

Abstract
Proteases play diverse and important roles in physiology and disease, including influencing critical processes in development, immune responses, and malignancies. Both the abundance and activity of these enzymes are tightly regulated and highly contextual; thus, in order to elucidate their specific impact on disease progression, better tools are needed to precisely monitor in situ protease activity. Current strategies for detecting protease activity are focused on functionalizing synthetic peptide substrates with reporters that emit detection signals following peptide cleavage. However, these activity-based probes lack the capacity to be turned on at sites of interest and therefore, are subject to off-target activation. Here we report a strategy that uses light to precisely control both the location and time of activity-based sensing. We develop photocaged activity-based sensors by conjugating photolabile molecules directly onto peptide substrates, thereby blocking protease cleavage by steric hindrance. At sites of disease, exposure to ultraviolet light unveils the nanosensors to allow proteases to cleave and release a reporter fragment that can be detected remotely. We apply this spatiotemporally-controlled system to probe secreted protease activity in vitro and tumor protease activity in vivo. In vitro, we demonstrate the ability to dynamically and spatially measure metalloproteinase activity in a 3D model of colorectal cancer. In vivo, veiled nanosensors are selectively activated at the primary tumor site in colorectal cancer xenografts to capture the tumor microenvironment-enriched protease activity. The ability to remotely control activity-based sensors may offer a valuable complement to existing tools for measuring biological activity.
Keywords: photoactivatble, proteases, activity-based biomarkers, nanosensors
TOC image
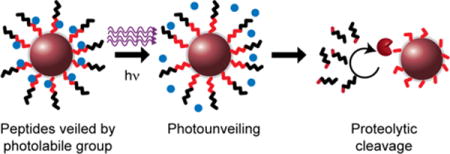
Biological function is context dependent, with diverse regulatory mechanisms that function at the genomic, transcriptional, and post-translational levels to modulate both the abundance and functional status of proteins.1,2 Therefore, the capacity to make dynamic measurements of protein function is crucial in achieving a thorough understanding of biological processes.3,4 Proteases are a key example of a protein family that needs to be studied at the activity level due to their extensive posttranslational modifications, presence of endogenous inhibitors (e.g., α2-macroglobulin), and pivotal roles played by these proteins in the bioregulation of healthy and disease processes.5–9 In the case of cancer biology, both the intratumoral localization and the dynamics of protease activity throughout disease progression are relevant to pathogenesis. Therefore, activity-based measurements that can capture this spatiotemporal heterogeneity may provide important insights.
Numerous techniques have been developed to measure protease activity in models of cancer, including activity-based probes (ABPs) that can assess levels of active enzymes by irreversible binding of a chemical probe.3,10–13 These probes enable the high-content analysis of enzymes, but applying these tools in vivo is technically challenging. Protease-driven imaging of diseased sites, where protease activity results in an increase in contrast, has also shown great promise for early and specific detection of tumor burden.14–19 Multiple groups have leveraged these two approaches for nanoparticle-based protease sensing, using scaffolds such as quantum dots and nanoparticles, to achieve improved sensitivity and targeting.20 Our group has previously reported a class of activity-based probes called ‘synthetic biomarkers’21–24 that produce a detection signal following protease cleavage similar to fluorogenic probes.16,17 In contrast to other platforms, however, our system is designed such that the liberated peptide fragments are concentrated in the urine, and detectable by a variety of analytical techniques ranging from mass spectrometry to single molecule assays.21–24 As the function of these systems are initiated by an active protease, the measurements collected reflects protease activity rather than abundance. While each these activity-based approaches are promising, they lack the ability to be remotely controlled.
In this study, we describe an approach to control the sensing of activity-based probes remotely. We used the synthetic biomarker system as a platform to demonstrate our approach for photoprotection, as it is founded on the general element of cleavage-based activity sensors, and because the framework is readily extensible to other techniques, such as fluorescence imaging methods.16,17 Specifically, we accomplish remote activation by veiling peptide substrates with small molecule photolabile groups, at residues adjacent to the enzyme-targeted scissile bond, which blocks protease activity due to steric hindrance. These photocaged sensors are delivered to sites of diseases where the photolabile molecule is removed by light (λ = 365 nm), allowing disease-associated proteases to cleave the peptide substrates. The extent of proteolytic activity is then directly measured by fluorescence or ELISA. Therefore, these particles leverage both activity-based monitoring and photolabile chemistry to act as SpatioTemporally Responsive Enzymatic Activity Monitors (STREAMs). We demonstrate the utility of this method to control the site and time at which biological protease activity is assayed in 3D in vitro tumor models and in a xenograft mouse model of cancer.
Results and Discussion
Development of spatiotemporally-responsive nanoparticle protease sensors
Matrix metalloproteinases (MMPs) represent an important protease family to study and assay as their activities are associated with numerous pathways in health and disease.5 Thus, we designed a veiled, MMP-sensitive nanosensor by conjugating the photolabile small molecule 1-(4,5-Dimethoxy-2-nitrophenyl) diazoethane (DMNPE) to protease-cleavable substrates (Fig. 1). DMNPE reacts with acidic groups,25,26 and by coupling it to an MMP substrate sequence containing free carboxylic acid side chains, serves as a removable barrier to block enzymatic cleavage. Furthermore, we hypothesized, based on previous studies,27 that DMNPE should be located within a few amino acids from the putative cleavage site in order to effectively block protease activity by steric hindrance. Based on these design criteria, we selected a peptide sequence that is sensitive to MMP activity28 (sequence: PLGLEEA) and contains carboxylic acid side chains adjacent to the scissile bond (G-L). We functionalized iron oxide nanoparticles (NPs, diameter ~ 100 nm; SI FIG. 1a) with fluorescein-conjugated peptide substrates (sequence: FAM-sk-PLGLEEA-GC; lower case = D-stereoisomer; name: C1) at a surface valency greater than 20 (Fig. 1a, SI Fig. 1b). The size of the nanoparticle is larger than the kidney filtration limit and therefore acts to prevent urinary filtration of the STREAMs construct prior to peptide cleavage for applications in vivo. DMNPE was selectively removed after photolysis in the presence of 365 nm light, making the peptide substrate available for cleavage by proteases and resulting in the release of reporters (Fig. 1). Thus, these constructs have the potential to enable spatiotemporal control of the accessibility of the substrate during measurements of protease activity. Since MMP activity is commonly implicated in cancer progression,5 we sought to test the utility of these SpatioTemporally Responsive Activity Monitors (STREAMs) in both in vitro and in vivo models of cancer.
Figure 1. Photoactivatable sensors of protease activity.
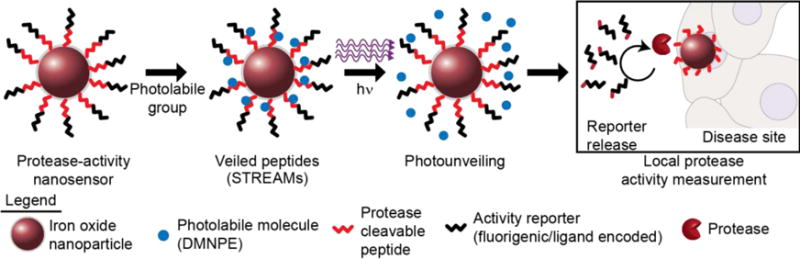
Photolabile groups can be directly coupled to peptide substrates and can be efficiently photolysed with 365 nm light to unveil enzyme cleavage site and enable local protease activity measurements. We apply this principle of SpatioTemporally Responsive Enzymatic Activity Monitors (STREAMs) to probe local protease activity in models of cancer.
