

This review by Dalal and Johnson focuses on the evolutionary rewiring of transcription regulators and the conservation of patterns of gene expression. They describe how preservation of gene expression patterns in the wake of extensive rewiring is a general feature of transcription circuit evolution.
Keywords: evolution, gene regulation, transcription
Abstract
Transcription regulators bind to cis-regulatory sequences and thereby control the expression of target genes. While transcription regulators and the target genes that they regulate are often deeply conserved across species, the connections between the two change extensively over evolutionary timescales. In this review, we discuss case studies where, despite this extensive evolutionary rewiring, the resulting patterns of gene expression are preserved. We also discuss in silico models that reach the same general conclusions and provide additional insights into how this process occurs. Together, these approaches make a strong case that the preservation of gene expression patterns in the wake of extensive rewiring is a general feature of transcription circuit evolution.
All cells respond to changes in their environment by altering expression of their genes. For a number of specific cases, we have a detailed, mechanistic explanation of how these expression changes occur and we understand, at least in broad strokes, how this process generally works across a wide variety of species. It is often assumed that gene expression patterns—and the underlying mechanisms that produce them—are logical, efficient, and orderly. Here, we review evidence—from both experimental and in silico analyses—that challenges this view. In its place, we suggest that an emphasis on the evolutionary history of transcription circuits better accounts for many of their properties, especially those that seem otherwise counterintuitive. In this review, we concentrate on one of those properties: the preservation of gene expression patterns across species despite wide-scale changes in the mechanisms that produce these patterns. Without insight from evolutionary studies, one might have assumed—incorrectly—that a deeply conserved pattern of gene expression would always be produced from a deeply conserved circuit.
The outputs of transcription circuits are often preserved across species, while the underlying mechanisms change
Many proteins are conserved throughout all branches of life. For example, the three enzymes that convert the sugar galactose into glucose are very similar among bacteria, archaebacteria, and eukaryotes (Caputto et al. 1950; Cardini et al. 1950; Isselbacher et al. 1956; Wilson and Hogness 1964; Frey 1996). Across this diverse group of organisms, the environmental signal that induces production of these enzymes—galactose—is also the same. Because the enzymes are conserved and their expression pattern is conserved, it would seem reasonable to assume that the underlying mechanism would also be conserved. As we see here, this is not the case; instead, the underlying circuitry changes much more rapidly over evolutionary time than do the enzymes themselves or their expression patterns. As we discuss in this review, this trend holds for many gene expression patterns, and we summarize evidence indicating that it is an inevitable consequence of the ways in which transcription circuits evolve.
The expression level of a given protein-coding gene depends on its rate of transcription, RNA processing, and translation and the rate of mRNA and protein degradation. For this review, we consider only the first step (transcriptional initiation) and only one aspect of this step: the association of transcription regulators (also called transcription factors) with cis-regulatory sequences. Here we define transcription regulators as sequence-specific DNA-binding proteins that control the expression of specific genes by binding to DNA sequences called cis-regulatory sequences (Fig. 1A). We consider a transcription circuit as the connections between a given transcription regulator and all of the genes that it regulates through direct binding to cis-regulatory sequences. Cis-regulatory sequences are typically short degenerate sequences that are relatively simple to gain or lose through mutation (Monod and Jacob 1961; Wray 2007; Payne and Wagner 2014). Losses by point mutation are simple to understand, but gains can also occur readily by point mutation: Given that cis-regulatory sequences are short and can act at many positions along DNA, there are many “near-miss” sequences available in a stretch of DNA that can be converted to functional cis-regulatory sequences by a simple point mutation. Indeed, gains are estimated to be nearly as frequent as losses (Force et al. 2005; Moses et al. 2006; Lynch 2007a; Tuch et al. 2008a).
Figure 1.
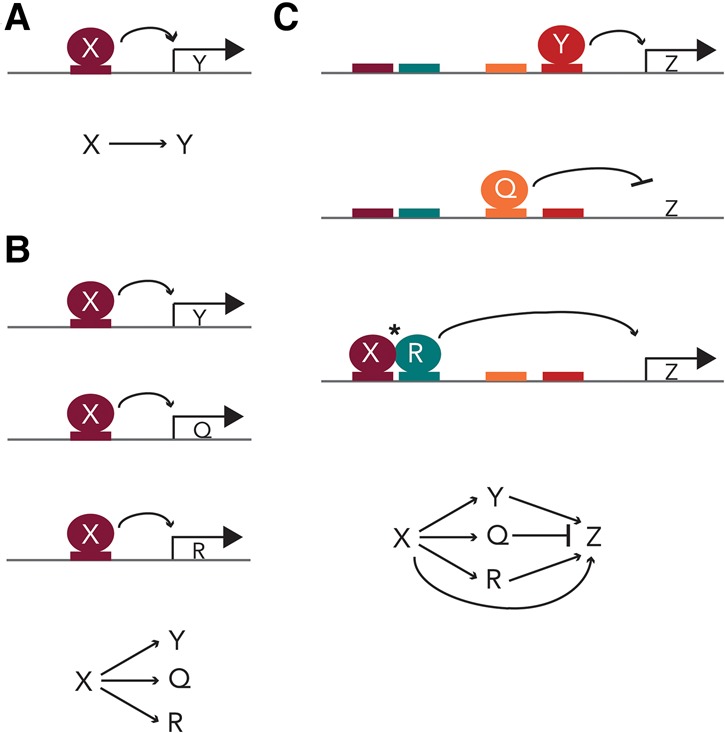
Transcription circuits can be described in many ways. (A) In the simplest cases, a transcription regulator, X (depicted as a burgundy circle), binds to its cognate cis-regulatory element (also depicted in burgundy) in the intergenic region of gene Y, inducing its expression. The interaction between X and Y is typically described with an arrow. (B) In simple circuits, a transcription regulator, X, can bind and induce the expression of multiple genes through cognate cis-regulatory elements in their intergenic regions. These circuits can also be depicted in network diagrams by arrows. (C) In most cases, intergenic regions contain multiple cis-regulatory elements so that multiple transcriptional regulators can combine to regulate (either induce or repress, as indicated by a blunt line) gene expression. Some regulators bind independently to their cis-regulatory sequences; others (X and R in this panel) assemble cooperatively, often through relatively weak protein–protein interactions (denoted by the asterisk).
