

Abstract
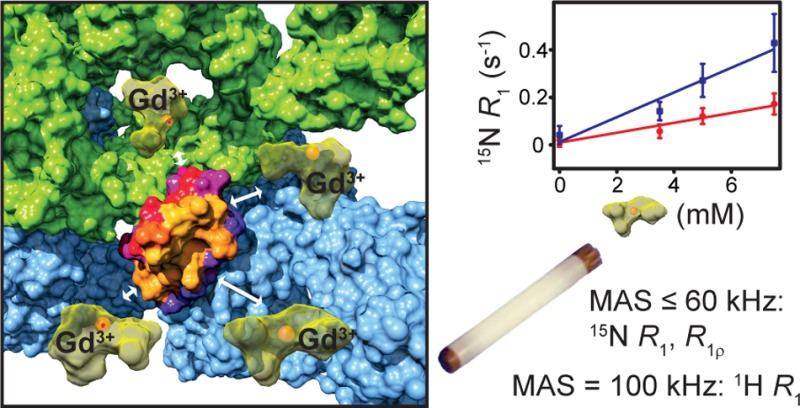
Solid-state NMR is becoming a viable alternative for obtaining information about structures and dynamics of large biomolecular complexes, including ones that are not accessible to other high-resolution biophysical techniques. In this context, methods for probing protein–protein interfaces at atomic resolution are highly desirable. Solvent paramagnetic relaxation enhancements (sPREs) proved to be a powerful method for probing protein–protein interfaces in large complexes in solution but have not been employed toward this goal in the solid state. We demonstrate that 1H and 15N relaxation-based sPREs provide a powerful tool for characterizing intermolecular interactions in large assemblies in the solid state. We present approaches for measuring sPREs in practically the entire range of magic angle spinning frequencies used for biomolecular studies and discuss their benefits and limitations. We validate the approach on crystalline GB1, with our experimental results in good agreement with theoretical predictions. Finally, we use sPREs to characterize protein–protein interfaces in the GB1 complex with immunoglobulin G (IgG). Our results suggest the potential existence of an additional binding site and provide new insights into GB1:IgG complex structure that amend and revise the current model available from studies with IgG fragments. We demonstrate sPREs as a practical, widely applicable, robust, and very sensitive technique for determining intermolecular interaction interfaces in large biomolecular complexes in the solid state.
Introduction
Knowledge of protein–protein interactions is essential for the understanding of many biological processes. However, atomic-resolution structural characterization of many important biomolecular complexes is impeded by their size, solubility, or ability to form crystals, preventing the application of standard methods such as solution NMR and X-ray crystallography. Solid-state NMR is an emerging alternative for studies of large protein assemblies,1−10 with new technologies and methods leading to continuously improved sensitivity and signal resolution for atomic-level structural information on large protein complexes.
Paramagnetic relaxation enhancement (PRE) occurs when an unpaired electron increases nuclear relaxation rates through dipolar interactions, which depend on the distance between the nucleus and the paramagnetic center. Strategies that make use of paramagnetic molecules help to alleviate the challenge of low sensitivity by enabling fast repetition of experiments and also provide a source of information about structures and dynamics.7,11−21 PREs have been used successfully in the solid state with the PREs obtained from paramagnetic tags attached to the proteins22 or by replacing non-paramagnetic ions with paramagnetic ions in metalloproteins.11,16,23,24 One potential disadvantage of such approaches is that introducing a non-native moiety into the protein can influence its structural integrity and/or dynamics, and such effects have to be considered carefully. In this context, employing an inert paramagnetic molecule dissolved in the solvent and which does not bind to the protein is less invasive and can provide long-range structural information with less potential for influencing the structure and dynamics of the studied system.25,26 If a paramagnetic compound, such as gadolinium diethylenetriaminepentaacetic acid bismethylamide (Gd(DTPA-BMA)), is added to the buffer surrounding the protein, the paramagnetic effects from such an agent, often called solvent PREs (sPREs), can be used to quantify solvent accessibility.27−30 sPREs have been used in solution NMR to gain additional restraints for structure calculation, prediction, and validation,31−33 to probe domain architecture,34 and to analyze protein–protein interactions.35