STREAMs are designed to leverage the strengths of numerous techniques, such that the unique combination of photolabile chemistry, nanoparticle formulation, and protease sensing enables STREAMs to perform the complex task of measuring in vivo enzyme activity with spatial and temporal control. Previous demonstrations of protease measurements in vivo lack external control (e.g. controlled triggering at the tumor site), and our addition of these traits with the STREAM platform may enable greater sensitivity and tumor contrast. Similarly, synthetic biomarkers are vulnerable to background activation in the circulation.29 The previous utilizations of DMNPE have been varied, ranging from caging nucleic acids (DNA30,31 and RNA25,27) to caging Ca2+32,33. To our knowledge, however, a general strategy for caging peptide substrates of proteases has not been previously described.
Chemical characterization of peptide-DMNPE conjugates
Prior to applying our STREAMs to assay for MMP activity, we sought to validate the chemical conjugation of the photolabile DMNPE group to the MMP substrate. DMNPE is comprised of a nitrophenyl group that is efficiently activated by 365 nm light, resulting in photolysis of the veiled substrate. DMNPE reacts with weak oxo-acids and thus can modify the glutamic acids that reside at the substrate’s P2′ and P3′ positions, located towards the C-terminal end of the scissile bond (Fig. 2a). The synthesis of the fluorescein-conjugated peptide (C1) was validated by MALDI mass spectrometry, which resulted in a major peak at 1461.43 m/z that corresponded with the calculated molecular weight of C1 (Fig. 2b). Next, we validated the location of the scissile bond (between the glycine and leucine) by incubating C1-NPs with recombinant MMP9 overnight and measured the size of the N-terminal cleavage fragment (Fig. 2c). DMNPE was incorporated into peptides using a modification of the approach of Friedman and coworkers for modifying insulin.34 To validate the coupling of DMNPE to the peptide, we used ESI-MS to analyze the conjugate because electrospray ionization does not lead to photolysis of DMNPE. Mass spectrometry analysis of the conjugate resulted in a mass shift associated with DMNPE coupled to the peptide (Fig. 2d, top). Next, we used MALDI – where ionization is based on UV light pulses – to simultaneously photolyse the DMNPE molecules and detect the uncaged peptide backbone. Indeed, the laser desorption resulted in a mass shift of the treated sample to yield a peak at the predicted peptide mass with no evidence of the parent mass, demonstrating that DMNPE could be efficiently photolysed and removed upon exposure to light (Fig. 2d, bottom).
Figure 2. Protecting groups can be coupled to amino acids adjacent to scissile bond.
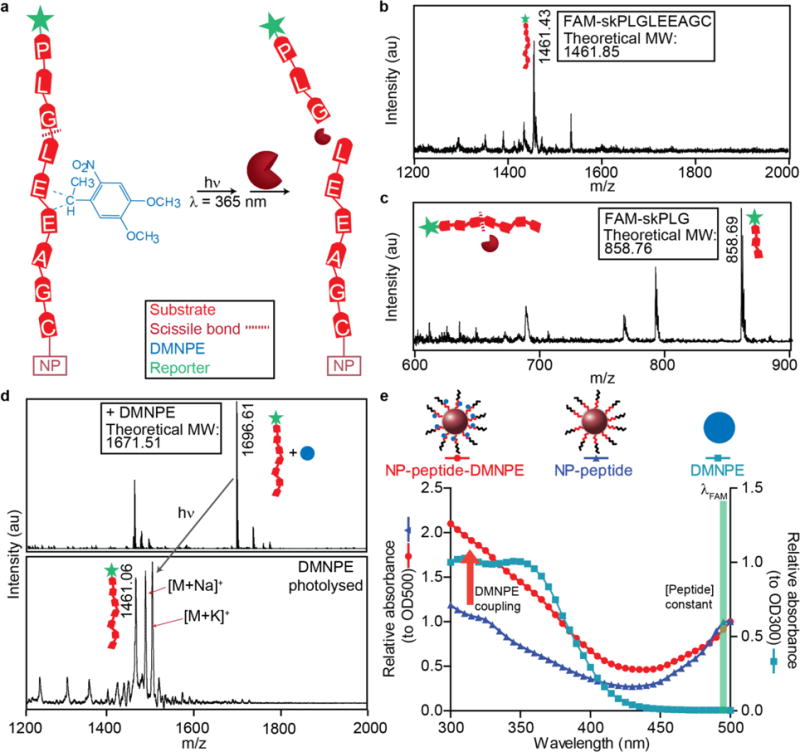
(a) The peptide backbone can be directly modified with a photolabile group (DMNPE; blue) at acidic residues. Adjacent to the peptide substrate, reporters that can be either fluorigenic or ligand-encoded (green) are released up cleavage. Activation by light removes the photolabile group and enables proteases to access the peptide. (b) Mass spectrometry analysis of the native peptide sequence. (c) Identification of the scissile bond by mass spectrometry analysis of MMP9 cleaved peptide fragment. (d) Coupling of a DMNPE molecule is confirmed by an m/z shift corresponding to the mass of one DMNPE molecule. Photolysis results in a mass shift back to the original mass of the native peptide. (e) Spectral characteristic of NP-peptides (triangles) and spectral shift with DMNPE coupled (circle) that approximately matches spectra of free DMNPE.
After successfully coupling the photolabile group to the MMP substrate/reporter backbone, we directly coupled the DMNPE groups to the conjugated C1-NPs. Uncoupled DMNPE was removed via spin filtration or FPLC, and successful conjugation of DMNPE was confirmed by shifts in absorbance values (Fig. 2e). Following conjugation of DMNPE with peptides, NPs should exhibit significant absorption at 300–350 nm, which would result in an overall absorbance shift, relative to that of unmodified NPs that should be reversed after photolysis. Consistent with this expectation, after light exposure, STREAMs exhibited an absorption peak that shifted back to overlap with that of pre-conjugated particles, demonstrating that DMNPE was released from the peptides (SI Fig. 2a).
STREAMs are protected from recombinant proteases until photoactivation
We next sought to test whether STREAMs could provide both spatial and temporal control of MMP activity measurements. We first evaluated whether the veiled nanoparticles would block protease cleavage until activation by light. Due to homoquenching of the fluorescent substrates once assembled on the NPs, protease activity can be monitored by measuring increases in sample fluorescence that occurs from peptide proteolysis (SI Fig. 2b). Nanoparticles were stable in physiological solution at 37 °C over 24 hours, as confirmed by a lack of fluorescent dequenching (SI Fig. 2c).
Proteolytic kinetics can be altered by presentation of peptides on surfaces.35,36 Therefore, measurements of proteolysis by recombinant enzymes were performed with the substrate on the particle, in the same formulation we used in vivo, to accurately capture differences due to presentation. We profiled proteolysis of this substrate by a panel of proteases consisting of MMPs, ADAMs, and blood-borne proteases. We observed that the unmodified substrate (C1-NP) was significantly cleaved by MMP13, 7, 1 and 9 (Fig. 3a, b, SI Fig. 3a). It is important to note, however, that some of the differences observed in enzyme-mediated substrate cleavage across enzymes may be due in part to the activity of the recombinant enzymes in vitro. Proteolysis by MMP7 was inhibited in the presence of Marimastat, an MMP inhibitor (SI Fig. 3b). Substrate concentration dependence on cleavage velocity was confirmed for MMP9 and MMP13, and data was fit to the Michelis-Menten equation, with catalytic efficiencies greater than 103 M−1 s−1 and 104 M−1 s−1, respectively (Fig 3c, d). In contrast, conjugation with DMNPE resulted in a marked reduction in proteolysis, protecting STREAMs from MMP13 and MMP9 activity (Fig. 3e, f). Stability of DMNPE-peptide-NP STREAM complexes was confirmed by testing samples two weeks post DMNPE-coupling for resistance to MMP9-mediated cleavage (SI Fig. 3c), where we observed equal levels of protection compared to freshly conjugated samples. Finally, we established that exposure of DMNPE veiled NPs to 365 nm light unveiled the scissile bond and rendered it susceptible to proteolytic cleavage by incubating NPs with MMP9 and MMP13 after increasing periods of exposure to light, which led to elevated proteolysis in a light exposure dependent manner (Fig. 3g, h). This dose response relationship between light exposure and enzyme-mediated proteolysis suggested that it should be possible to tune the fraction of photolabile groups that are released, and thus enable graded control for use in dynamic and repeated measurements. Furthermore, to extend the utility of our approach, we demonstrate unveiling of STREAMs with two photon excitation, which should enable deeper tissue penetration due to the near-infrared optical window (SI Fig. 4). These results highlight STREAMs as a framework for adding spatiotemporal control to protease-activity measurements.