The potential for rapid gains and losses of cis-regulatory sequences is one of several features contributing to the evolutionary rewiring of transcription circuits (Carroll 2000; Wray 2003; Tuch et al. 2008a). By evolutionary rewiring, we refer to genetic differences in regulator–target gene connections from one species to another when the regulators and the target genes are conserved in both species. The connections occur through direct binding of the regulator to cis-regulatory sequences controlling expression of the target gene. The term “transcriptional rewiring” is sometimes used in the literature to indicate a change in the transcriptional program as development proceeds, but we use the term in a different sense, referring to genetic differences in transcription circuits between one species and another. Despite extensive evolutionary rewiring among even closely related species (Tsong et al. 2006; Borneman et al. 2007; Martchenko et al. 2007; Tuch et al. 2008b; Lavoie et al. 2009; Weirauch and Hughes 2010; Tirosh et al. 2011; Nocedal and Johnson 2015; Villar et al. 2015; Nocedal et al. 2017), the output from these circuits often remains relatively constant. In the next sections, we discuss specific examples from fungi that illustrate this idea.
Sex determination in ascomycete fungi
The ascomycetes include Saccharomyces cerevisiae, used in baking and brewing; Kluyveromyces lactis, used in cheese production; and Candida albicans, the most prevalent fungal pathogen of humans. They span a nominal evolutionary distance of 300 million years and encompass hundreds of individual species. All (or nearly all) exist in three cell types: a, α, and a/α cells, where a cells mate with α cells to form a/α cells (for review, see Ni et al. 2011). The difference between these three cell types results from the expression of sets of cell type-specific genes, one set unique to each of the three cell types. For example, the a-specific genes are expressed only in a cells, and it is the expression of these genes that allows them to mate with α cells. There are approximately seven a-specific genes, with the exact number depending on the species; they are conserved across the ascomycete lineage, and their functions have been extensively studied (Dohlman and Thorner 2001). In S. cerevisiae, the a-specific genes are expressed constitutively and repressed in α and a/α cells by the transcription regulator MATα2 (Strathern et al. 1980; Johnson and Herskowitz 1985); this protein binds directly to its cis-regulatory sequence, which is found upstream of each a-specific gene (Fig. 2A). In C. albicans, an entirely different regulatory scheme is used (Fig. 2B): Here, the a-specific genes are activated by the transcription regulator MATa2, which is made only in a cells (Tsong et al. 2003). Thus, these two species use opposite mechanisms of control: negative in S. cerevisiae and positive in C. albicans. Moreover, the regulatory proteins themselves are different from one another; the repressor (MATα2 in S. cerevisiae) is a homeodomain protein, and the activator (MATa2 in C. albicans) is a HMG domain protein (Weirauch and Hughes 2011). In species branching between S. cerevisiae and C. albicans, some a-specific genes are regulated by both mechanisms (Fig. 2C; Baker et al. 2012). Moreover, the particular a-specific genes under such dual control vary from one species to the next. Thus, there are a large number of ways to regulate the a-specific genes, ranging from purely negative to purely positive and including various dual combinations (Fig. 2). Despite all of these differences, the general circuit output has remained the same: Only a cells express the a-specific genes. Although it is currently impossible to rule out every alternative possibility, a sound working hypothesis holds that there is no special advantage in regulating the a-specific genes one way or another; what is important is they be expressed only in a cells.
Figure 2.
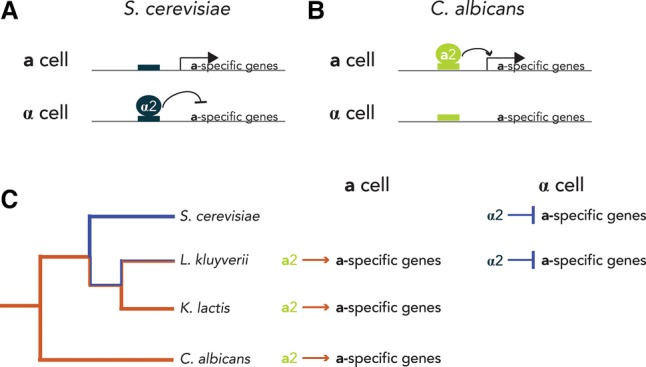
Transcription circuits can evolve through intermediates. (A) In S. cerevisiae, the a-specific genes are regulated via negative control. Here the transcription regulator MATα2 (blue circle) represses the a-specific genes in α cells by binding to its cis-regulatory element (blue box) in the promoter regions of a-specific genes. In a cells, the a-specific genes are constitutively expressed. (B) In contrast, C. albicans uses positive regulation, in which the transcription regulator MATa2 (lime-green circle) induces the a-specific genes in a cells via direct binding. (C) In species intermediate to S. cerevisiae and C. albicans, dual forms of regulation are observed. For example, in Lachancea kluyveri, MATa2 induces the a-specific genes in a cells (orange arrow), and MATα2 represses the a-specific genes in α cells (blue bars).
Ribosomal protein production in fungi
The mechanism underlying the regulation of ribosomal protein genes also changes over evolutionary times. Because ribosome production is closely tied to growth control in most species, one might expect that the expression circuitry underlying ribosome regulation would also be deeply conserved. However, the regulatory proteins that control the ribosomal protein genes vary greatly across fungal species (Tanay et al. 2005; Tuch et al. 2008b; Lavoie et al. 2010). Although some of these changes might alter the response of ribosomal genes to growth conditions, they appear far more extensive than one might expect for small, species-specific modifications.
