes, smaller than 2μm. Reduced particle size decreases analyte peak dispersion, thus enhancing peak resolution. Sub-2μm particles produce sharper peaks while decreasing sample loading over conventional HPLC. These advantages of sub-2μm particles have been reviewed by Nguyen and Schug [46]. However, very high pressure is required to push mobile phase through a column packed with smaller diameter particles. Standard HPLC pumping systems have traditionally had maximum pressure levels of approximately 6,000 psi (~420 Atmospheres) while UHPLC pumps are designed to handle pressures in excess of 15,000 psi (~1,000 Atmospheres). These fundamental aspects and practical requirements of UHPLC have been reviewed [60]. Owing to the fact that most HPLC pump manufacturers are offering pumping systems capable of maintaining the high back pressure levels required for the technique, UHPLC has come increasingly into use for biomonitoring in complex biological matrices.
The improvements offered by UHPLC in linear velocity, column efficiency and peak resolution coupled with improvements in data acquisition rates made possible by more powerful computers have made HPLC-MS methods more capable of determining multiple analytes in a single chromatographic analysis. In the past, one of the main technological limitations of mass spectrometers used in HPLC analysis has been the rate of data acquisition and the dwell time of monitoring the response at specific masses. Detector sampling rates must be rapid enough to obtain a sufficient number of data points across the analyte peak [61]. Low data acquisition rates have been known for many years to lead to poor chromatographic peak integration and poor reproducibility of peak area determinations [61]. Rapid data acquisition is necessary in order to minimize chromatographic peak distortion, which can be a problem with spectral data collected from increasingly narrow chromatographic peaks characteristic of UHPLC when linked to tandem mass spectrometry.
While nearly half of the methods reviewed here make use of small-bore, micro- or capillary columns, none pair a reduced diameter, shorter column with sub-2μm particle separation medium as seen in true UHPLC applications. UHPLC offers advantages for greater peak resolution and rapid analysis. When used in combination with the improved techniques for multiple analyte extraction and concentration described in the methods reviewed here, UHPLC has the potential for simultaneous analysis of multiple anticancer drugs in urine or other biological fluids.
5.0. Conclusions
The first anticancer drug determinations were developed for pharmaceutical applications including analyses of biological materials found in animal pre-clinical and human clinical trials, and required mg/L sensitivity. Early HPLC anticancer drug determinations were limited by non-specific ultraviolet, fluorometric or electrochemical detection techniques. Gas chromatographic techniques required cumbersome analyte derivatization for sensitive and specific determination of non-volatile, thermolabile anticancer drugs. HPLC-MS now provides a powerful tool for drug quantification in biological matrices. Early sample preparation techniques using either liquid-liquid extraction or non-polar alkane C18 media are now used in combination for more efficient analyte extraction and concentration from urine. Improvements in solid phase extraction media provide for polar RP and hydrophilic/lipophilic interactions to extract and retain a wider range of analytes having varied polarities, pKa and LogP values. After sample preparation, most remaining interfering or co-eluting substances are eliminated in the chromatographic column or are filtered from the analysis stream by ion selection in the tandem mass spectrometer. These improvements in HPLC-MS/MS methodology have led to methods for simultaneous determination of multiple anticancer drugs in urine at μg/L detection levels, which are needed for workplace exposure studies. These methods may be expected to serve as useful tools for characterizing anticancer drug exposure in healthcare workers.
Acknowledgments
The authors wish to thank Kenneth L. Cheever and Anne Vonderheide for their assistance as reviewers in the preparation of this manuscript.
Footnotes
Publisher's Disclaimer: Disclaimers
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the National Institute for Occupational Safety and Health (NIOSH) or the Centers for Disease Control and Prevention (CDC). Mention of company names and/or products does not constitute an endorsement by NIOSH.
Conflict of interest
The authors hereby report that we have no conflict of interest with the material reported in this paper. The authors alone are responsible for the content and writing of this paper.