While paramagnetic agents are often used to speed up acquisition in solid-state NMR,36 there are few examples where sPREs have been used to study solvent accessibility in solid-state NMR, and, to our knowledge, they have not yet been employed for characterization of protein–protein interfaces in biomolecular complexes. However, sPREs have been used, e.g., to identify crystal contacts in a heavily deuterated crystalline sample of the α-spectrin domain of SH3.17 In that work, irregularly high 1H sPREs for residues in close proximity to exchangeable hydroxyl protons (<3.5 Å) were observed, leading to major challenges in the interpretation of distances between nuclei and protein surface.17 In another study, 13C R1-based sPREs were used to identify regions with increased solvent accessibility in Aβ1–40 fibrils.37 Because of still-active spin diffusion, which leads to the partial averaging of 13C R1 rates over several sites,38 the obtained sPREs were only qualitative in nature.37 Overall, elimination of spin diffusion is a prerequisite for obtaining quantitative site-specific sPREs. Suppression of spin diffusion can be achieved through either dilution of the strong dipolar proton–proton network through deuteration17 or fast magic angle spinning (MAS) or a combination of the two approaches.39
Here, we explored sPREs derived from 1H R1, 15N R1, and 15N R1ρ measurements in order to develop a practical approach for applying sPREs to characterize protein–protein interfaces in large complexes in the solid state. We considered several factors that can influence applicability of the approach, including signal-to-noise ratio, site-specific nature of measurements, sensitivity and dynamic range of the employed probe, and accessibility of specialized equipment.
To test the suitability of solid-state sPREs to map solvent accessibility, we have performed measurements on the B1 domain of immunoglobulin-binding protein G (GB1) in three different environments: isolated GB1 in solution (GB1free), GB1 in a crystal on its own (GB1cryst), and finally, GB1 in a precipitated complex with full-length IgG (GB1IgG). Protein G produced by group G and C streptococcci40 is part of the bacterial defense strategy against antibodies that enables the bacteria to escape detection by the host immune system.41 The high affinity between GB1 and IgG is commonly exploited in numerous biotechnological applications, such as immunosorbent assays or affinity purification of antibodies. Insights into molecular aspects of the complex can guide and support therapeutic strategies as well as bioengineering efforts. Differences in the solvent accessibility from sPREs revealed details of binding of GB1 to IgG and evidence for previously not observed additional interactions.
Results and Discussion
Overview of the Different sPRE Approaches
In solution NMR, sPREs can be obtained by measuring relaxation rates in a sample with increasing concentration of a paramagnetic dopant. The slope of the line obtained from fitting the relaxation rates as a function of dopant concentration yields the sPREs. The same approach can be used in the solid state, but with an individually prepared sample for each dopant concentration. The most popular paramagnetic dopant used in solid-state NMR applications is CuEDTA. However, complexes using EDTA as a chelator were shown to bind preferentially to some proteins due to their overall negative charge and thus introduce undesired bias in sPRE applications.42,43 Even though CuEDTA does not bind to either GB1 or GB1:IgG complex, in order to increase general applicability of the approach, we have decided to use a neutral probe for most of our measurements. Toward this aim, we employed Gd(DTPA-BMA), which is one of the most popular stable neutral paramagnetic probes for sPRE applications in solution NMR and a popular intravenous MRI contrast agent. An additional benefit of using this dopant instead of CuEDTA is that Gd3+ is much more efficient in inducing PREs compared to Cu2+, which means that much smaller concentrations of the dopant can be used to obtain a similar effect.44 Reducing the required dopant concentration aids, e.g., to minimize the rf induced heating.
We found that Gd(DTPA-BMA) can be added to hydrated protein samples in solid-state NMR after they were prepared in the required solid form, e.g., crystal, sediment, or precipitate, without need for co-crystallization of the proteins with the paramagnetic agent as it has been suggested previously.17 We did not observe any significant deviations from a linear relationship between relaxation rates and dopant concentrations under the conditions and concentrations explored in this study. The advantage in measuring sPREs with this approach lies in the fact that the sPREs do not need to be modeled explicitly (e.g., fitting correlation times, etc.), consequently allowing a more straightforward comparison between sPREs derived from different types of measurements in solution and solid state. However, global scaling of the data is required to allow comparison of two data sets (see below).