Sugar metabolism
Extensive transcription rewiring is also observed in the regulation of genes needed to metabolize sugars. As described at the beginning of this review, galactose transcriptionally induces the synthesis of the same three enzymes (a kinase, a transferase, and an epimerase) across a wide variety of species (Caputto et al. 1950; Cardini et al. 1950; Isselbacher et al. 1956; Wilson and Hogness 1964; Frey 1996). In S. cerevisiae, the galactose-mediated induction of the three enzymes (Gal1, Gal7, and Gal10) is positively controlled (>1000-fold) by the transcription regulator Gal4, which binds to cis-regulatory sequences upstream of each gene (Giniger et al. 1985). An unmistakable ortholog of Gal4 (also called Gal4) is found in C. albicans but it does not regulate the GAL genes (Martchenko et al. 2007). Instead, it regulates genes involved in glucose acquisition (Askew et al. 2009). In C. albicans, the induction of the three Gal enzymes by galactose is more modest (sixfold to 12-fold) and is controlled by the regulators Rtg1 and Rtg3 (Brown et al. 2009; Dalal et al. 2016). In the intermediate species K. lactis, Gal4 regulates production of the three enzymes, but the induction range is intermediate (125-fold to 150-fold) between S. cerevisiae and C. albicans (Rubio-Texeira 2005; Hittinger and Carroll 2007). Although the quantitative aspects of galactose-mediated induction have changed across species, the general circuit output (transcriptional induction of the three Gal enzymes by galactose) has remained the same despite a switch in the key transcription regulators.
Nucleotide metabolism
In S. cerevisiae, the transcription regulator Ppr1 induces the expression of four URA genes in response to uracil starvation (Loison et al. 1980; Losson and Lacroute 1983; Roy et al. 1990). These genes encode deeply conserved enzymes that synthesize uracil de novo (Lacroute 1968; Floyd and Jones 1985). C. albicans has a Ppr1 ortholog but it does not regulate the URA genes (Tebung et al. 2016). Instead, Ppr1 regulates the breakdown of allantoin into ammonia and carbon dioxide, enabling C. albicans to use allantoin as a sole nitrogen source (Vogels and Van der Drift 1976; Tebung et al. 2016). Currently, it is not known how the C. albicans URA genes are regulated.
For both uracil synthesis and allantoin catabolism, the basic circuit output is preserved between S. cerevisiae and C. albicans: In the absence of uracil, the genes necessary to synthesize uracil are induced. In the presence of allantoin, the genes necessary to catabolize allantoin are induced. These outputs are maintained despite the switch in the role of Ppr1.
Meiosis and sporulation in the ascomycetes
The observation that circuit output can remain the same despite a reshuffling of the key transcription regulators is not limited to small, well-defined transcription circuits (Fig. 1B). For example, the transcription regulator Ndt80 activates hundreds of sporulation and meiosis genes in S. cerevisiae (Hepworth et al. 1998) and is required to complete meiosis (Xu et al. 1995). Ndt80 is needed for meiosis in multiple ascomycete species (Nocedal et al. 2017), including K. lactis (120 million years diverged from S. cerevisiae) and Pichia pastoris (210 million years diverged from S. cerevisiae). ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) analysis of Ndt80 across multiple species shows that, in all species, the Ndt80 regulon is large and consists predominantly of deeply conserved genes; however, the genes controlled by Ndt80 differ greatly across their entire species, with little overlap between them. The large-scale movement of target genes in and out of Ndt80 control involves hundreds of gains and losses of cis-regulatory sequences. Again, the overall circuit output is preserved (Ndt80 is needed for meiosis and sporulation) despite these wide-scale changes.
Examples from animals
Ascomycete yeasts are a convenient model system for understanding the evolution of transcription circuits (Hughes and de Boer 2013; Muñoz et al. 2016). However, do the conclusions hold for nonyeast species? Studies in flies, some of which predate the yeast work discussed above, led to similar ideas regarding transcription circuit flexibility. Analysis of the stripe 2 enhancer of even-skipped (eve), an important developmental gene in Drosophila melanogaster, provides one of the earliest examples. eve encodes a homeodomain protein that is expressed in precise spatial (stripe) and temporal patterns during embryonic development (Nüsslein-Volhard and Wieschaus 1980; Macdonald et al. 1986; Patel et al. 1994). The cis-regulatory sequences are grouped into a series of modular enhancers, each controlling a particular stripe or combination of stripes (Small et al. 1992). The module that specifies the second stripe of eve expression (moving from anterior to posterior) is “read” (through direct DNA binding) by the maternal morphogen Bicoid and the gap proteins Hunchback, Krüppel, and Giant. Ludwig et al. (1998) compared the 480-base-pair (bp) stripe 2 module between D. melanogaster and Drosophila pseudoobscura, noting that there was relatively little primary sequence conservation. However, when the D. pseudoobscura enhancer was introduced into D. melanogaster, the D. melanogaster spatiotemporal pattern of striped gene expression was reproduced. Thus, despite major sequence differences between the enhancers (which changed the order and number of cis-regulatory sequences), circuit output (expression of eve in stripe 2) was preserved. When enhancers from more distantly related flies were examined, the differences were even more pronounced, with little DNA sequence similarity despite a conserved output (Hare et al. 2008). Although this example is not strictly rewiring as defined at the beginning of this review, it beautifully illustrates the flexibility of cis-regulatory sequences in producing a conserved output.
Analysis of the circuit controlling differentiation in the D. melanogaster eye provides another example. Here, the transcription regulator dPax2 is expressed specifically in cone cells, where it is required for proper differentiation of retinal cells into ommatidia (Fu and Noll 1997; Evans et al. 2012). A 362-bp enhancer (called the Sparkling enhancer) is responsible for this cone cell-specific expression (Fu and Noll 1997; Fu et al. 1998; Flores et al. 2000); it contains multiple cis-regulatory sequences for the Notch effector Suppressor of Hairless, the Ets family effectors of EGFR/MAPK signaling, and the Runx family transcription regulator Lozenge. All three regulatory inputs occur at the level of binding of the regulator to the enhancer, and all are necessary for cone cell-specific expression of dPax2 (Flores et al. 2000). When the sparkling enhancer was compared across Drosophila species, primary sequence conservation was very low (Fig. 3; Evans et al. 2012); in particular, the distribution and number of cis-regulatory sequences were different from species to species. For example, one species might have a high-affinity binding site for a transcription regulator, where another species might have a series of low-affinity binding sites for that same regulator. However, the sparkling enhancer from other species, when moved into D. melanogaster, still yielded the D. melanogaster expression pattern: expression only in cone cells (Swanson et al. 2010, 2011). Thus, although the DNA sequence and arrangement of cis-regulatory sites in the Sparkling enhancer vary from species to species, it retains the ability to specify a particular expression pattern in not only its cognate species but D. melanogaster as well.