Figure 3. STREAM sensing of recombinant proteases.
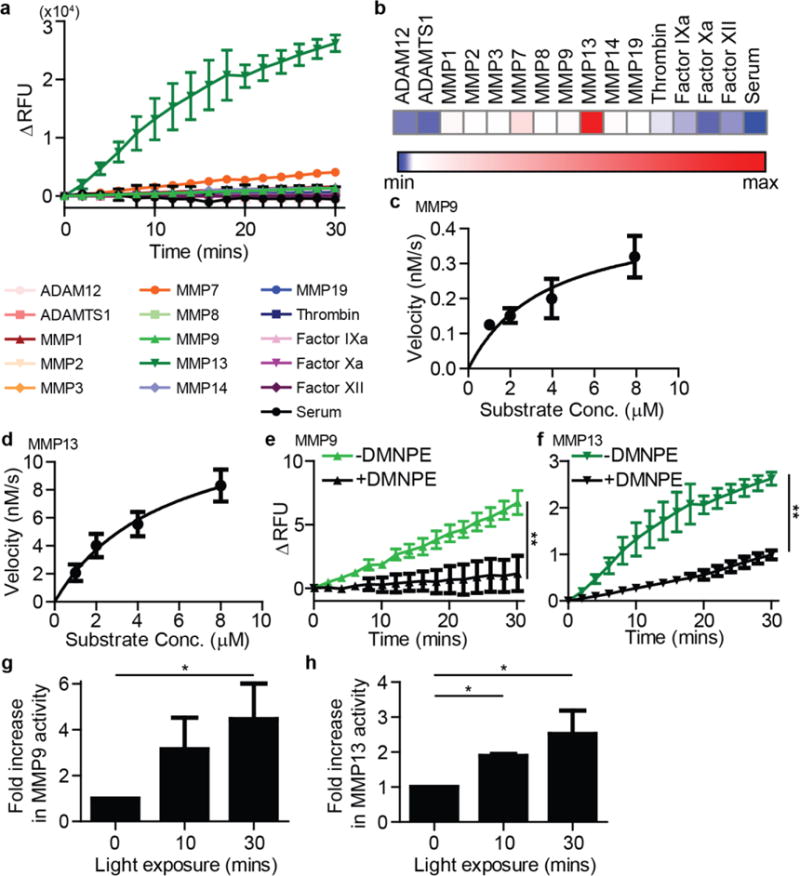
(a) Fluorescence dequenching measurements of protease cleavage by multiple enzymes targeting C1-NPs. MMP13, 7, 1, 9 and 14 are able to cleave the substrate, with MMP13 as the most efficient. (b) Heatmap of cleavage velocity of the different proteases. (c) Michaelis-Menten analysis of MMP9 cleavage of C1-NPs. (d) Michaelis-Menten analysis of MMP13 cleavage of C1-NPs. (e) Dequenching measurements of MMP9 cleavage against unmodified C1-NPs and DMNPE-veiled C1-NPs. (f) Dequenching measurements of MMP9 cleavage against unmodified C1-NPs and DMNPE-veiled C1-NPs. (g) Light activation of particles and subsequent increase in MMP9 activity. (h) Light activation of particles and subsequent increase in MMP13 activity. (all experiments: n = 2–3; all error bars: ± SD; e/f: **P<0.01, 2way ANOVA; g/h: *P<0.05, one-tail, Student’s t-test; light exposure: 8 mW/cm2).
To validate that our approach is generalizable to alternative substrates, we applied the STREAMs principle to a second peptide sequence. Additionally, the reporter for this additional sequence was designed to be orthogonal to the original sequence (containing a near IR dye as opposed to fluorescein). Coupling of DMNPE to this second substrate (RLVGEGC) reduced proteolysis by plasmin, which was recovered by UV exposure (SI Fig. 5). The ability to produce STREAMs with orthogonal reporters for multiple substrate targets may enable multiplexing for future applications. Additionally, coupling this approach with alternate modes for multiplexing for analyte detection should enable simulatenous monitoring of several substrates.37
STREAMs are spatiotemporally-responsive protease sensors in 3D cancer models
To investigate whether STREAM constructs might be applied in more complex settings, we assayed their performance as proteolysis sensors in a 3D cancer model in vitro. We selected the LS174T cell line, which has been used extensively for in vivo cancer models and is known to secrete active MMPs (including MMP2, 9).38 In order to confirm that our nanosensors were responsive to secreted proteases, fluorigenic C1-NPs were incubated with conditioned media from LS174T cells grown on tissue culture plastic, which resulted in peptide cleavage and a dose dependent increase in fluorescence that was specific for the L-amino acid version of the protease sensor. By contrast, control nanoparticles conjugated to D-amino acid stereoisomers, which are not cleavable by proteases, were not cleaved by cell-secreted proteases present in conditioned media (SI Fig. 6a, b). We additionally measured protease activity derived from the CCD-18Co cell line, which is a line of non-transformed cells isolated from normal colon tissue that has been used previously as a control in cancer studies.39 Protease cleavage from these cells, while detectable, was significantly lower compared to LS174T cells (SI Fig. 6c).
Next, we sought to probe the activation of the nanosensors in a 3D ECM environment.40 LS174T cells were embedded in collagen I together with veiled or unmodified nanosensors (Fig. 4a). The constructs were monitored for protease activity by collecting the supernatant and measuring liberated peptide fragments under different conditions: L-amino acid peptide substrates were compared to D-amino acid counterparts to measure nonspecific background, and the role of DMPNE veiling was measured. On the first day, constructs bearing L-amino acid sensors released significantly more fluorescent peptides than those with D-amino acid nanoparticles. Additionally, DMNPE-veiled, L-amino acid sensors produced significantly less peptide fluorescence compared to unmodified L-amino acid sensors, indicating that the photolabile groups shielded the NPs from proteolytic cleavage in the context of cell-secreted proteases (Fig. 4b).
Figure 4. STREAMs embedded in cancer tissue models for protease sensing.
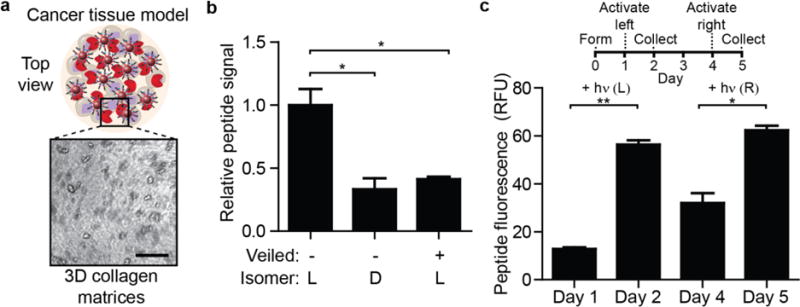
(a) 3D collagen tissues containing embedded colorectal cancer cells established as an in vitro model of the tumor microenvironment. Cells inside the collagen gel can be visualized and are homogeneously distributed (scale bar: 200 μm). C1-NPs (veiled or unmodified, L and D stereoisomers) were also embedded. (b) One day after forming the gel, the surrounding media was assessed for peptide fluorescence. Veiled substrates had significantly lower rates of proteolysis as did D-stereoisomer peptides compared to gels that contained L-stereoisomer particles (*P<0.05, two-tail Student’s t-test, n = 3, s.e.m.). (c) Spatial and temporal activation of STREAMs in cancer collagen tissue. The left half of gels was exposed to light on day 1 and total peptide signal was measured in collected supernatant. Three days later, the right half of gels was activated and peptide signal was measured (**P<0.01, *P<0.05, two-tail, paired Student’s t-test; n = 3, s.e.m.; light exposure: 30s at 200 mW/cm2).
