

Abstract
Inflammatory myofibroblastic tumor (IMT) is a rare tumor in the central nervous system (CNS), mostly being extracranial. Approximately 100 sporadic cases have been reported in the literature. The rarity of the tumor, its various histopathological characteristics, and its variable aggressive course render it difficult to diagnose and treat. IMT is generally a histological diagnosis which is rarely suspected preoperatively. It mimics other intracranial tumors such as giant cell tumor, hemangiopericytoma, anaplastic meningioma, plasmacytoma, and lymphoma. Rarely, it can present with a clinical picture which mimics a benign infective process, Rosai-Dorfman disease, or an idiopathic hypertrophic pachymeningitis. High index of suspicion is required as total resection of this lesion is mandatory to prevent recurrence. Here, we describe a case of a 10-year-old child which initially presented with clinical features mimicking chronic suppurative otitis media and radiological presentation of a small intracranial abscess. He was initially treated by an ENT surgeon who started him on intravenous antibiotics, but the patient was lost to follow up. He returned after 2 months with a large lesion at the same location. Histological examination revealed multiple spindle cells with plasma cells and lymphocytes scattered among these spindle cells. The spindle cells were immunopositive for smooth muscle actin and negative for epithelial membrane antigen, S100, and CD34.
KEYWORDS: Anaplastic lymphoma kinase, inflammatory myofibroblastic tumor, inflammatory pseudotumor, plasma cell granuloma
INTRODUCTION
Inflammatory myofibroblastic tumor (IMT) is a rare neoplasm composed of myofibroblastic spindle cells, accompanied by an inflammatory infiltrate of plasma cells, lymphocytes, and eosinophils. It occurs primarily in soft tissue and the viscera of children and young adults. It usually follows a benign clinical course and is characterized by nonneoplastic polyclonal proliferation of mature plasma cells and other mononuclear cells. IMT in the central nervous system (CNS) tends to arise mainly from meningeal structures. It has a high frequency of recurrence and malignant transformation compared with IMT not affecting the CNS. IMT-CNS is generally a histological diagnosis which is rarely suspected preoperatively. It mimics other intracranial tumors such as giant cell tumor, hemangiopericytoma, and anaplastic meningioma. Here, we describe a case of IMT which initially presented with clinical features mimicking chronic suppurative otitis media with a small intracranial contrast-enhancing space occupying-lesion mimicking accesses.
CASE REPORT
A 10-year-old child presented with ear discharge and localized mastoid tenderness. Computed tomography (CT) brain revealed a small isointense extra-axial lesion involving the left transverse-sigmoid junction with intense homogeneous contrast enhancement. He was initially treated by an ENT surgeon who started him on intravenous antibiotics and inserted a grommet. The patient was then lost to follow up. He returned after 2 months with increasing headache and severe mastoid tenderness. CECT and contrast-enhanced magnetic resonance imaging brain showed an intensely contrast-enhancing lesion around 5 cm × 3 cm at the left transverse-sigmoid junction [Figures 1 and 2]. The child was operated in left lateral position and retromastoid suboccipital craniectomy was done. Intraoperatively, tumor was eroding occipital bone and was vascular, grayish red, nonsuckable, and attached to tentorium which was grossly thickened. Total resection was done and attachment to tentorium was thoroughly coagulated. Morphological appearance was like meningioma. Histological examination revealed multiple spindle cells with plasma cells and lymphocytes scattered among these spindle cells [Figure 3]. The spindle cells were diffusely immunopositive for vimentin and smooth muscle actin (SMA) but negative for epithelial membrane antigen, S100 negative, and CD34 negative [Figure 4]. The cells stained negative for anaplastic lymphoma kinase (ALK) [Figure 5]. Postoperative CT confirmed complete removal. No radiotherapy was given and the child is still recurrence free after 4 months.
Figure 1.