References
- 1. (NIOSH publication No: 2004-165).NIOSH Alert: preventing occupational exposures to antineopolastic and other hazardous drugs in health care settings. 2004 Cincinnati, OH. [Google Scholar]
- 2.Connor TH, McDiarmid MA. Preventing occupational exposures to antineoplastic drugs in health care settings. CA Cancer J Clin. 2005;56:354–365. doi: 10.3322/canjclin.56.6.354. [DOI] [PubMed] [Google Scholar]
- 3.BLS (U.S. Bureau of Labor Statistics) Occupational Employment Statistics Homepage OES Data. 2011 May; [2011] Available: www.bls.gov/oes (accessed 03.02.2017)
- 4.Schierl R, Bohlandt A, Nowak D. Guidance values for surface monitoring of antineoplastic drugs in German pharmacies. Ann Occup Hyg. 2009;53:1–9. doi: 10.1093/annhyg/mep050. [2009] [DOI] [PubMed] [Google Scholar]
- 5.Connor TH, DeBord G, Pretty JR, Oliver MS, Roth TS, Lees PSJ, Krieg EF, Rogers B, Escalante CP, Toennis CA, Clark JC, Johnson B, McDiarmid MA. Evaluation of antineoplastic drug exposure of health care workers at three university-based US cancer centers. J Occup Environ Med. 2010;52:1019–1027. doi: 10.1097/JOM.0b013e3181f72b63. [DOI] [PubMed] [Google Scholar]
- 6.Siderov J, Kirsa S, McKauchlan R. Reducing workplace cytotoxic surface contamination using a closed-system drug transfer device. J Oncol Pharm Pract. 2010;16:19–25. doi: 10.1177/1078155209352543. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida J, Koda S, Nishida S, Yoshida T, Miyajima K, Kumagai S. Association between occupational exposure levels of antineoplastic drugs and work environment in five hospitals in Japan. J Oncol Pharm Pract. 2011;17:29–38. doi: 10.1177/1078155210380485. [DOI] [PubMed] [Google Scholar]
- 8.Sessink PJM, Connor TH, Jorgenson JA, Tyler TG. Reduction in surface contamination with antineoplastic drugs in 22 hospital pharmacies in the US following implementation of a closed-system drug transfer device. Journal of Oncology Pharmacy Practice. 2011;17:39–48. doi: 10.1177/1078155210361431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sessink PJM, Leclercq GM, Wouters DM, Halbardier L, Hammad C, Kassoul N. Environmental contamination, product contamination and workers exposure using a robotic system for antineoplastic drug preparation. Journal of Oncology Pharmacy Practice. 2014;21:118–127. doi: 10.1177/1078155214522840. [DOI] [PubMed] [Google Scholar]
- 10.Sottani C, Porro B, Imbriani M, Minoia C. Occupational exposure to antineoplastic drugs in four Italian health care settings. Toxicol Lett. 2012;213:107–115. doi: 10.1016/j.toxlet.2011.03.028. [DOI] [PubMed] [Google Scholar]
- 11.Hon CY, Teschke K, Shen H. Health care workers knowledge, perceptions, and behaviors regarding antineoplastic drugs: Survey from British Columbia, Canada. J Occupat Environ Hyg. 2015;12:669–677. doi: 10.1080/15459624.2015.1029618. [DOI] [PubMed] [Google Scholar]
- 12.Kopp B, Schierl R, Nowak D. Evaluation of working practices and surface contamination with antineoplastic drugs in outpatient oncology health care settings. Int Arch Occup Environ Health. 2013;86:47–55. doi: 10.1007/s00420-012-0742-z. [DOI] [PubMed] [Google Scholar]
- 13.Merger D, Tanguay C, Langlois E, Lefebvre M, Bussieres JF. Multicenter study of environmental contamination with antineoplastic drugs in 33 Canadian hospitals. Int Arch Occup Environ Health. 2014;87:307–313. doi: 10.1007/s00420-013-0862-0. [DOI] [PubMed] [Google Scholar]