1H relaxation for sPRE in the solid state is one of the most sensitive probes to paramagnetic effects. For 1H relaxation measurements, to maximize sensitivity, one would like to maximize concentration of the protons in the sample.7,45,46,37,38 On the other hand, to suppress the rate-averaging spin diffusion, one needs to minimize the concentration of protons or average out the 1H–1H dipolar couplings by fast MAS. For different levels of protonation, different spinning frequencies are optimal. For example, deuterated 100% back-exchanged samples at 60 kHz spinning provide the best compromise between resolution and sensitivity.45 However, the 1H–1H spin diffusion is not sufficiently suppressed under these conditions to enable site-specific measurements.47 Due to both cost and practical considerations, we decided to use deuterated GB1 with 100% back-exchanged protons and fully protonated natural abundance IgG. The presence of more protons in the system required very fast spinning to sufficiently suppress 1H–1H spin diffusion for site-specific measurements of 1H sPREs (Figure 4a). We have previously demonstrated that high-quality spectra can be obtained for 100% back-exchanged [U-2H,13C,15N]GB1 in complex with natural abundance IgG using as little as 15 μg of labeled protein in a 0.81 mm rotor with ∼100 kHz MAS.7 Furthermore, recent studies report that, at >100 kHz, spinning frequencies, 1H–1H spin diffusion is significantly slowed down even in fully protonated samples, especially for protons with large differences in their chemical shifts.47,48 This is in accordance with the observation of large differences in site-specific 1H R1 (Figure 4a) in our experiments, suggesting that, at 100 kHz spinning, 1H–1H spin diffusion is sufficiently slowed down—if not completely suppressed—to at least allow characterization of the protein–protein interfaces from sPREs (e.g., T18H and E19H are separated by about 1 ppm, and their sPREs differ by a factor of ∼2). The sufficient suppression of the proton spin diffusion is corroborated by the absence of the unusually high 1H sPREs for amide protons in the proximity of hydroxyl sites that were observed in the presence of residual spin diffusion17 (e.g., T18H, which one might expect to be influenced in this way, has a rather low sPRE).
Figure 4.
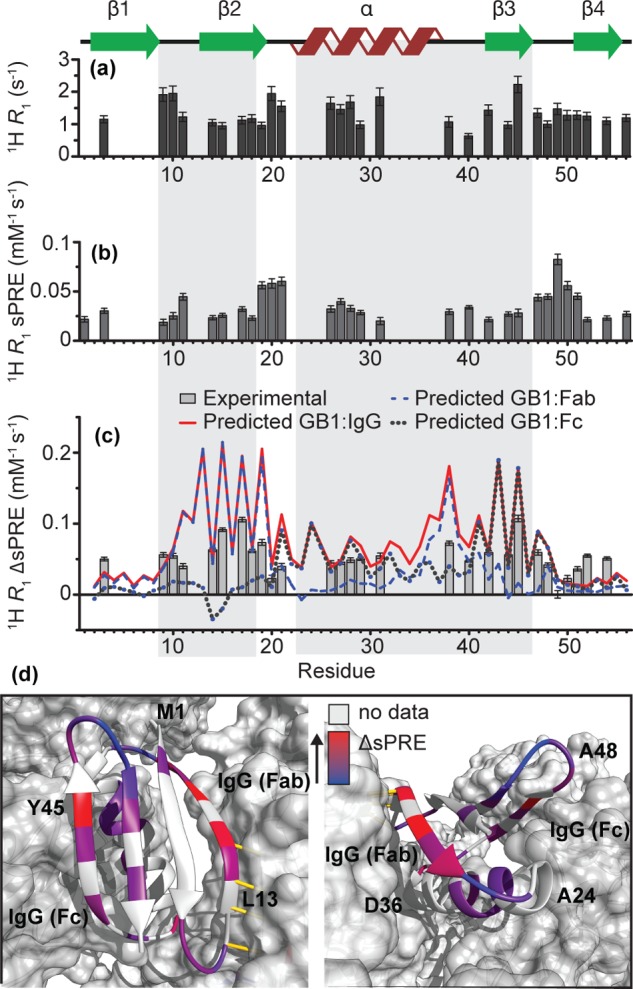
1H R1 relaxation rates (a), 1H R1 sPREs (b), and 1H R1 ΔsPREs (c) for GB1IgG. Lines indicate scaled ΔsPREs calculated from back-predicted sPREs: GB1:IgG complex model (red continuous), GB1:Fab complex model (dashed blue), and GB1:Fc complex model (dotted gray). The scaling procedure is the same as in Figure 3. The expected binding sites are highlighted as in Figure 3. (d) 1H R1 ΔsPREs plotted onto the structure of GB1 in complex with full-length IgG. Residues for which data are not available either due to severe overlap, missing peak, or insufficient signal-to-noise are indicated in gray.