In order to correlate regions of light-activation with protease activity measurements, we included light-activated rhodamine dye to visualize regions exposed to light (SI Fig. 7a). To explore the ability to monitor protease activity with spatial and temporal control, only the left half of the gels was illuminated. After 24 hours, the supernatant surrounding the gels contained higher levels of peptide fluorescence, suggesting that restricted light activation unveiled peptides and made them available for proteolysis (Fig. 4c). Similarly, when the opposite side of the cancer tissue model was illuminated three days later, we observed a significant increase in fluorescent reporters released. By contrast, unmodified sensors did not exhibit significant changes in peptide fluorescence after UV exposure (SI Fig. 7b). Collectively, these results demonstrate that STREAMs can be used to spatially probe enzyme activity in engineered constructs.
STREAMs are protected from in vivo proteases until photoactivated
Having established that STREAMs can be used to spatially and temporally detect cancer cell-derived MMP activity in situ, we derived a method to measure protease activity in vivo. First, we assayed whether DMNPE-veiled STREAMs were protected in the context of the enzyme milieu present in living animals. To this end, we adapted the STREAM paradigm for use with the synthetic biomarker platform recently developed in our lab, which provides a urinary readout of in vivo proteolysis.21 Synthetic biomarkers are comprised of peptide-reporter tandem conjugates that are coupled to a nanoparticle core. These protease nanosensors are infused intravenously and passively accumulate at sites of disease. Proteolysis of the peptide substrate liberates the reporter, which accumulates in the urine and can be quantified by mass spectrometry or ELISA.21–23
For the in vivo studies, we utilized our previous approaches for engineering ligand-encoded urinary reporters and companion ELISAs.23,24 This urinary reporter is comprised of a poly(ethylene-glycol) element (PEG; 5 kDa) that efficiently clears into the urine,24 and bears a fluorescein group, and a biotin, enabling detection in the urine via a sandwich ELISA for the reporter (sequence: Biotin-PEG(5kDa)-(KFAM)-PLGLEEA-GC; reporter: Biotin-PEG(5kDa)-(KFAM); name: V1).22,23 This reporter element is released upon proteolysis and clears into the urine for quantification (SI Fig. 8). The custom sandwich ELISA exhibited high sensitivity, as it detected low picomolar concentrations of the reporter (SI Fig. 9a). This peptide-reporter element is coupled to PEGylated (20 kDa) NPs and modified with DMNPE in the same manner as in vitro STREAMs. All in vivo experiments were performed with the V1 substrate coupled to nanoparticles.
To assay their performance in vivo, equivalent concentrations (by peptide) of unmodified synthetic biomarkers and STREAM synthetic biomarkers were injected intravenously into healthy Swiss Webster mice (SI Fig. 9b), and urine was collected thirty minutes after nanoparticle infusion. We observed a significant decrease in the reporter release from STREAM synthetic biomarkers (>4 fold) in healthy mice (Fig. 5a). To confirm that the protecting group modification was the source of the dampened urinary signal, in a separate cohort of animals, we infused STREAMs that were pre-activated ex vivo to induce photolysis of DMNPE (SI Fig. 9c), and observed that the majority of the signal reduction associated with veiled peptides was lost (~3 fold recovery). The observation that veiled particles yield a lower urine signal in healthy animals suggested that STREAM synthetic biomarkers are protected from cleavage in circulation. We validated this hypothesis by incubating C1-NPs with recombinant thrombin, an ubiquitous plasma protease essential for blood clotting, and noted reduced cleavage of the substrate (SI Fig. 10). Thus, the application of STREAMs to protease-sensitive synthetic biomarkers had the potential to enable improved specificity in protease measurements by localizing the sites of activation.
Figure 5. In vivo STREAMs for urinary measurements of protease activity.
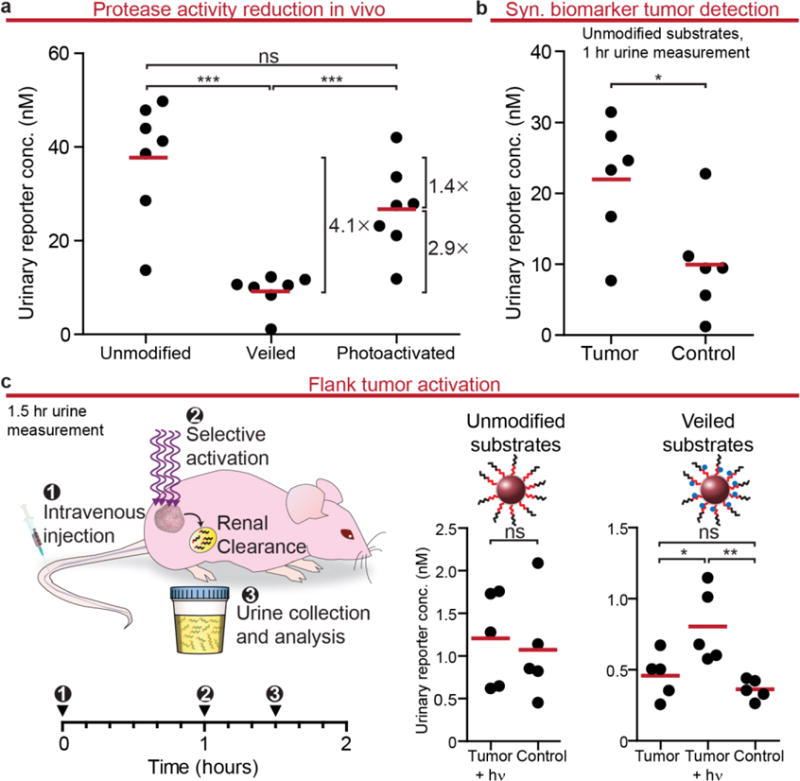
(a) V1-NPs were veiled with DMNPE and injected into healthy mice. This resulted in ~4 fold decrease in signal compared to unmodified substrates. Ex vivo activation and subsequent infusion into mice resulted in a signal increase of ~3 fold (***P<0.001, two-tail Student’s t-test). (b) Urinary reporter concentrations from tumor mice were significantly greater than healthy mice confirming that V1-NPs could be used as synthetic biomarkers of cancer (*P<0.05, two-tail Student’s t-test). (c) One hour after NP injection, mice were voided of urine and STREAMs were activated at the tumor. Urine was collected thirty minutes after. Approximately two-fold increase could be detected with the addition of light at the tumor. The same protocol was followed using unmodified substrates. There was no significant difference between the tumor animals and the control animals with unmodified substrates being exposed to light at this 1.5 hour time point owing to rapid depletion of available substrates (*P<0.05, **P<0.01, two-tail Student’s t-test; light exposure: 30s at 200 mW/cm2).
Photoactivated STREAMs measure protease activity in the tumor microenvironment
With the adaptation of STREAMs for use in vivo, we sought to apply this platform to interrogate protease activity of the tumor microenvironment. Since the novel peptide sequence (V1) had yet to be validated within the synthetic biomarker framework to detect cancer, we first tested its capacity to distinguish healthy mice from those bearing bilateral flank human colorectal cancer xenografts. Our previous work identified an optimal time frame in which to perform urinary measurements to achieve signal sepearation between tumor-bearing and healthy mice.23,29 At early timepoints (minutes), signal is primarily generated by blood-borne protease activity as nanoparticles need longer periods in order to accumulate at the tumor site via the enhanced permeability and retention (EPR) effect.41–43 At later timepoints (hours), the vast majority of administered substrates have been consumed in both tumor and healthy controls, dampening any distinguishable signal between the two groups.29 Therefore, with an optimized timepoint of one hour post administration of V1-NPs, a significantly higher reporter signal was present in the urine of tumor-bearing mice one hour after infusion, validating the use of this peptide as a synthetic biomarker for cancer (Fig. 5b).