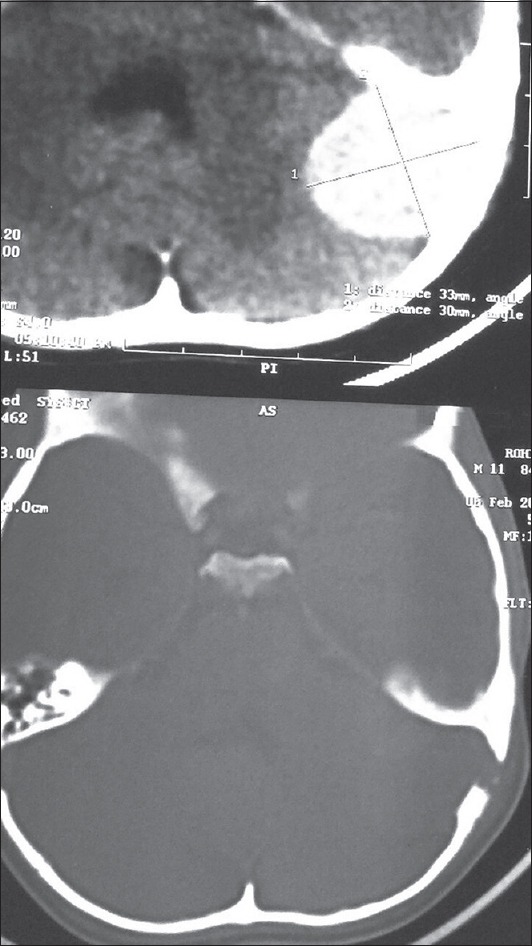
Contrast-enhanced computed tomography showing an intensely contrast-enhancing lesion at the left transverse-sigmoid junction causing destruction of the occipital bone
Figure 2.
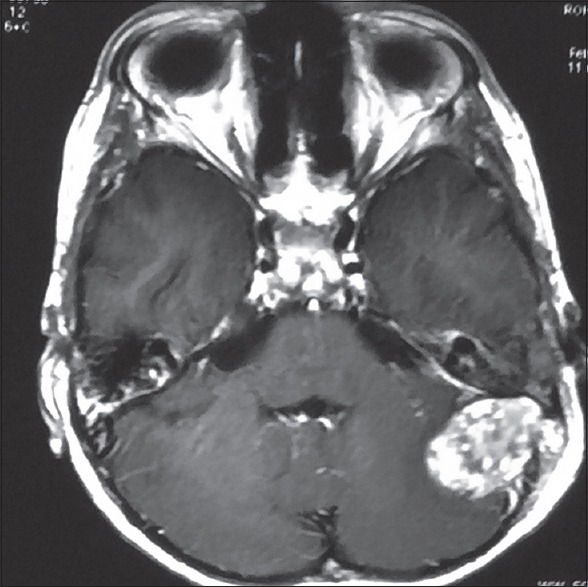
Preoperative axial contrast-enhanced magnetic resonance imaging showing a gadolinium-enhancing extra-axial mass of the left transverse-sigmoid junction
Figure 3.
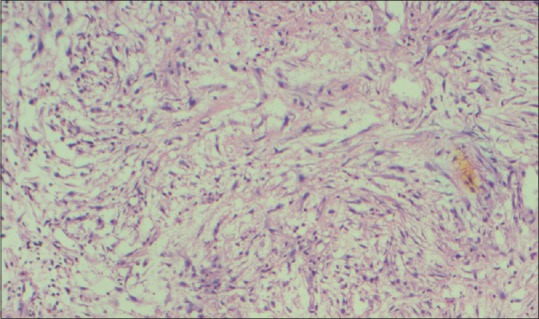
Histological appearance showing spindle cells with diffuse lymphocytes and plasmocytes infiltrate
Figure 4.
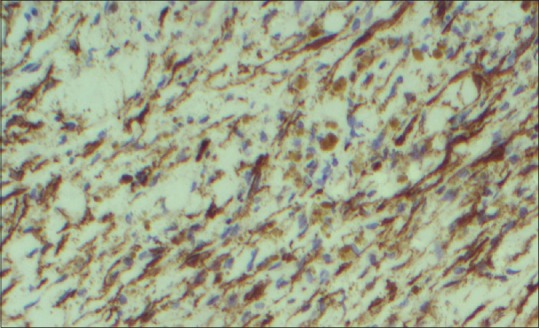
Spindle cells showing diffuse immunopositivity for smooth muscle actin
Figure 5.