Employing 1H’s for sPRE measurements will therefore require specialized and still not widely available ultrafast MAS probes. In addition, experiments at >100 kHz spinning frequencies necessitate use of rotors with very small volumes. The decrease of signal-to-noise ratio due to the small sample volume can be, to a large extent, offset by detecting signal on protons.46 However, 100% back-exchanged perdeuterated samples can be used effectively for 1H-detected experiments already at 60 kHz, in which case larger volume rotors can be used.45 Finally, for systems with extensive slow dynamics, sometimes adequate 1H resolution is difficult to achieve, even at the highest spinning frequencies, requiring use of 13C- or 15N-detected experiments, in which case larger volume rotors are desirable. For the above reasons, it is worth exploring other probes for sPREs that can be utilized at slower spinning frequencies in larger volume rotors.
In the case of 15N, at spinning frequencies >20 kHz, proton-driven spin diffusion is sufficiently slowed down to enable site-specific measurements of 15N R1 even in fully protonated systems.49−51,56 In the solid state, protein 15N T1’s are very long (20–40 s), so, in spite of lower sensitivity of 15N to paramagnetic effects compared to 1H, a high dynamic range of the relaxation rates is available, and relatively small changes can be detected. Consequently, large variations in 15N sPREs can be observed, just as in the case of 1H, but the measurements can be performed even at moderate spinning frequencies (as low as 10 kHz49−51 if minimal rate averaging can be tolerated). A disadvantage of using 15N R1 for sPREs is that the long relaxation times require long (i.e., many seconds) relaxation delays for adequate sampling of the relaxation rates, resulting in long overall experimental times. Alternatively, we examined the applicability of the typically much shorter 15N T1ρ times (on the order of dozens to hundreds of milliseconds9,10,52−55) as the basis for sPREs. Below we demonstrate that 15N R1ρ sPREs are sufficiently sensitive to characterize protein–protein interfaces, with the emerging picture virtually identical to the one obtained from 15N R1. The much shorter required relaxation delays in 15N R1ρ experiments permit considerably faster performance compared to 15N R1 sPRE acquisition, further allowing higher signal-to-noise ratios in the available experimental time. For example, measurements for one concentration of paramagnetic dopant for GB1IgG took 3–4 days in the case of 15N R1 (estimation of 15N R1 from only two points, which took 5 days, was practically possible for the diamagnetic variant with the measurement of a full curve being prohibitively long8) and 18–24 h in the case of 15N R1ρ. As a side note, 1H T1ρ typically is too short to provide a reliable quantitative sPRE probe (or at least not in a range where no significant line broadening is observed).