We sought to detect the levels of tumor-associated protease activity in vivo, via transdermal activation of STREAMs. We first needed to confirm that light penetration through skin is adequate to activate STREAMs. To this end, we developed an agarose gel embedded with recombinant MMP9 and STREAMs (SI Fig. 11a) with similar transmittance at 365 nm as skin (10% vs. 17%;44 SI Fig. 11b). A brief light exposure (1 minute) of the tissue model resulted in dramatic increase in proteolytic cleavage of the sensors, suggesting that transdermal activation is feasible in vivo (SI Fig. 11c).
Using the in vivo tumor model employed above, we implanted bilateral flank human colorectal tumors and injected veiled STREAM synthetic biomarkers intravenously. In this approach, the STREAMs are protected from cleavage in blood and other organs, including the tumor, unless selectively unveiled by exposure to light. Thus, by shining light on tumor-bearing flanks, subsequent reporter release should be mediated by the elevated protease concentration in the vicinity of the tumor (Fig. 5c). To test this hypothesis, one hour after injection, urine was voided to eliminate reporters that had already accumulated by non-specific protease cleavage. STREAMs were activated by illuminating the tumor site for 30 seconds per flank, and urine was collected again thirty minutes after exposure. Unmodified synthetic biomarkers, following this protocol, were unable to distinguish between tumor and healthy animals, due to rapid depletion of the substrate within the first hour and to greater noise generated by blood-borne protease cleavage. This result that unprotected synthetic biomarkers, using this substrate, are unable to distinguish between tumor and healthy mice at late timepoints is supported by previous work, which characterizes the importance of the timepoint for urine measurement.29 This waning sensitivity is due to a diminished signal separation that occurs over time, as this class of substrates are susceptible to cleavage by background proteases. Alternate substrates that are more resistant to background proteases would not suffer from this drawback.29 Therefore, another benefit of the STREAMs approach is that it provides greater temporal flexibility in when urine samples are collected, as the kinetics of the experiment are externally controlled by initiating activation with light. In contrast to unmodified synthetic biomarkers, a significantly higher signal was present in the urine of tumor-bearing mice after light activation of STREAM synthetic biomarkers when compared to the non-illuminated cohort (2.1 fold). This finding suggests that STREAMs were activated at the tumor by light and cleaved by tumor-associated proteases. The urine signals obtained from the light activated group were also significantly higher than the STREAM derived signal observed in healthy animals without light treatment (2.6 fold; Fig. 5c). This signal enhancement is consistent with our previous work, but in the case of STREAMs, is associated with proteases in the tumor bed as opposed to tumor-derived proteases secreted into the bloodstream.29 In order to test whether UV exposure itself had an impact on the proteolysis of unmodified substrates, we tested urine in mice with and without light exposure and observed no significant differences of the urine signals collected in each case (SI Fig. 12). Collectively, STREAM synthetic biomarkers enable the tissue specific detection of protease activity in vivo with simple quantifications in the urine.
One important aspect of our approach to consider is the choice of light source and the wavelength used for unveiling. We use a power density of approximately 200 mW/cm2 for a 30 second exposure. This dosing is similar to or lower than the power used in other examples of in vivo photoactivation that maintain cellular viability, 44–46 and thus have been cited as demonstrations of the safety of this approach for brief exposures. As photolabile chemistry advances to improve quantum yield of photolysis, these power requirements will diminish. Additionally, the use of UVA light (320–400 nm) vs UVB light (280–320 nm) is of importance as UVA light is a relatively poor tumor initiating agent,47 and UVA light is used clinically as a therapeutic for skin disease. Importantly, we demonstrated that our system is compatible with two-photon unveiling, which should benefit potential in vivo applications (SI Fig. 3). Furthermore, our group has shown that implantable light sources can be used to probe previously inaccessible tumors.48 For immediate applications, STREAMs have the potentional to help guide the development of therapeutics, as well as profile the invasive potential of tumors. As one example, there has been a growing interest in developing therapeutics antibodies that are unveiled in the tumor microenvironment due to proteolytic stimuli.49 By measuring activity in patient derived xenografts, STREAMs could be used to identify optimal substrates that can mask therapeutics, such that their specific release occurs only at tumor sites. This capacity may instill the STREAM platform with the potential to stratify protease-activated therapeutics based on tumor type and specific protease activity in vivo.
Conclusions
The STREAM approach is a simple, modular strategy for modifying peptide substrates with photolabile protecting groups, comprised of a nanoparticle backbone decorated with protease-sensitive peptides, which are veiled by photolabile small molecules. STREAMs show significant resistance to proteolysis prior to light activation. We demonstrate spatial and temporal control over STREAMs in vitro in 3D tumor models, and in human cancer xenografts in vivo. STREAMs are readily applicable to numerous clinical and basic biological questions and can be integrated with other protease activity technologies, as well as other enzymes. Overall, the application of photolabile protecting groups to mediate the remote control of activity-based sensors in vivo and in vitro provides a powerful and sought after tool for use in a broad range of biological measurements that can complement the myriad existing methods for external modulation of biological systems.50–53
Materials and Methods
Synthesis of peptides/reporters and NPs
Fluorescein-conjugated peptides (MMP sensitive, C1: FAM-sk-PLGLEEA-GC) were either synthesized by the Koch Institute Proteomics Core or the Tufts University Peptide Core. D-amino acid controls were also synthesized, where the substrate sequence was all D-stereoisomers. Peptides for in vivo studies that contain a ligand-encoded reporter for urinary clearance and subsequent ELISA detection were synthesized by CPC Scientific, Inc (V1: Biotin-PEG(5kDa)-(KFAM)-PLGLEEA-GC). The PEG 5 kDa reporter is efficiently cleared by the renal system into the urine and can be quantifed by ELISA for the conjugated ligands. The alternate substrate to show STREAMs extensibility was synthesized at Tufts University Peptide Core (sequence: eGvndneeGffsarKsRLVGEGC). VT750 (Perkin Elmer) was conjugated to the free lysine prior to coupling to DMNPE. DMNPE can indeed react with numerous glutamic acids throughout the tandem peptide, necestating a high DMNPE:peptide excess of 100:1.
Nanoparticles were formed by reacting iron(III) chloride hexahydrate and iron(II) chloride tetrahydrate with dextran as previously described.43 Nanoparticles were aminated by reacting with ammonium hydroxide. Size measurements were performed by dynamic light scattering (Malvern Instruments Nano ZS90) revealed a mean diameter less than 100 nm. NPs were reacted with a 500-fold molar excess of N-succinimidyl iodoacetate (SIA) (Pierce) for one hour at room temperature in 50 mM sodium borate, pH 8.3, 5 mM EDTA to provide thiol reactive handles. Excess SIA was removed either by fast-protein liquid chromatography (FPLC, GE Healthcare) or by spin-filters (MWCO = 30 kDa, Millipore). SIA-NPs were reacted with peptide substrate-reporter complexes at a 1:95 ratio in the borate buffer overnight at room temperature. For the in vivo particles, mPEG thiol (20 kDa, Laysan) was also reacted with at a 20 molar excess ratio to NPs to provide stability and prevent phagocytic uptake. After purification and buffer exchange into PBS, peptide-reporter valency was quantified by absorbance. For strong quenching, valency greater than 20 was needed. NP-peptide-reporter complexes were stored at 4 °C.