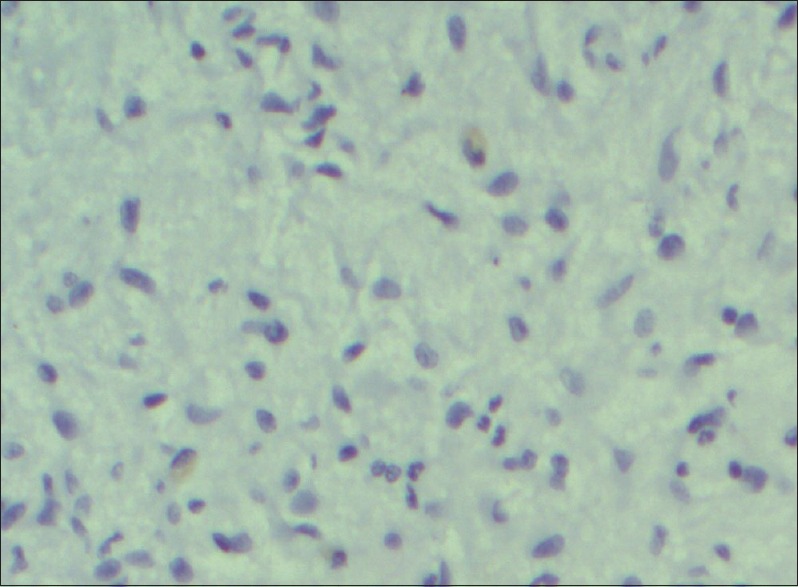
Spindle cells showing negative staining for anaplastic lymphoma kinase
DISCUSSION
IMT is a distinctive myofibroblastic spindle cell lesion accompanied by an inflammatory infiltrate of plasma cells, lymphocytes, and eosinophils. Approximately 100 sporadic cases have been reported in the literature.[1,2] Because of varied histological features, it is also known as inflammatory pseudotumor, plasma cell granuloma (PCG), pseudosarcomatous myofibroblastic proliferation, inflammatory myofibrohistiocytic proliferation, etc., In the 2002 World Health Organization classification of soft tissue tumors, these lesions were renamed “inflammatory myofibroblastic tumors” and allocated to the soft tissue tumor category.[3] ALK protein overexpression in myofibroblastic cells was found in 35% to 60% of IMT cases, suggesting neoplastic nature of these tumors rather than a reactive process.[4,5,6] The neuroimaging findings and clinical course of IMT-CNS are nonspecific, such that differential diagnosis is important in the case of meningeal lesions, especially with meningioma or sarcoma. Radiologically, two patterns are seen: nodular pattern and an en plaque-like pattern. There is no correlation between radiological findings and the histopathological features. Histopathologically, there are two variants: PCG or fibrohistiocytic (FHC) variant. The PCG variant is composed mainly of plasma cells and lymphocytic infiltration whereas the latter variant is rich in spindle myofibroblasts mixed with few inflammatory cells. Spindle-shaped mesenchymal cells of IMTs express SMA in all cases. Immunostaining for SMA was found to helpfully discriminate myofibroblastic cells and to make a differential diagnosis. Those cases with both leptomeningeal and brain involvement are difficult to distinguish from lymphoproliferative disorders such as plasmacytoma or infectious conditions such as patchy hypertrophic meningitis. In IMT, cells are heterogeneously positive for lymphoid markers, CD3 or CD20. The confirmation of the presence of SMA-positive myofibroblasts is potentially important for the differential diagnoses from idiopathic hypertrophic pachymeningitis or meningioma. The infiltration of polyclonal plasma cells and other mixed inflammatory cells is a helpful feature when differentiating IMT-CNS and lymphoma or plasmacytoma. Sixty percent of IMT-CNS tumors arise from the meninges with or without parenchymal invasion and only 24% from intra-axial or intraventricular location. The present case had attachment to tentorium and was classified as the meningeal type. Eighty-two percent of cases present as a single intracranial lesion, 3% as a solitary spinal lesion, and 16% as multiple synchronous or metachronous lesions.[7]
IMT-CNS has a high recurrence rate and hence total resection is mandatory. After incomplete resection, 40% recurrence rate is seen within 2 years.[8,9] Recurrence rate after gross total resection for ALK-positive and ALK-negative cases is around 33% and 9%, respectively. Most recurrences in ALK-positive patients occur within 2 years after surgery. Tumor cells which express ALK are more likely to recur after total resection. Therefore, patients need close follow-up. ALK is a tyrosine kinase receptor that is normally expressed in the developing CNS. The ALK expression in IMT of the CNS is specific to the FHC variant.