sPREs: Solution vs Crystal
First, we set out to explore the applicability of sPREs by investigating reduced solvent accessibility in GB1 crystals. To that end, we used experimental 15N R1-based sPREs of free GB1 in solution (GB1free) to provide a baseline for observing changes in solvent accessibility due to protein–protein interactions and crystal contacts. The 15N R1-based sPREs shown in Figure 1a inform on the solvent accessibility of the protein in the absence of intermolecular interactions, and regions that are well protected from solvent access in isolated GB1 can be identified as most of β1, parts of the α-helix, and parts of β4. In contrast, the most accessible regions are the outer beta strands β2 and β3. This is in good agreement with scaled theoretical sPREs calculated from an available structure of isolated GB1 using a previously described grid-based approach30,33,56 (see Experimental Section). The predicted values reproduce the experimental sPREs well, with the exception of β2 and Y45, where sPREs are underestimated. The discrepancy for β2 could potentially be explained by the previously reported large-amplitude motions of the strand involving rotations around its long axis, which render the amide nitrogens more solvent accessible.57 We note that ultimately conformational dynamics should be taken into account when calculating sPREs from structures.
Figure 1.
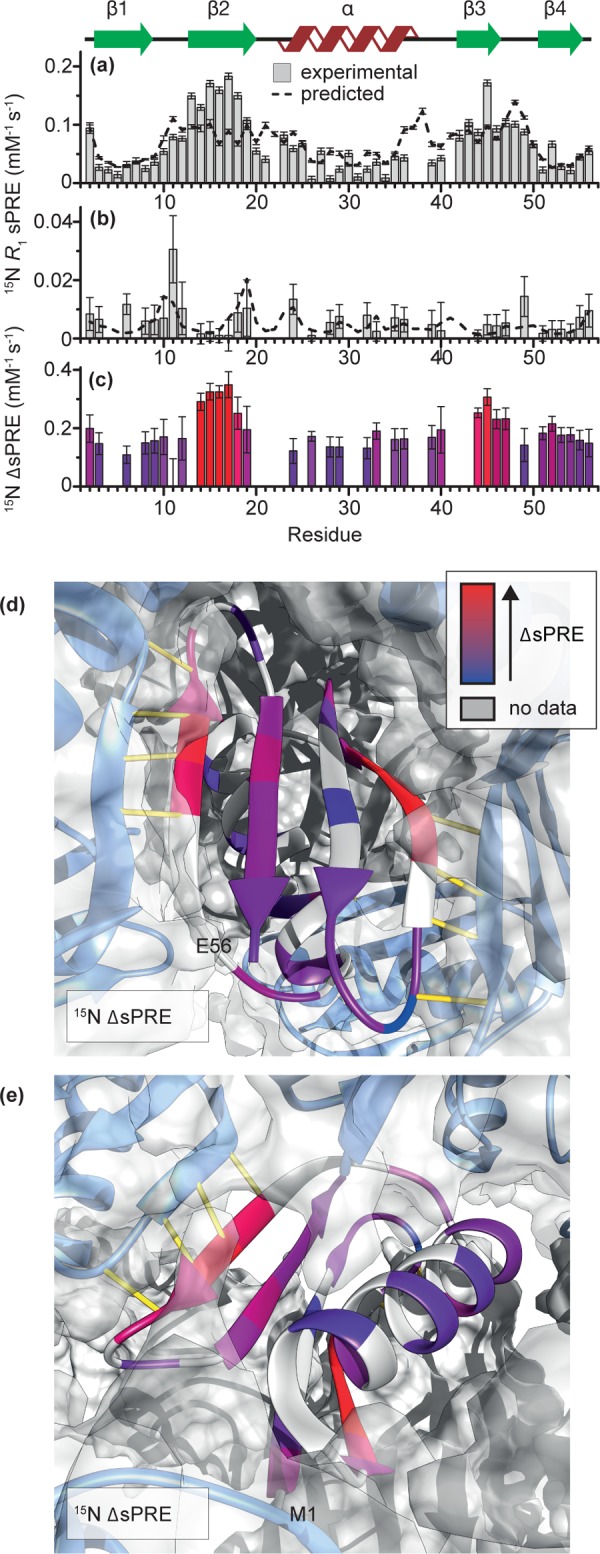
Experimental 15N R1 solvent PREs for GB1 (a) in solution and (b) in a crystal (b). (c) Experimental 15N R1 ΔsPREs for GB1 crystal (i.e., difference between, normalized to averages, 15N R1 sPREs in solution and in crystal) as a function of residue, and (d,e) projected onto structure of GB1 in a lattice. Dashed lines in (a,b) indicate 15N sPREs calculated from structures: solution NMR structure of isolated GB1 (PDB ID: 3gb1)58 in (a) and GB1 in a lattice (PDB ID: 2qmt)59 in (b). Each set of predicted sPREs were scaled by a global factor equal to the ratio of the averages of the predicted sPREs and experimental sPREs. Yellow lines in (d,e) represent intermolecular hydrogen bonds. Residues for which data are not available either due to severe overlap, missing peak, or insufficient signal-to-noise are indicated in gray.