Conjugation of DMNPE to peptides and peptide-NPs
Peptides were coupled to DMNPE either before or after conjugation to NPs. DMNPE was generated using the DMNPE generation kit (Life Technologies) according to manufacturer protocols. DMNPE was then allowed to react with peptides in a 50:50 DMSO to PBS ratio overnight on a shaker with excess DMNPE. After the reaction was completed, excess DMNPE was removed either by high-pressure liquid chromatography (HPLC) or by FPLC/spin filters (if peptide was already coupled to NPs). Confirmation of modification was either verified by absorbance changes (DMNPE has a max absorbance around 350 nm) or by mass spectrometry.
Mass spectrometry analysis of peptide-DMNPE
After purification by HPLC, peptide-DMNPE was analyzed by mass spectrometry by ESI-MS. DMNPE (MW = 209.66 Da) presence was confirmed by a mass shift from the peptide mass. Typical MALDI analysis cannot be used to detect DMNPE as the MALDI laser operates at the same wavelength as DMNPE max absorbance. Therefore, to demonstrate, that DMNPE can be removed by light treatment, the MALDI analysis was performed on the same peptide-DMNPE complex showing a mass shift back to the original peptide mass.
In vitro recombinant protease assays
C1-NP complexes sensitive to MMP cleavage were mixed with 1% (wt/vol) BSA (Sigma) and incubated with recombinant proteases (MMPs and ADAMs: Enzo Life Sciences; Clotting proteases: Haematologic Technologies) in a final volume of 100 μL in enzyme specific buffers (MMP buffer: 50 mM Tris, 150 mM NaCl, 5 mM CaCl2, 1μM ZnCl2, pH 7.5; Clotting proteases: PBS) in a 384 well plate for time-lapse fluorimetry to measure dequenching from homoquenched peptides at 37 °C (SpectroMax Gemini EM Microplate Reader). For the metalloproteases, enzymes were diluted 1:10 in enzyme specific buffer and for clotting proteases, enzymes were diluted 1:100. Cleavage heatmap was generated using GENE-E (Broad Institute). Michaelis-Menten constants were determined by assessing initial cleavage velocities at different substrate concentrations. The MMP inhibitor Marimatstat (Tocris) was added to mixture at 100 μM final concentration. To identify the cleavage position by MMP9, C1-NPs were incubated with MMP9 overnight and the N-terminal cleavage fragment was isolated and analyzed by MALDI. The sequence corresponding to the dominant peak was identified and the final amino acid was in that sequence represents the P1 position (towards the N-terminal end from scissile bond). For protease resistance assays, various DMNPE:peptide ratios were reacted overnight and purified prior to being added to proteases.
Light activation of peptides
Light activation of peptides for biochemical studies was performed using a CL-1000 UV Crosslinker (UVP, 8 mW/cm2). Power density was measured by an OAI 306 UV power meter at 365 nm. Typical exposure time for these studies was 10–30 minutes. For activation in cell and animal studies, Lumen Dynamics UV system with 365 nm fiber light guide was used (OmniCure 1000, 200 mW/cm2). For in vivo activation at the tumor site, mice were anesthetized and the light was guided through an optical cable and placed approximately 3 cm from the flank tumor. Each flank tumor was exposed for 30 seconds.
Two-photon unveiling was performed at the KI Microscopy Core with a multiphoton microscope (Olympus FV-1000MP) operating at 690 nm with a Spectra-Physics Deepsea Tia-sapphire laswer at power 1 W using a 25× objective with 1.05 NA. Samples were placed in glass bottom 384 well plates. Images were captured at 840 nm.
Cell culture and secreted protease activity assay
LS174T and CCD-18Co (ATCC CRL-1459) cells were cultured in Eagle’s Minimal Essential Medium (ATCC) supplemented with 10% FBS (Gibco) and 1% penicillin-streptomycin (CellGro). Cells were passaged when confluence reached 80%. To isolate secreted proteases, after cells were plated, cells were washed and replaced in serum-free media. Conditioned media was collected 24 hours later and exposed to C1-NPs to measure fluorescence dequenching.
3D tissue engineering models
LS174T cells were encapsulated in 2.5 mg/mL collagen hydrogels (Rat tail collagen Type I, Corning). Imaging was done on Nikon Eclipse Ti Inverted Microscopes and Zeiss Stereoscope Discovery v20. When protease activity was measured, surrounding media was serum-free.
Agarose gel assay
Agarose (Type I-A, Sigma) was dissolved in MMP9 specific buffer (1% w/v) and heated. As the gel mixture was cooling, gel solution was transferred into a 96 well plate and mixed with STREAMs and recombinant MMP9. After gelation, the gels were activated (as above) and fluorescence dequenching through cleavage was monitored using time-lapse fluorimetry.
In vivo wildtype animal studies
The in vivo STREAM synthetic biomarkers (V1-NPs) were diluted to 1 μM in sterile PBS. Wild-type, female Swiss Webster mice (4–6 wk, Taconic) were infused intravenously via the tail vein. Immediately after infusion, mice were placed in an in-house devised urine collector with a 96 well plate base. To quantify level of protection, unmodified synthetic biomarkers were also injected. Additionally, for a third group, STREAMs were activated prior to injection. Thirty minutes post-injection, urine was collected and stored at −80 °C.
For analysis, urine was diluted from 100× to 10,000× in PBS BSA (1%). Reporter concentration was quantified by a custom designed and characterized ELISA as described by our group previously.22,23 Briefly, α-FITC antibodies (GeneTex) were used as the capture antibody at the bottom of a high-binding 96 well plate. NeutrAvidin-HRP (Pierce) was used as the detection antibody to recognize the N-terminal biotin on the reporter. Bound HRP was exposed to Ultra-TMB (Pierce) substrate and the reaction was allowed to progress. The reaction was quenched when the ladder could be visualized using 1 M HCl. Absorbance was measured at 450 nm using a plate reader (Molecular Devices SpectraMax Plus).
Flank tumor model of colorectal cancer
Female NCr Nude mice (4–6 week, Taconic) were inoculated subcutaneously with 3 × 106 LS174T cells per flank and allowed to grow. Two weeks after inoculation, the mice were infused with the STREAMs. Tumor bearing mice and age-matched controls were infused with STREAM synthetic biomarkers and placed in urine collectors. After 1 hour, the mice were voided of urine. A fraction of these animals were exposed to light over the flank tumors as described above. All animals were infused with 0.5 mL of PBS subcutaneously to increase urine production at 1 hour. The animals were placed back into urine collectors. Urine from all animals was collected 30 minutes later and analyzed as described above. Unmodified synthetic biomarkers were also infused in a different cohort of mice and a similar set of operations was performed.
Supplementary Material
Acknowledgments
We thank H. Fleming (MIT) for critical reading and editing of the manuscript, A. Warren (MIT) for insightful discussions and technical guidance, K. Moynihan (MIT) for discussion on two photon unveiling, Jeff Wyckoff for assistance with two photon experiments, and the Koch Biopolymers & Proteomics Core for assistance. This work was supported in part by the MIT Deshpande Center for Technological Innovation, the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute, and the Koch Institute Frontier Research Program through the Koch Institute Frontier Research Fund. J.S.D. thanks the National Science Foundation Graduate Research Fellowship Program for support. G.A.K. holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund. S.N.B. is a Howard Hughes Medical Institute Investigator.