Figure 3.
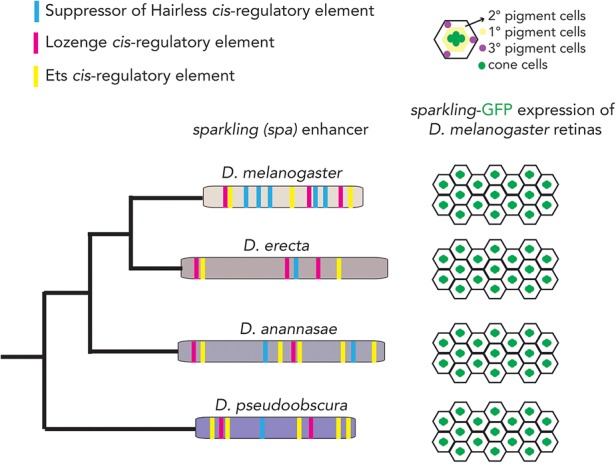
Transcription circuits can evolve in ways that preserve their output. The D. melanogaster sparkling enhancer (from the intergenic region of dPax2) and orthologous enhancers from related Drosophila species are shown at the left. There is very little primary sequence similarity among enhancers, as depicted by the different background colors. Binding sites for the transcription regulators Lozenge, Suppressor of Hairless, and Ets are depicted as magenta, cyan, and yellow boxes, respectively. As shown at the right, expression from this enhancer yields cone cell-specific expression in D. melanogaster. Despite the low overall DNA sequence similarity, cone cell-specific expression is maintained when orthologous sparkling enhancers are expressed in D. melanogaster.
Studies in mammalian species have also revealed surprising plasticity in transcription circuits. For example, C/EBPα, a bZIP transcription regulator, is expressed in many cells and is essential for proper differentiation in various tissues, including the liver (Graves et al. 1986; Landschulz et al. 1988; Xanthopoulos et al. 1989; Williams et al. 1991; Scott et al. 1992; Ohlsson et al. 2016). ChIP-seq analysis of this regulator in liver tissue of five vertebrate species (humans, mice, dogs, opossums, and chickens, spanning nominally 80 million years of evolution) showed that CEBPα binds to tens of thousands of target genes in every species. However, only a few dozen of these binding connections were conserved across all five species (Schmidt et al. 2010). It is difficult to rigorously document the role of C/EBPα in liver development in all of these species; it is also difficult to determine which C/EBPα target gene connections (as determined by ChIP) are important for this process. However, despite the apparently large changes in C/EBPα target-binding patterns across species (occurring primarily through gains and losses of its cis-regulatory sequence), liver development proceeds in all five species (Schmidt et al. 2010).
Taken together, the case studies illustrate that the general output of transcription circuits is often preserved across large evolutionary distances in yeast, flies, and vertebrates despite extensive changes in the way that key regulators are connected to their target genes. This principle seems to apply to both small (mating, galactose metabolism, and nucleotide metabolism) and large (meiosis, ribosome production, and liver development) transcription circuits.
How extensive is transcriptional rewiring?
Thus far, we argued here that transcription rewiring is common, and, in this section, we review evidence (predominantly from full-genome experimental and computational studies) that gives a rough idea of the frequency. Based on several independent studies in ascomycetes, it is estimated that, on average, ∼15% of the connections between a transcription regulator and its target genes in S. cerevisiae will be preserved in C. albicans (for example, see Borneman et al. 2007; Tuch et al. 2008b; Habib et al. 2012; Sarda and Hannenhalli 2015; Nocedal et al. 2017). Of course, the exact number depends on the particular regulator examined (as well as the methodologies used), but this rough average is a reasonable starting place for appreciating the overall extent of transcriptional rewiring. It is important to note that these studies do not distinguish between rewiring that is neutral and rewiring that is adaptive.
Other studies have examined the extent of evolutionary transcriptional rewiring across groups of animal species. By combining results from several studies, Carvunis et al. (2015) estimated that the rate of evolutionary rewiring (normalized to years of divergence) was approximately the same across insects, birds, and mammals. In very rough terms, for two species diverged by 100 million years, the binding pattern of a transcriptional regulator in one species is ∼10% preserved in the other. This number is of the same order of magnitude as that observed across fungal species and again represents a useful first approximation. We emphasize that this is a rough overall average, with individual transcriptional regulators showing considerable variation around this approximation. We also note that as methodologies improve for more accurately mapping functional regulator target gene connections, these numbers will continue to be refined.
Insights from in silico approaches
In previous sections, we described experimental studies of transcription circuits across multiple species. Here we review in silico approaches that independently lead to many of the same conclusions, often predating the experimental work. For example, Ciliberti et al. (2007a,b) set up simulations so that any gene (A or X in Fig. 4, top) can activate, repress, or have no effect on the expression of another gene, Y, as determined by the cis-regulatory sequences near gene Y. The construction of large in silico circuits from these simpler networks showed that they often had hundreds of “neighbors” where the output of the transcription circuit was preserved. A neighbor is simply a new circuit that differs from its precursor by one regulatory connection, caused by either a loss or gain of a cis-regulatory sequence. The analysis demonstrated that a given output can be produced by many different underlying transcription circuits, all of which can be formed one step at a time from the ancestral circuit. Because most of the steps leading to new neighbors do not destroy the output of the circuit, multistep exploration of many circuit configurations can occur without a loss of function. Thus, one can start with any given circuit configuration, change one interaction at a time, and reach many other circuit configurations, all while preserving general circuit output (Ciliberti et al. 2007a; Payne and Wagner 2014). These models predict the existence of multiple circuit architectures underlying the same output, an idea that is strongly supported by the experimental work.
Figure 4.