The 15N R1-based sPREs measured in GB1 crystals (GB1cryst) present a quite different picture (Figure 1b): in contrast to GB1free, the outer β2 and β3 strands are much more protected in the crystal compared to the most solvent accessible residues in loop 1. This is consistent with the fact that, in crystals, GB1 forms extended β-sheets stabilized by intermolecular hydrogen bonds between β2 and β3.59,60 Moreover, the scaled theoretical 15N sPREs calculated for GB1 in a lattice agree reasonably well with the experimental sPREs except for T11 and T49, which are located in the loops, and L6. In contrast to the previous study on crystalline SH3,17 we do not observe the unexpectedly high relaxation rates for sites in close proximity to hydroxyl groups (unless these sites are solvent accessible, in which case we do observe high PREs). Absence of this effect in our experiments suggests that the assumption of the absence of spin diffusion in a perdeuterated sample with 10% back-exchanged protons at moderate spinning frequency (24 kHz)17 might not have been entirely justified. Residual spin diffusion due to locally higher density of exchangeable protons and moderate spinning frequency can easily explain the anomalously high sPREs observed by Linser et al.17 Under conditions suggested in the present work, proton-driven spin diffusion between nitrogens is extremely well suppressed, which abolishes any effect of the exchangeable hydroxyl protons on the relaxation rates of amide sites in the vicinity.
Theoretical sPREs can be used to validate models by comparing the experimental sPREs to the ones predicted from the model. However, even in the absence of a model, intermolecular interactions can be detected by identifying sites with increased protection from the solvent due to these interactions. We propose to simply use the difference between experimental sPREs in the absence and presence of intermolecular interactions, i.e., for GB1 free in solution (GB1free) and GB1 in the assembly (GB1cryst or GB1IgG in the latter part of the paper), respectively. Remarkably, the difference sPREs (ΔsPREs) provide a powerful way to detect intermolecular interactions. In general, one will need to take into account any conformational changes upon binding whose effect cannot be distinguished from reduced solvent accessibility due to binding without additional data. GB1 does not undergo any large backbone conformational changes either in crystal or in the GB1:IgG complex,7,8,61 so no further correction is required. In cases where secondary chemical shifts indicate conformational changes upon binding, solving the structure of the considered protein in a complex will be prerequisite for quantification of the protein interface using sPREs (though qualitative information about the interactions still can be obtained in the absence of such a structure). Because of the “built-in” compensation for solvent accessibility patterns due to the conformation of the molecule, the effect of intermolecular interactions is effectively “amplified” in ΔsPREs.
The potential of ΔsPREs is illustrated by the experimental 15N R1 ΔsPREs for GB1cryst shown in Figure 1c–e, which highlight the increased protection of β2 and β3 due to the presence of intermolecular hydrogen bonds in the crystal. At the same time, other subtler features become apparent, such as better protection of the N-terminus compared to the C-terminus or slightly higher protection of the C-terminal end of the helix compared to the N-terminal end. Note that to minimize the bias and to account for the different dynamic ranges of the data sets that are subtracted to yield ΔsPREs, they were scaled by a global factor equal to the ratio of the rate averages of the two data sets (see the Experimental Section). For visualization purposes, a constant (equal to the absolute value of the minimum ΔsPREs) is added so that all experimental ΔsPREs have the same sign.