- 14.Odraska P, Dolezalova L, Kuta J, Oravec M, Piler P, Synek S, Blaha L. Association of surface contamination by antineoplastic drugs with different working conditions in hospital pharmacies. Arch Environ Occupat Hlth. 2014;69:148–158. doi: 10.1080/19338244.2013.763757. [DOI] [PubMed] [Google Scholar]
- 15.Viegas S, Pádua M, Veiga AC, Carolino E, Gomes M. Antineoplastic drugs contamination of workplace surfaces in two Portuguese hospitals. Environ Monit Assess. 2014;186:7807–7818. doi: 10.1007/s10661-014-3969-1. [DOI] [PubMed] [Google Scholar]
- 16.Berruyer M, Tanguay C, Caron NJ, Lefebvre M, Bussières M. Multicenter study of environmental contamination with antineoplastic drugs in 36 Canadian hospitals: A 2013 follow-up study. J Occup Environ Hyg. 2015;12:87–94. doi: 10.1080/15459624.2014.949725. [DOI] [PubMed] [Google Scholar]
- 17.Hon CY, Teschke K, Shen H, Demers PA, Venners S. Antineoplastic drug contamination in the urine of Canadian healthcare workers. International Archives of Occupational and Environmental Health. 2015;88:933–941. doi: 10.1007/s00420-015-1026-1. [DOI] [PubMed] [Google Scholar]
- 18.Stokvis E, Rosing H, Beijnen JH. Liquid chromatography-mass spectrometry for the quantitative bioanalysis of anticancer drugs. Mass Spectrom Rev. 2005;24:887–917. doi: 10.1002/mas.20046. [DOI] [PubMed] [Google Scholar]
- 19.Turci R, Sottani C, Spagnoli G, Minoia C. Biological and environmental monitoring of hospital personnel exposed to antineoplastic agents: a review of analytical methods. J Chromatogr B. 2003;789:169–209. doi: 10.1016/s1570-0232(03)00100-4. [DOI] [PubMed] [Google Scholar]
- 20.Nussbaumer S, Bonnabry P, Veuthey JL, Fleury-Souverain S. Analysis of anticancer drugs: A review. Talanta. 2011;85:2265–2289. doi: 10.1016/j.talanta.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 21.Sottani C, Turci R, Perbellini L, Minoia C. Liquid–liquid extraction procedure for trace determination of cyclophosphamide in human urine by high-performance liquid chromatography tandem mass spectrometry. Rapid Commun Mass Spectrom. 1998;12:1063–1068. doi: 10.1002/(SICI)1097-0231(19980831)12:16<1063::AID-RCM287>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 22.Hedmer M, Höglund P, Cavallin-Ståhl E, Albin M, Jönsson BAG. Validation of urinary excretion of cyclophosphamide as a biomarker of exposure by studying its renal clearance at high and low plasma concentrations in cancer patients. Int Arch Occup Environ Health. 2008;81:285–293. doi: 10.1007/s00420-007-0211-2. [DOI] [PubMed] [Google Scholar]
- 23.Pretty JR, Connor TH, Spasojevic I, Kurtz KS, McLaurin JL, B’Hymer C, Debord DG. Sampling and mass spectrometric analytical methods for five antineoplastic drugs in the healthcare environment. J Oncol Pharm Pract. 2012;18:23–36. doi: 10.1177/1078155210389215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anilanmert B, Sertler S, Cavus F, Cengiz S. Validated method for occupational cyclophosphamide monitiring using LC-MS/MS and a Poroshell column. Chem Papers. 2015;69:1395–1401. [Google Scholar]
- 25.Wick C, Slawson MH, Jorgenson JA, Tyler LS. Using a closed-system protective device to reduce personnel exposure to antineoplastic agents. Amer J Hlth-Sys Pharm. 2003;60:2314–2320. doi: 10.1093/ajhp/60.22.2314. [DOI] [PubMed] [Google Scholar]