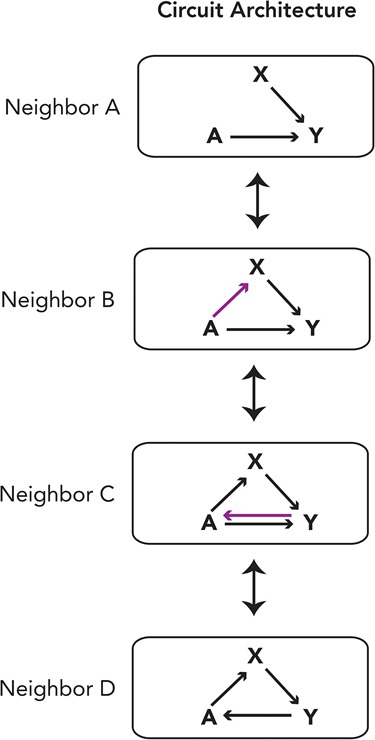
Transcription circuits can evolve one interaction at a time. There are many ways to preserve the output of a transcription circuit (induction of Y). These variants can be categorized and clustered by their circuit architecture. Four different neighboring circuit variants, all comprising three proteins (A, X, and Y), are displayed. The circuits are the same except for one wiring interaction (shown in purple) that varies among them. With additional components, one can easily imagine how changing wiring interactions one at a time can yield many different circuits.
We do not know the detailed evolutionary history of any extant circuit but we do have a reasonably detailed history of one—the a-specific gene circuit in ascomycete yeasts. The evolution of the circuit proceeded through intermediates that preserved the overall output of the circuit (Baker et al. 2012), a result consistent with the in silico analysis.
Rewiring that preserves output: neutral or adaptive?
Three different mechanisms can, in principle, account for the cases discussed in this review, where the output of a transcription circuit was preserved despite extensive changes in the connections between the transcription regulators and their target genes. (1) The changes could have made the circuit function “better” in those species where they occurred, (2) the changes could have resulted from a tradeoff that made something else in the cell function better, or (3) the circuit could have changed neutrally, with no particular immediate advantage for the new configuration. For the circuits discussed in this review, it is not currently possible to rigorously distinguish among these three possibilities; however, it is possible to make a few generalizations. First, neutral evolution is likely to be responsible for many of the circuit differences observed because (1) there are strong theoretical bases for this statement, (2) there are many experimental results consistent with the idea, and (3) there are currently no experimental results that rule it out as a broad explanation for many of the observed changes. Neutral evolution is predicted to be especially important in small effective population sizes where such changes are expected to predominate over adaptive changes (Lynch et al. 2006; Lynch 2007a,b). As discussed above, neutral evolution expands the number of circuit configurations that can be explored; indeed, the term “constructive neutral evolution” is used sometimes to convey this point (Stoltzfus 1999; Gray et al. 2010; Doolittle 2013). This idea can readily account for the change in the eve stripe 2 and Sparkling enhancers that have accumulated across fly species and the differences in the way that the a-specific genes are regulated across the ascomycete yeasts. The alternative hypothesis would hold that, for each species, there is a special advantage of its particular configuration over the others. Although this hypothesis cannot be rigorously excluded, it requires many specific assumptions, none of which currently have experimental support.
On the other hand, the analysis of GAL gene regulation shows that although the overall circuit output of these genes is maintained, the quantitative features of GAL gene induction are different from species to species (Rubio-Texeira 2005), ranging from 1000-fold induction of only a few genes in S. cerevisiae (St John and Davis 1981) to 10-fold induction of many genes in C. albicans (Dalal et al. 2016). Rtg1 and Rtg3 (the GAL regulators in C. albicans) are likely to be the ancestral GAL regulators, so it is a reasonable hypothesis that the increase in the magnitude of GAL gene induction and the reduction of the regulon size resulted at least in part from the rewiring of the GAL genes from Rtg1 and Rtg3 to Gal4. It seems likely that this change in the dynamics and structure of the GAL regulon was ultimately adaptive (in any case, there is a clear species difference). However, to date, there is no evidence that rigorously demonstrates this idea.
How far from the ancestral role do transcriptional regulators fall?
Although the number of case studies is limited (and probably not statistically significant), the transcription regulators that have been most carefully studied in fungi have maintained a loose connection with their ancestral roles. For example, although the genes regulated by Gal4 are completely different in S. cerevisiae (GAL genes) and C. albicans (glucose utilization genes), Gal4 remains associated with sugar metabolism (Ren et al. 2000; Askew et al. 2009). In the case of Ppr1, the difference is between pyrimidine biosynthesis in S. cerevisiae (Loison et al. 1981) and purine catabolism in C. albicans (Tebung et al. 2016); hence, Ppr1 has remained associated with nucleotide metabolism. For Ndt80, the shift seems more dramatic: from regulating meiosis and sporulation in S. cerevisiae (Xu et al. 1995) to biofilm formation in C. albicans (Nobile et al. 2012). However, in both cases, there is regrouping of individual cells to form higher-order structures, suggesting some overall relationship between the two processes. We emphasize again that this is a very small sample size with dubious statistical significance; however, the idea that, despite extensive rewiring, transcription regulators may be constrained from traversing too far from their original roles is an intriguing one (Fig. 5).
Figure 5.