Footnotes
Supporting Information Available. Supplementary figures 1–12. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mata J, Marguerat S, Bähler J. Post-Transcriptional Control of Gene Expression: A Genome-Wide Perspective. Trends Biochem Sci. 2005;30:506–514. doi: 10.1016/j.tibs.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y-C, Peterson SE, Loring JF. Protein Post-Translational Modifications and Regulation of Pluripotency in Human Stem Cells. Cell Res. 2014;24:143–160. doi: 10.1038/cr.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cravatt BF, Wright AT, Kozarich JW. Activity-Based Protein Profiling: From Enzyme Chemistry to Proteomic Chemistry. Annu Rev Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 4.Kobe B, Kemp BE. Active Site-Directed Protein Regulation. Nature. 1999;402:373–376. doi: 10.1038/46478. [DOI] [PubMed] [Google Scholar]
- 5.López-Otín C, Bond JS. Proteases: Multifunctional Enzymes in Life and Disease. J Biol Chem. 2008;283:30433–30437. doi: 10.1074/jbc.R800035200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khersonsky O, Tawfik DS. Enzyme Promiscuity: A Mechanistic and Evolutionary Perspective. Annu Rev Biochem. 2010;79:471–505. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- 7.Fortelny N, Cox JH, Kappelhoff R, Starr AE, Lange PF, Pavlidis P, Overall CM. Network Analyses Reveal Pervasive Functional Regulation Between Proteases in the Human Protease Web. PLoS Biol. 2014;12:e1001869. doi: 10.1371/journal.pbio.1001869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orgaz JL, Pandya P, Dalmeida R, Karagiannis P, Sanchez-Laorden B, Viros A, Albrengues J, Nestle FO, Ridley AJ, Gaggioli C, et al. Diverse Matrix Metalloproteinase Functions Regulate Cancer Amoeboid Migration. Nat Commun. 2014;5 doi: 10.1038/ncomms5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmalfeldt B, Prechtel D, Härting K, Späthe K, Rutke S, Konik E, Fridman R, Berger U, Schmitt M, Kuhn W, et al. Increased Expression of Matrix Metalloproteinases (MMP)-2, MMP-9, and the Urokinase-Type Plasminogen Activator Is Associated with Progression from Benign to Advanced Ovarian Cancer. Clin Cancer Res. 2001;7:2396–2404. [PubMed] [Google Scholar]
- 10.Bachovchin DA, Brown SJ, Rosen H, Cravatt BF. Identification of Selective Inhibitors of Uncharacterized Enzymes by High-Throughput Screening with Fluorescent Activity-Based Probes. Nat Biotechnol. 2009;27:387–394. doi: 10.1038/nbt.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans MJ, Cravatt BF. Mechanism-Based Profiling of Enzyme Families. Chem Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]
- 12.Bachovchin DA, Koblan LW, Wu W, Liu Y, Li Y, Zhao P, Woznica I, Shu Y, Lai JH, Poplawski SE, et al. Nat Chem Biol. advance online publication; 2014. A High-Throughput, Multiplexed Assay for Superfamily-Wide Profiling of Enzyme Activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kato D, Boatright KM, Berger AB, Nazif T, Blum G, Ryan C, Chehade KAH, Salvesen GS, Bogyo M. Activity-Based Probes That Target Diverse Cysteine Protease Families. Nat Chem Biol. 2005;1:33–38. doi: 10.1038/nchembio707. [DOI] [PubMed] [Google Scholar]
- 14.Miller MA, Meyer AS, Beste MT, Lasisi Z, Reddy S, Jeng KW, Chen CH, Han J, Isaacson K, Griffith LG et al. ADAM-10 and -17 Regulate Endometriotic Cell Migration via Concerted Ligand and Receptor Shedding Feedback on Kinase Signaling. Proc Natl Acad Sci. 2013;110:E2074–E2083. doi: 10.1073/pnas.1222387110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen C-H, Miller MA, Sarkar A, Beste MT, Isaacson KB, Lauffenburger DA, Griffith LG, Han J. Multiplexed Protease Activity Assay for Low-Volume Clinical Samples Using Droplet-Based Microfluidics and Its Application to Endometriosis. J Am Chem Soc. 2013;135:1645–1648. doi: 10.1021/ja307866z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitney M, Savariar EN, Friedman B, Levin RA, Crisp JL, Glasgow HL, Lefkowitz R, Adams SR, Steinbach P, Nashi N, et al. Ratiometric Activatable Cell-Penetrating Peptides Provide Rapid In Vivo Readout of Thrombin Activation. Angew Chem Int Ed. 2013;52:325–330. doi: 10.1002/anie.201205721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hilderbrand SA, Weissleder R. Near-Infrared Fluorescence: Application to in Vivo Molecular Imaging. Curr Opin Chem Biol. 2010;14:71–79. doi: 10.1016/j.cbpa.2009.09.029. [DOI] [PubMed] [Google Scholar]
- 18.Weissleder R, Tung C-H, Mahmood U, Bogdanov A. In Vivo Imaging of Tumors with Protease-Activated near-Infrared Fluorescent Probes. Nat Biotechnol. 1999;17:375–378. doi: 10.1038/7933. [DOI] [PubMed] [Google Scholar]
- 19.Olson ES, Jiang T, Aguilera TA, Nguyen QT, Ellies LG, Scadeng M, Tsien RY. Activatable Cell Penetrating Peptides Linked to Nanoparticles as Dual Probes for in Vivo Fluorescence and MR Imaging of Proteases. Proc Natl Acad Sci. 2010;107:4311–4316. doi: 10.1073/pnas.0910283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Welser K, Adsley R, Moore BM, Chan WC, Aylott JW. Protease Sensing with Nanoparticle Based Platforms. Analyst. 2010;136:29–41. doi: 10.1039/c0an00429d. [DOI] [PubMed] [Google Scholar]
- 21.Kwong GA, von Maltzahn G, Murugappan G, Abudayyeh O, Mo S, Papayannopoulos IA, Sverdlov DY, Liu SB, Warren AD, Popov Y, et al. Mass-Encoded Synthetic Biomarkers for Multiplexed Urinary Monitoring of Disease. Nat Biotechnol. 2013;31:63–70. doi: 10.1038/nbt.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin KY, Kwong GA, Warren AD, Wood DK, Bhatia SN. Nanoparticles That Sense Thrombin Activity as Synthetic Urinary Biomarkers of Thrombosis. ACS Nano. 2013 doi: 10.1021/nn403550c. 130909231146001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warren AD, Kwong GA, Wood DK, Lin KY, Bhatia SN. Point-of-Care Diagnostics for Noncommunicable Diseases Using Synthetic Urinary Biomarkers and Paper Microfluidics. Proc Natl Acad Sci. 2014;111:3671–3676. [Google Scholar]
- 24.Warren AD, Gaylord ST, Ngan KC, Dumont Milutinovic M, Kwong GA, Bhatia SN, Walt DR. Disease Detection by Ultrasensitive Quantification of Microdosed Synthetic Urinary Biomarkers. J Am Chem Soc. 2014 doi: 10.1021/ja505676h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shah S, Jain PK, Kala A, Karunakaran D, Friedman SH. Light-Activated RNA Interference Using Double-Stranded siRNA Precursors Modified Using a Remarkable Regiospecificity of Diazo-Based Photolabile Groups. Nucleic Acids Res. 2009;37:4508–4517. doi: 10.1093/nar/gkp415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williger B-T, Reich R, Neeman M, Bercovici T, Liscovitch M. Release of Gelatinase A (Matrix Metalloproteinase 2) Induced by Photolysis of Caged Phosphatidic Acid in HT 1080 Metastatic Fibrosarcoma Cells. J Biol Chem. 1995;270:29656–29659. doi: 10.1074/jbc.270.50.29656. [DOI] [PubMed] [Google Scholar]