- 26.Sottani C, Tranfo G, Faranda P, Minoia C. Highly sensitive high-performance liquid chromatography/selective reaction monitoring mass spectrometry method for the determination of cyclophosphamide and ifosfamide in urine of health care workers exposed to antineoplastic agents. Rapid Commun Mass Spectrom. 2005;19:2794–2800. doi: 10.1002/rcm.2116. [DOI] [PubMed] [Google Scholar]
- 27.Maeda S, Miyawaki K, Matsumoto S, Oishi M, Miwa Y, Kurokawa N. Evaluation of environmental contaminations and occupational exposures involved in preparation of chemotherapeutic drugs. Yakugaku Zasshi. 2010;130:903–910. doi: 10.1248/yakushi.130.903. [DOI] [PubMed] [Google Scholar]
- 28.Canal-Raffin M, Khennoufa K, Martinez B, Goujon Y, Folch C, Ducint D, Titier K, Brochard P, Verdun-Esquer C, Molimard M. Highly sensitive LC–MS/MS methods for urinary biological monitoring of occupational exposure to cyclophosphamide, ifosfamide, and methotrexate antineoplastic drugs and routine application. J Chromatogr B. 2016;1038:109–117. doi: 10.1016/j.jchromb.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 29.B’Hymer C, Cheever KL. Evaluation of a Procedure for the Simultaneous Quantification of 4-Ketocyclophosphamide, Cyclophosphamide, and Ifosfamide in Human Urine. J Chromatogr Sci. 2010;48:328–333. doi: 10.1093/chromsci/48.5.328. [DOI] [PubMed] [Google Scholar]
- 30.Kasel D, Jetter A, Harlfinger S, Gebhardt W, Fuhr U. Quantification of cyclophosphamide and its metabolites in urine using liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2004;18:1472–1478. doi: 10.1002/rcm.1508. [DOI] [PubMed] [Google Scholar]
- 31.Dubbelman AC, Tibben M, Rosing H, Gebretensae A, Nan L, Gorman SH, Robertson P, Schellens JHM, Beijnen JH. Development and validation of LC-MS/MS assays for the quantification of bendamustine and its metabolites in human plasma and urine. J Chromatogr B. 2012;893–894:92–100. doi: 10.1016/j.jchromb.2012.02.039. [DOI] [PubMed] [Google Scholar]
- 32.Hendrikx JJMA, Rosing H, Schinkel AH, Schellens JHM, Beijnen JH. Combined quantification of paclitaxel, docetaxel and ritonavir in human feces and urine using LC-MS/MS. Biomed Chromatogr. 2014;28:302–310. doi: 10.1002/bmc.3021. [DOI] [PubMed] [Google Scholar]
- 33.Sottani C, Tranfo G, Bettinelli M, Faranda P, Spagnoli M, Minoia C. Trace determination of anthracyclines in urine: a new high-performance liquid chromatography/tandem mass spectrometry method for assessing exposure of hospital personnel. Rapid Commun Mass Spectrom. 2004;18:2426–2436. doi: 10.1002/rcm.1642. [DOI] [PubMed] [Google Scholar]
- 34.Ndaw S, Denis F, Marsan P, d’Almeida A, Robert A. Biological monitoring of occupational exposure to 5-fluorouracil: Urinary α-fluoro-β-alanine assay by high performance liquid chromatography tandem mass spectrometry in health care personnel. J Chromatogr B. 2010;878:2630–2634. doi: 10.1016/j.jchromb.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 35.Sottani C, Zucchetti M, Zaffaroni M, Bettinelli M, Minoia C. Validated procedure for simultaneous trace level determination of the anti-cancer agent gemcitabine and its metabolite in human urine by high-performance liquid chromatography with tandem mass spectrometry. Rapid Commun Mass Spectrom. 2004;18:1017–1023. doi: 10.1002/rcm.1436. [DOI] [PubMed] [Google Scholar]