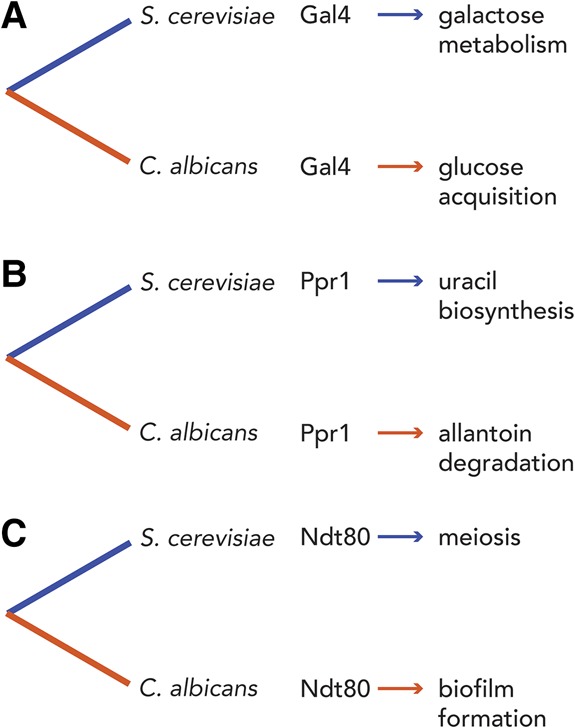
Rewired transcription regulators remain associated with similar processes. Here we show three deeply conserved transcription regulators and the different functions that they have become associated with as they have gained and lost interactions with target genes between S. cerevisiae (blue lines) and C. albicans (orange lines). In all three cases, the DNA-binding specificity of the regulator remains unchanged across the two species. (A) Gal4 induces genes needed to metabolize galactose in S. cerevisiae and genes needed for glucose acquisition in C. albicans. (B) Ppr1 induces genes necessary to synthesize uracil in S. cerevisiae and genes necessary to metabolize allantoin in C. albicans. (C) Ndt80 is necessary to complete meiosis in S. cerevisiae and form biofilms in C. albicans.
Conclusions
Here we reviewed case studies across yeasts, flies, and mammals in which the overall output of a transcription circuit has been preserved across species despite many changes in the connections between transcription regulators and their target genes. We also pointed out that this feature was predicted from in silico experiments, where simulations showed that transcription circuits could traverse multiple configurations while still preserving a specific output. The convergence of theoretical and experimental approaches makes a strong case that this rapid evolutionary movement is an inherent feature of transcription circuits and that, in a sense, transcription circuits should be viewed as works in progress rather than perfected solutions. Indeed, there are likely to be many different circuit configurations for a given output. Although many of the explorations of alternative configurations probably occur neutrally, they produce a continuous level of useful variation on which selection can subsequently act.
Acknowledgments
We thank Nairi Hartooni for comments on the manuscript, Ishita Shah for advice on the figures, and Ethel Enoex-Godonoo for help editing the manuscript. Work from the authors’ laboratory was supported by grants R01 AI049187 and R01 GMO37049 from the National Institutes of Health.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.303362.117.
References
- Askew C, Sellam A, Epp E, Hogues H, Mullick A, Nantel A, Whiteway M. 2009. Transcriptional regulation of carbohydrate metabolism in the human pathogen Candida albicans. PLoS Pathog 5: e1000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker C, Booth L, Sorrells T, Johnson A. 2012. Protein modularity, cooperative binding, and hybrid regulatory states underlie transcriptional network diversification. Cell 151: 80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borneman AR, Gianoulis TA, Zhang ZD, Yu H, Rozowsky J, Seringhaus MR, Wang LY, Gerstein M, Snyder M. 2007. Divergence of transcription factor binding sites across related yeast species. Science 317: 815–819. [DOI] [PubMed] [Google Scholar]
- Brown V, Sabina J, Johnston M. 2009. Specialized sugar sensing in diverse fungi. Curr Biol 19: 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputto R, Leloir LF, Cardini CE, Paladini AC. 1950. Isolation of the coenzyme of the galactose phosphate-glucose phosphate transformation. J Biol Chem 184: 333–350. [PubMed] [Google Scholar]
- Cardini CE, Paladini AC, Caputto R, Leloir LF. 1950. Liver uridine phosphorylase. Acta Physiol Lat Am 1: 57–63. [PubMed] [Google Scholar]
- Carroll SB. 2000. Endless forms: the evolution of gene regulation and morphological diversity. Cell 101: 577–580. [DOI] [PubMed] [Google Scholar]
- Carvunis A-R, Wang T, Skola D, Yu A, Chen J, Kreisberg JF, Ideker T. 2015. Evidence for a common evolutionary rate in metazoan transcriptional networks. Elife 4: e11615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciliberti S, Martin OC, Wagner A. 2007a. Innovation and robustness in complex regulatory gene networks. Proc Natl Acad Sci 104: 13591–13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciliberti S, Martin OC, Wagner A. 2007b. Robustness can evolve gradually in complex regulatory gene networks with varying topology. PLoS Comput Biol 3: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalal CK, Zuleta IA, Mitchell KF, Andes DR, El-Samad H, Johnson AD. 2016. Transcriptional rewiring over evolutionary timescales changes quantitative and qualitative properties of gene expression. Elife 5: e18981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohlman HG, Thorner JW. 2001. Regulation of G protein-initiated signal transduction in yeast: paradigms and principles. Annu Rev Biochem 70: 703–754. [DOI] [PubMed] [Google Scholar]
- Doolittle WF. 2013. Is junk DNA bunk? A critique of ENCODE. Proc Natl Acad Sci 110: 5294–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans NC, Swanson CI, Barolo S. 2012. Sparkling insights into enhancer structure, function, and evolution. Curr Top Dev Biol 98: 97–120. [DOI] [PubMed] [Google Scholar]
- Flores GV, Duan H, Yan H, Nagaraj R, Fu W, Zou Y, Noll M, Banerjee U. 2000. Combinatorial signaling in the specification of unique cell fates. Cell 103: 75–85. [DOI] [PubMed] [Google Scholar]
- Floyd EE, Jones ME. 1985. Isolation and characterization of the orotidine 5′-monophosphate decarboxylase domain of the multifunctional protein uridine 5′-monophosphate synthase. J Biol Chem 260: 9443–9451. [PubMed] [Google Scholar]
- Force A, Cresko WA, Pickett FB, Proulx SR, Amemiya C, Lynch M. 2005. The origin of subfunctions and modular gene regulation. Genetics 170: 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey PA. 1996. The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J 10: 461–470. [PubMed] [Google Scholar]
- Fu W, Noll M. 1997. The Pax2 homolog sparkling is required for development of cone and pigment cells in the Drosophila eye. Genes Dev 11: 2066–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, Duan H, Frei E, Noll M. 1998. shaven and sparkling are mutations in separate enhancers of the Drosophila Pax2 homolog. Development 125: 2943–2950. [DOI] [PubMed] [Google Scholar]