CONCLUSION
Because of its variable clinical presentation but highly aggressive course, possibility of IMT should always be kept in mind. Total resection should be achieved as these tumors recur often rapidly. Confirmation of the FHC variant by histopathology warrants searching for ALK expression. Such patients can be offered adjuvant therapy such as radiotherapy.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Häusler M, Schaade L, Ramaekers VT, Doenges M, Heimann G, Sellhaus B. Inflammatory pseudotumors of the central nervous system: Report of 3 cases and a literature review. Hum Pathol. 2003;34:253–62. doi: 10.1053/hupa.2003.35. [DOI] [PubMed] [Google Scholar]
- 2.Tresser N, Rolf C, Cohen M. Plasma cell granulomas of the brain: Pediatric case presentation and review of the literature. Childs Nerv Syst. 1996;12:52–7. doi: 10.1007/BF00573857. [DOI] [PubMed] [Google Scholar]
- 3.Fletcher CD, Unni K, Mertens F. World Health Organization Classification of Tumors. Lyon, France: IARC Press; 2002. Pathology and Genetics, Tumors of Soft Tissue and Bone; pp. 91–3. [Google Scholar]
- 4.Cook JR, Dehner LP, Collins MH, Ma Z, Morris SW, Coffin CM, et al. Anaplastic lymphoma kinase (ALK) expression in the inflammatory myofibroblastic tumor: A comparative immunohistochemical study. Am J Surg Pathol. 2001;25:1364–71. doi: 10.1097/00000478-200111000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Coffin CM, Patel A, Perkins S, Elenitoba-Johnson KS, Perlman E, Griffin CA. ALK1 and p80 expression and chromosomal rearrangements involving 2p23 in inflammatory myofibroblastic tumor. Mod Pathol. 2001;14:569–76. doi: 10.1038/modpathol.3880352. [DOI] [PubMed] [Google Scholar]
- 6.Cessna MH, Zhou H, Sanger WG, Perkins SL, Tripp S, Pickering D, et al. Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its mesenchymal mimics: A study of 135 cases. Mod Pathol. 2002;15:931–8. doi: 10.1097/01.MP.0000026615.04130.1F. [DOI] [PubMed] [Google Scholar]
- 7.Buccoliero AM, Caldarella A, Santucci M, Ammannati F, Mennonna P, Taddei A, et al. Plasma cell granuloma – An enigmatic lesion: Description of an extensive intracranial case and review of the literature. Arch Pathol Lab Med. 2003;127:e220–3. doi: 10.5858/2003-127-e220-PCGEL. [DOI] [PubMed] [Google Scholar]
- 8.Pascual-Gallego M, Yus-Fuertes M, Jorquera M, Gonzalez-Mat A, Ortega L, Martinez-Martinez A, et al. Recurrent meningeal inflammatory myofibroblastic tumor: A case report and literature review. Neurol India. 2013;61:644–9. doi: 10.4103/0028-3886.125273. [DOI] [PubMed] [Google Scholar]
- 9.Denis DJ, Elayoubi K, Weil AG, Berthelet F, Bojanowski MW. Inflammatory myofibroblastic tumors of the central nervous system that express anaplastic lymphoma kinase have a high recurrence rate. Surg Neurol Int. 2013;4:70. doi: 10.4103/2152-7806.112614. [DOI] [PMC free article] [PubMed] [Google Scholar]