- 36.Turci R, Fiorentino ML, Sottani C, Minoia C. Determination of methotrexate in human urine at trace levels by solid phase extraction and high-performance liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2000;14:173–179. doi: 10.1002/(SICI)1097-0231(20000215)14:3<173::AID-RCM862>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 37.Rule G, Chapple M, Henion J. A 384-Well Solid-Phase Extraction for LC/MS/MS Determination of Methotrexate and Its 7-Hydroxy Metabolite in Human Urine and Plasma. Anal Chem. 2001;73:439–443. doi: 10.1021/ac000897i. [DOI] [PubMed] [Google Scholar]
- 38.Bluett J, Riba-Garcia I, Hollywood K, Verstappen SMM, Barton A, Unwin RD. A HPLC-SRM-MS based method for the detection and quantification of methotrexate in urine at doses used in clinical practice for patients with rheumatological disease: a potential measure of adherence. Analyst. 2015;140:1981–1987. doi: 10.1039/c4an02321h. [DOI] [PubMed] [Google Scholar]
- 39.Barbieri A, Sabatini L, Indiveri P, Bonfiglioli R, Lodi V, Violante FS. Simultaneous determination of low levels of methotrexate and cyclophosphamide in human urine by micro liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid Commun Mass Spectrom. 2006;20:1889–1893. doi: 10.1002/rcm.2527. [DOI] [PubMed] [Google Scholar]
- 40.Sottani C, Rinaldi P, Leoni E, Poggi G, Teragni C, Delmonte A, Minoia C. Simultaneous determination of cyclophosphamide, ifosfamide, doxorubicin, epirubicin and daunorubicin in human urine using high-performance liquid chromatography/electrospray ionization tandem mass spectrometry: bioanalytical method validation. Rapid Commun Mass Spectrom. 2008;22:2645–2659. doi: 10.1002/rcm.3657. [DOI] [PubMed] [Google Scholar]
- 41.Fabrizi G, Fioretti M, Mainero Rocca L. Dispersive solid-phase extraction procedure coupled to UPLC-ESI-MS/MS analysis for the simultaneous determination of thirteen cytotoxic drugs in human urine. Biomed Chromatogr. 2016;30:1297–1308. doi: 10.1002/bmc.3684. [DOI] [PubMed] [Google Scholar]
- 42.Mallet CR, Lu ZL, Fisk R, Mazzeo JR, Neue UD. Performance of an ultra-low elution-volume 96-well plate: drug discovery and development applications. Rapid Commun Mass Spectrom. 2003;17:163–170. doi: 10.1002/rcm.888. [DOI] [PubMed] [Google Scholar]
- 43.Rubino FM, Verduci C, Buratti M, Fustinoni S, Campo L, Omodeo-Salè E, Giglio M, Iavicoli S, Brambilla G, Colombi A. Assay of urinary α-fluoro-β-alanine by gas chromatography-mass spectrometry for the biological monitoring of occupational exposure to 5-fluorouracil in oncology nurses and pharmacy technicians. Biomed Chromatogr. 2006;20:257–266. doi: 10.1002/bmc.559. [DOI] [PubMed] [Google Scholar]
- 44.Sessink PJM, Timmersmans JL, Anzion RBM, Bos RP. Assessment of occupational exposure of pharmaceutical plant workers to 5-fluorouracil: Determination of α-fluoro-β-alanine in urine. J Occupat Med. 1994;36:79–83. [PubMed] [Google Scholar]
- 45.Kosjek T, Heath E. Occurrence, fate and determination of cytostatic pharmaceuticals in the environment. Trends Anal Chem. 2011;30:1065–1087. [Google Scholar]
- 46.Nguyen HP, Schug KA. The advantages of ESI-MS detection in conjunction with HILIC mode separations: Fundamentals and applications. J Sep Sci. 2008;31:1465–1480. doi: 10.1002/jssc.200700630. [DOI] [PubMed] [Google Scholar]