- Giniger E, Varnum SM, Ptashne M. 1985. Specific DNA binding of GAL4, a positive regulatory protein of yeast. Cell 40: 767–774. [DOI] [PubMed] [Google Scholar]
- Graves BJ, Johnson PF, McKnight SL. 1986. Homologous recognition of a promoter domain common to the MSV LTR and the HSV tk gene. Cell 44: 565–576. [DOI] [PubMed] [Google Scholar]
- Gray MW, Lukes J, Archibald JM, Keeling PJ, Doolittle WF. 2010. Irremediable complexity? Science 330: 920–921. [DOI] [PubMed] [Google Scholar]
- Habib N, Wapinski I, Margalit H, Regev A, Friedman N. 2012. A functional selection model explains evolutionary robustness despite plasticity in regulatory networks. Mol Syst Biol 8: 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare EE, Peterson BK, Iyer VN, Meier R, Eisen MB. 2008. Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet 4: e1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepworth SR, Friesen H, Segall J. 1998. NDT80 and the meiotic recombination checkpoint regulate expression of middle sporulation-specific genes in Saccharomyces cerevisiae. Mol Cell Biol 18: 5750–5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger CT, Carroll SB. 2007. Gene duplication and the adaptive evolution of a classic genetic switch. Nature 449: 677–681. [DOI] [PubMed] [Google Scholar]
- Hughes TR, de Boer CG. 2013. Mapping yeast transcriptional networks. Genetics 195: 9–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isselbacher KJ, Anderson EP, Kurahashi K, Kalckar HM. 1956. Congenital galactosemia, a single enzymatic block in galactose metabolism. Science 123: 635–636. [DOI] [PubMed] [Google Scholar]
- Johnson AD, Herskowitz I. 1985. A repressor (MATα 2 product) and its operator control expression of a set of cell type specific genes in yeast. Cell 42: 237–247. [DOI] [PubMed] [Google Scholar]
- Lacroute F. 1968. Regulation of pyrimidine biosynthesis in Saccharomyces cerevisiae. J Bacteriol 95: 824–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landschulz WH, Johnson PF, McKnight SL. 1988. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science 240: 1759–1764. [DOI] [PubMed] [Google Scholar]
- Lavoie H, Hogues H, Whiteway M. 2009. Rearrangements of the transcriptional regulatory networks of metabolic pathways in fungi. Curr Opin Microbiol 12: 655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie H, Hogues H, Mallick J, Sellam A, Nantel A, Whiteway M. 2010. Evolutionary tinkering with conserved components of a transcriptional regulatory network. PLoS Biol 8: e1000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loison G, Losson R, Lacroute F. 1980. Constitutive mutants for orotidine 5 phosphate decarboxylase and dihydroorotic acid dehydrogenase in Saccharomyces cerevisiae. Curr Genet 2: 39–44. [DOI] [PubMed] [Google Scholar]
- Loison G, Jund R, Nguyen-Juilleret M, Lacroute F. 1981. Evidence for transcriptional regulation of dihydroorotic acid dehydrogenase in Saccharomyces cerevisiae. Curr Genet 3: 119–123. [DOI] [PubMed] [Google Scholar]
- Losson R, Lacroute F. 1983. Plasmids carrying the yeast OMP decarboxylase structural and regulatory genes: transcription regulation in a foreign environment. Cell 32: 371–377. [DOI] [PubMed] [Google Scholar]
- Ludwig MZ, Patel NH, Kreitman M. 1998. Functional analysis of eve stripe 2 enhancer evolution in Drosophila: rules governing conservation and change. Development 125: 949–958. [DOI] [PubMed] [Google Scholar]
- Lynch M. 2007a. The origins of genome architecture. Sinauer Associates, Sunderland, MA. [Google Scholar]
- Lynch M. 2007b. The evolution of genetic networks by non-adaptive processes. Nat Rev Genet 8: 803–813. [DOI] [PubMed] [Google Scholar]
- Lynch M, Koskella B, Schaack S. 2006. Mutation pressure and the evolution of organelle genomic architecture. Science 311: 1727–1730. [DOI] [PubMed] [Google Scholar]
- Macdonald PM, Ingham P, Struhl G. 1986. Isolation, structure, and expression of even-skipped: a second pair-rule gene of Drosophila containing a homeo box. Cell 47: 721–734. [DOI] [PubMed] [Google Scholar]
- Martchenko M, Levitin A, Hogues H, Nantel A, Whiteway M. 2007. Transcriptional rewiring of fungal galactose-metabolism circuitry. Curr Biol 17: 1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Jacob F. 1961. Teleonomic mechanisms in cellular metabolism, growth, and differentiation. Cold Spring Harb Symp Quant Biol 26: 389–401. [DOI] [PubMed] [Google Scholar]
- Moses AM, Pollard DA, Nix DA, Iyer VN, Li X-Y, Biggin MD, Eisen MB. 2006. Large-scale turnover of functional transcription factor binding sites in Drosophila. PLoS Comput Biol 2: e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz A, Santos Muñoz D, Zimin A, Yorke JA. 2016. Evolution of transcriptional networks in yeast: alternative teams of transcriptional factors for different species. BMC Genomics 17: 826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni M, Feretzaki M, Sun S, Wang X, Heitman J. 2011. Sex in fungi. Annu Rev Genet 45: 405–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobile CJ, Fox EP, Nett JE, Sorrells TR, Mitrovich QM, Hernday AD, Tuch BB, Andes DR, Johnson AD. 2012. A recently evolved transcriptional network controls biofilm development in Candida albicans. Cell 148: 126–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocedal I, Johnson AD. 2015. How transcription networks evolve and produce biological novelty. Cold Spring Harb Symp Quant Biol 80: 265–274. [DOI] [PubMed] [Google Scholar]
- Nocedal I, Mancera E, Johnson AD. 2017. Gene regulatory network plasticity predates a switch in function of a conserved transcription regulator. Elife 6: e23250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nüsslein-Volhard C, Wieschaus E. 1980. Mutations affecting segment number and polarity in Drosophila. Nature 287: 795–801. [DOI] [PubMed] [Google Scholar]