- 27.Jain PK, Shah S, Friedman SH. Patterning of Gene Expression Using New Photolabile Groups Applied to Light Activated RNAi. J Am Chem Soc. 2011;133:440–446. doi: 10.1021/ja107226e. [DOI] [PubMed] [Google Scholar]
- 28.Devel L, Rogakos V, David A, Makaritis A, Beau F, Cuniasse P, Yiotakis A, Dive V. Development of Selective Inhibitors and Substrate of Matrix Metalloproteinase-12. J Biol Chem. 2006;281:11152–11160. doi: 10.1074/jbc.M600222200. [DOI] [PubMed] [Google Scholar]
- 29.Kwong GA, Dudani JS, Carrodeguas E, Mazumdar EV, Zekavat SM, Bhatia SN. Mathematical Framework for Activity-Based Cancer Biomarkers. Proc Natl Acad Sci. 2015 doi: 10.1073/pnas.1506925112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schroeder A, Goldberg MS, Kastrup C, Wang Y, Jiang S, Joseph BJ, Levins CG, Kannan ST, Langer R, Anderson DG. Remotely Activated Protein-Producing Nanoparticles. Nano Lett. 2012;12:2685–2689. doi: 10.1021/nl2036047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monroe WT, McQuain MM, Chang MS, Alexander JS, Haselton FR. Targeting Expression with Light Using Caged DNA. J Biol Chem. 1999;274:20895–20900. doi: 10.1074/jbc.274.30.20895. [DOI] [PubMed] [Google Scholar]
- 32.DelPrincipe F, Egger M, Niggli E. Calcium Signalling in Cardiac Muscle: Refractoriness Revealed by Coherent Activation. Nat Cell Biol. 1999;1:323–329. doi: 10.1038/14013. [DOI] [PubMed] [Google Scholar]
- 33.Soeller C, Jacobs MD, Donaldson PJ, Cannell MB, Jones KT, Ellis-Davies GCR. Application of Two-Photon Flash Photolysis to Reveal Intercellular Communication and Intracellular Ca2+ Movements. J Biomed Opt. 2003;8:418–427. doi: 10.1117/1.1582468. [DOI] [PubMed] [Google Scholar]
- 34.Jain PK, Karunakaran D, Friedman SH. Construction of a Photoactivated Insulin Depot. Angew Chem Int Ed. 2013;52:1404–1409. doi: 10.1002/anie.201207264. [DOI] [PubMed] [Google Scholar]
- 35.Trzcinska R, Balin K, Kubacki J, Marzec ME, Pedrys R, Szade J, Silberring J, Dworak A, Trzebicka B. Relevance of the Poly(ethylene Glycol) Linkers in Peptide Surfaces for Proteases Assays. Langmuir. 2014;30:5015–5025. doi: 10.1021/la500457q. [DOI] [PubMed] [Google Scholar]
- 36.Adjémian J, Anne A, Cauet G, Demaille C. Cleavage-Sensing Redox Peptide Monolayers for the Rapid Measurement of the Proteolytic Activity of Trypsin and Α-Thrombin Enzymes. Langmuir. 2010;26:10347–10356. doi: 10.1021/la100397g. [DOI] [PubMed] [Google Scholar]
- 37.Abbasi AZ, Amin F, Niebling T, Friede S, Ochs M, Carregal-Romero S, Montenegro J-M, Rivera Gil P, Heimbrodt W, Parak WJ. How Colloidal Nanoparticles Could Facilitate Multiplexed Measurements of Different Analytes with Analyte-Sensitive Organic Fluorophores. ACS Nano. 2011;5:21–25. doi: 10.1021/nn1034026. [DOI] [PubMed] [Google Scholar]
- 38.Brand K, Baker AH, Perez-Cantó A, Possling A, Sacharjat M, Geheeb M, Arnold W. Treatment of Colorectal Liver Metastases by Adenoviral Transfer of Tissue Inhibitor of Metalloproteinases-2 into the Liver Tissue. Cancer Res. 2000;60:5723–5730. [PubMed] [Google Scholar]
- 39.Takawa M, Masuda K, Kunizaki M, Daigo Y, Takagi K, Iwai Y, Cho HS, Toyokawa G, Yamane Y, Maejima K, et al. Validation of the Histone Methyltransferase EZH2 as a Therapeutic Target for Various Types of Human Cancer and as a Prognostic Marker. Cancer Sci. 2011;102:1298–1305. doi: 10.1111/j.1349-7006.2011.01958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Infanger DW, Lynch ME, Fischbach C. Engineered Culture Models for Studies of Tumor-Microenvironment Interactions. Annu Rev Biomed Eng. 2013;15:29–53. doi: 10.1146/annurev-bioeng-071811-150028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang AZ, Langer R, Farokhzad OC. Nanoparticle Delivery of Cancer Drugs. Annu Rev Med. 2012;63:185–198. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]
- 42.Hrkach J, Hoff DV, Ali MM, Andrianova E, Auer J, Campbell T, Witt DD, Figa M, Figueiredo M, Horhota A, et al. Preclinical Development and Clinical Translation of a PSMA-Targeted Docetaxel Nanoparticle with a Differentiated Pharmacological Profile. Sci Transl Med. 2012;4:128ra39–ra128ra39. doi: 10.1126/scitranslmed.3003651. [DOI] [PubMed] [Google Scholar]
- 43.Park J-H, von Maltzahn G, Zhang L, Derfus AM, Simberg D, Harris TJ, Ruoslahti E, Bhatia SN, Sailor MJ. Systematic Surface Engineering of Magnetic Nanoworms for in Vivo Tumor Targeting. Small Weinh Bergstr Ger. 2009;5:694–700. doi: 10.1002/smll.200801789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tong R, Chiang HH, Kohane DS. Photoswitchable Nanoparticles for in Vivo Cancer Chemotherapy. Proc Natl Acad Sci. 2013;110:19048–19053. doi: 10.1073/pnas.1315336110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li L, Tong R, Chu H, Wang W, Langer R, Kohane DS. Aptamer Photoregulation in Vivo. Proc Natl Acad Sci. 2014;111:17099–17103. doi: 10.1073/pnas.1420105111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee TT, García JR, Paez JI, Singh A, Phelps EA, Weis S, Shafiq Z, Shekaran A, del Campo A, García AJ. Light-Triggered in Vivo Activation of Adhesive Peptides Regulates Cell Adhesion, Inflammation and Vascularization of Biomaterials. Nat Mater. 2015;14:352–360. doi: 10.1038/nmat4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bowden GT. Prevention of Non-Melanoma Skin Cancer by Targeting Ultraviolet-B-Light Signalling. Nat Rev Cancer. 2004;4:23–35. doi: 10.1038/nrc1253. [DOI] [PubMed] [Google Scholar]
- 48.Bagley AF, Hill S, Rogers GS, Bhatia SN. Plasmonic Photothermal Heating of Intraperitoneal Tumors through the Use of an Implanted Near-Infrared Source. ACS Nano. 2013;7:8089–8097. doi: 10.1021/nn4033757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EEM, Liang TW, Wong C, Bessette PH, Kamath K, Moore SJ, et al. Tumor-Specific Activation of an EGFR-Targeting Probody Enhances Therapeutic Index. Sci Transl Med. 2013;5:207ra144–ra207ra144. doi: 10.1126/scitranslmed.3006682. [DOI] [PubMed] [Google Scholar]
- 50.Deisseroth K. Optogenetics. Nat Methods. 2011;8:26–29. doi: 10.1038/nmeth.f.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee SE, Sasaki DY, Park Y, Xu R, Brennan JS, Bissell MJ, Lee LP. Photonic Gene Circuits by Optically Addressable siRNA-Au Nanoantennas. ACS Nano. 2012;6:7770–7780. doi: 10.1021/nn301744x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chien Y-H, Chou Y-L, Wang S-W, Hung S-T, Liau M-C, Chao Y-J, Su C-H, Yeh C-S. Near-Infrared Light Photocontrolled Targeting, Bioimaging, and Chemotherapy with Caged Upconversion Nanoparticles in Vitro and in Vivo. ACS Nano. 2013;7:8516–8528. doi: 10.1021/nn402399m. [DOI] [PubMed] [Google Scholar]
- 53.Lee S, Park H, Kyung T, Kim NY, Kim S, Kim J, Heo WD. Reversible Protein Inactivation by Optogenetic Trapping in Cells. Nat Methods. 2014;11:633–636. doi: 10.1038/nmeth.2940. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.