- 47.Hemstrom P, Irgum K. Hydrophilic interaction chromatography. J Sep Sci. 2006;29:1784–1821. doi: 10.1002/jssc.200600199. [DOI] [PubMed] [Google Scholar]
- 48.Jian W, Edom RW, Xu Y, Weng N. Recent advances in application of hydrophilic interaction chromatography for quantitative bioanalysis. J Sep Sci. 2010;33:681–697. doi: 10.1002/jssc.200900692. [DOI] [PubMed] [Google Scholar]
- 49.Zimmer D. Introduction to quantitative liquid chromatography-tandem mass spectrometry (LC-MS-MS) Chromatographia. 2003;57:S325–S332. [Google Scholar]
- 50.Goddlett DR, Gale DC, Guiles S, Crowther JB. Mass spectrometry in pharmeceutical analysis. In: Meyers RA, editor. Encyclopedia of Analytical Chemistry: Applications, Theory and Instrumentation. John Wiley & Sons; NY: 2001. pp. 7209–7278. [Google Scholar]
- 51.B’Hymer C, Cheever KL. Biomarker and Metabolite Analysis by HPLC-MS. In: Cazes J, editor. Encyclopedia of Chromatography. CRC Press; Boca Rotan, Florida: 2010. pp. 238–246. [Google Scholar]
- 52.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem. 2003;75:3019–3030. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- 53.Hendrikx JJMA, Rosing H, Schinkel AH, Schellens JHM, Beijnen JH. Quantification of taxanes in biological matrices: A review of bioanalytical assays and recommendations for development of new assays. Bioanalysis. 2014;6:993–1010. doi: 10.4155/bio.14.48. [DOI] [PubMed] [Google Scholar]
- 54.Varma D, Jansen SA, Ganti S. Chromatography with higher pressure, smaller particles and higher temperature: a bioanalytical perspective. Bioanalysis. 2010;2:2019–2034. doi: 10.4155/bio.10.148. [DOI] [PubMed] [Google Scholar]
- 55.Turci R, Sottani C, Schierl R, Minoia C. Validation protocol and analytical quality in biological monitoring of occupational exposure to antineoplastic drugs. Tox Lett. 2006;162:256–262. doi: 10.1016/j.toxlet.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 56.Rosing H, Man WY, Doyle E, Bult A, Beijen JH. Bioanalytical liquid chromatographic method validation. A review of current practices and procedures. J Liq Chrom Rel Technol. 2000;23:329–354. [Google Scholar]
- 57.US Food and Drug Administration. Guidance for industry: bioanalytical method validation. US Food and Drug Administration; Rockford, Maryland: May, 2001. http://www.fda.gov/downloads/Drugs/Guidance/ucm070107.pdf (accessed 03.02.2017) [Google Scholar]
- 58.International Conference on Harmonization (ICH) Harmonized Tripartite Guideline, Guidance on Validation of Analytical Procedures. Canary Warf; London, UK: 2011. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002662.pdf (accessed 03.02.2017) [Google Scholar]
- 59.EURACHEM. The fitness of purpose of analytical methods: a laboratory guide to method validation and related topics. (2nd) 2014 Leoben, Austria. https://www.eurachem.org/images/stories/Guides/pdf/MV_guide_2nd_ed_EN.pdf (accessed 03.02.2017)
- 60.Wu NJ, Clausen AM. Fundamental and practical aspects of ultrahigh pressure liquid chromatography for fast separations. J Sep Sci. 2007;30:1167–1182. doi: 10.1002/jssc.200700026. [DOI] [PubMed] [Google Scholar]
- 61.Holland JF, Enke CG, Allison J, Stults JT, Pinkston JD, Newcome B, Watson JT. Mass-Spectrometry on the Chromatographic Time Scale – Realistic Expectations. Anal Chem. 1983;55:997A–1012A. [Google Scholar]