- Ohlsson E, Schuster MB, Hasemann M, Porse BT. 2016. The multifaceted functions of C/EBPα in normal and malignant haematopoiesis. Leukemia 30: 767–775. [DOI] [PubMed] [Google Scholar]
- Patel NH, Condron BG, Zinn K. 1994. Pair-rule expression patterns of even-skipped are found in both short- and long-germ beetles. Nature 367: 429–434. [DOI] [PubMed] [Google Scholar]
- Payne JL, Wagner A. 2014. The robustness and evolvability of transcription factor binding sites. Science 343: 875–877. [DOI] [PubMed] [Google Scholar]
- Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, et al. 2000. Genome-wide location and function of DNA binding proteins. Science 290: 2306–2309. [DOI] [PubMed] [Google Scholar]
- Roy A, Exinger F, Losson R. 1990. Cis- and trans-acting regulatory elements of the yeast URA3 promoter. Mol Cell Biol 10: 5257–5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Texeira M. 2005. A comparative analysis of the GAL genetic switch between not-so-distant cousins: Saccharomyces cerevisiae versus Kluyveromyces lactis. FEMS Yeast Res 5: 1115–1128. [DOI] [PubMed] [Google Scholar]
- Sarda S, Hannenhalli S. 2015. High-throughput identification of cis-regulatory rewiring events in yeast. Mol Biol Evol 32: 3047–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S, et al. 2010. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328: 1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott LM, Civin CI, Rorth P, Friedman AD. 1992. A novel temporal expression pattern of three C/EBP family members in differentiating myelomonocytic cells. Blood 80: 1725–1735. [PubMed] [Google Scholar]
- Small S, Blair A, Levine M. 1992. Regulation of even-skipped stripe 2 in the Drosophila embryo. EMBO J 11: 4047–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John TP, Davis RW. 1981. The organization and transcription of the galactose gene cluster of Saccharomyces. J Mol Biol 152: 285–315. [DOI] [PubMed] [Google Scholar]
- Stoltzfus A. 1999. On the possibility of constructive neutral evolution. J Mol Evol 49: 169–181. [DOI] [PubMed] [Google Scholar]
- Strathern JN, Spatola E, McGill C, Hicks JB. 1980. Structure and organization of transposable mating type cassettes in Saccharomyces yeasts. Proc Natl Acad Sci 77: 2839–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson CI, Evans NC, Barolo S. 2010. Structural rules and complex regulatory circuitry constrain expression of a Notch- and EGFR-regulated eye enhancer. Dev Cell 18: 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson CI, Schwimmer DB, Barolo S. 2011. Rapid evolutionary rewiring of a structurally constrained eye enhancer. Curr Biol 21: 1186–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay A, Regev A, Shamir R. 2005. Conservation and evolvability in regulatory networks: the evolution of ribosomal regulation in yeast. Proc Natl Acad Sci 102: 7203–7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebung WA, Choudhury BI, Tebbji F, Morschhäuser J, Whiteway M. 2016. Rewiring of the Ppr1 zinc cluster transcription factor from purine catabolism to pyrimidine biogenesis in the Saccharomycetaceae. Curr Biol 26: 1677–1687. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Wong KH, Barkai N, Struhl K. 2011. Extensive divergence of yeast stress responses through transitions between induced and constitutive activation. Proc Natl Acad Sci 108: 16693–16698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsong AE, Miller MG, Raisner RM, Johnson AD. 2003. Evolution of a combinatorial transcription circuit: a case study in yeasts. Cell 115: 389–399. [DOI] [PubMed] [Google Scholar]
- Tsong AE, Tuch BB, Li H, Johnson A. 2006. Evolution of alternative transcriptional circuits with identical logic. Nature 443: 415–420. [DOI] [PubMed] [Google Scholar]
- Tuch BB, Li H, Johnson AD. 2008a. Evolution of eukaryotic transcription circuits. Science 319: 1797–1799. [DOI] [PubMed] [Google Scholar]
- Tuch BB, Galgoczy DJ, Hernday AD, Li H, Johnson AD. 2008b. The evolution of combinatorial gene regulation in fungi. PLoS Biol 6: e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar D, Berthelot C, Aldridge S, Rayner TF, Lukk M, Pignatelli M, Park TJ, Deaville R, Erichsen JT, Jasinska AJ, et al. 2015. Enhancer evolution across 20 mammalian species. Cell 160: 554–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogels GD, Van der Drift C. 1976. Degradation of purines and pyrimidines by microorganisms. Bacteriol Rev 40: 403–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Hughes TR. 2010. Conserved expression without conserved regulatory sequence: the more things change, the more they stay the same. Trends Genet 26: 66–74. [DOI] [PubMed] [Google Scholar]
- Weirauch MT, Hughes TR. 2011. A catalogue of eukaryotic transcription factor types, their evolutionary origin, and species distribution. Subcell biochem 52: 25–73. [DOI] [PubMed] [Google Scholar]
- Williams SC, Cantwell CA, Johnson PF. 1991. A family of C/EBP-related proteins capable of forming covalently linked leucine zipper dimers in vitro. Genes Dev 5: 1553–1567. [DOI] [PubMed] [Google Scholar]
- Wilson DB, Hogness DS. 1964. The enzymes of the galactose operon in Escherichia coli. I. Purification and characterization of uridine diphosphogalactose 4-epimerase. J Biol Chem 239: 2469–2481. [PubMed] [Google Scholar]
- Wray GA. 2003. Transcriptional regulation and the evolution of development. Int J Dev Biol 47: 675–684. [PubMed] [Google Scholar]
- Wray GA. 2007. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet 8: 206–216. [DOI] [PubMed] [Google Scholar]
- Xanthopoulos KG, Mirkovitch J, Decker T, Kuo CF, Darnell JE. 1989. Cell-specific transcriptional control of the mouse DNA-binding protein mC/EBP. Proc Natl Acad Sci 86: 4117–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Ajimura M, Padmore R, Klein C, Kleckner N. 1995. NDT80, a meiosis-specific gene required for exit from pachytene in Saccharomyces cerevisiae. Mol Cell Biol 15: 6572–6581. [DOI] [PMC free article] [PubMed] [Google Scholar]