sPREs in GB1:IgG Complex
Currently only structures of protein G domains with immunoglobulin G (IgG) fragments are available. Interactions between the Fab fragment of IgG and protein G domains have been investigated by X-ray crystallography62 and solution NMR.63 The crystal structure showed that the main interactions between the Fab fragment and GB3 correspond to residues 10–18 of GB1 and a minor contact between the Fab fragment and residues 33 and 37 of GB1 (for clarity we use GB1 residue numbering throughout this paragraph). The solution NMR analysis identified chemical shift perturbations (CSPs) in residues 9–17 (also 7, 19, 36, 37, 38, 40, 43, and 53).63 In solution NMR studies of GB2 and the Fc fragment of IgG, residues in regions 23–36 and 40–46, which are located in β3 and the α-helix of GB1, were found to be involved in the interaction.64 This is in agreement with a crystal structure of GB2 in complex with the Fc fragment of IgG, where the residues involved in binding correspond to residues 27, 28, 31, 32, 35, 40, 42, and 43 of GB1.65 It is worth noting that residues involved in binding to the Fab fragment (residues 9–18) were not affected by the interaction with the Fc fragment.65 In our previous solid-state NMR study of the complex of GB1 bound to full-length human IgG, we established that GB1 binds to both Fab and Fc fragments of IgG simultaneously.7 Here we use the sPRE methodology validated above on the GB1 crystal to obtain further insights into the GB1-IgG interactions.
In the range explored by us, 15N R1 or R1ρ relaxation rates vs Gd(DTPA-BMA) concentration in a precipitated GB1:IgG complex show a good linear relationship (see Figure 2a,b and Supporting Information (SI) Figure 3). The sPREs obtained from slopes of such trends are shown in Figure 2c (15N R1 sPREs) and Figure 2d (15N R1ρ sPREs). A direct comparison of predicted and experimental sPREs (SI Figure 1) shows that interactions with both Fc and Fab fragments must be present. To further analyze the increased protection due to protein–protein interactions, we focus on ΔsPREs in the discussion. As we mentioned above, this is preferred to direct analysis of sPREs because ΔsPREs mostly suppress pattern of protection from solvent due to conformation of the studied protein, leaving one with a pattern mostly based on intermolecular interactions (unless the protein undergoes significant conformational change upon binding where the analysis becomes much more involved).
Figure 2.
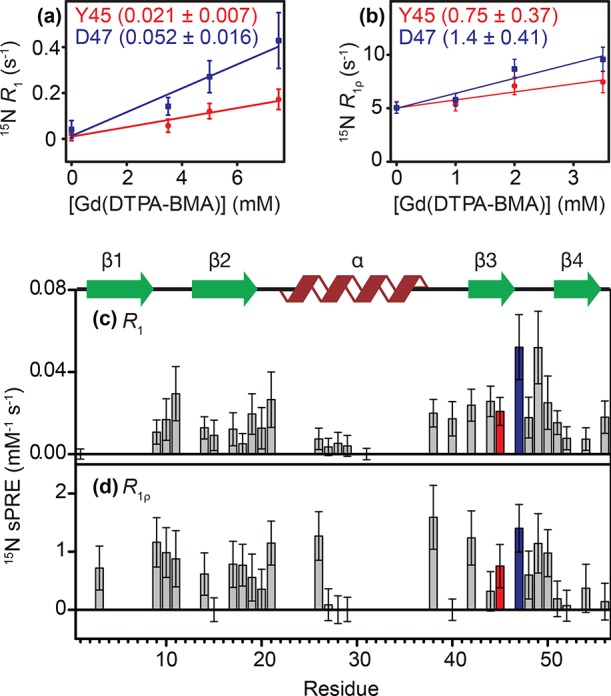
Examples of linear fits for sPREs for GB1 in complex with IgG (GB1IgG) based on (a) 15N R1 and (b) 15N R1ρ relaxation rates. (c) 15N R1 sPREs and (d) 15N R1ρ sPREs for GB1 in the precipitated complex with IgG.
Figure 3a,b shows experimental 15N R1 and R1ρ ΔsPREs for GB1IgG. Both data sets reveal a rather similar pattern, which differs somewhat from ΔsPREs for GB1cryst. Overall, as expected due to the shorter T1ρ times compared to T1 times, the dynamics range of R1ρ ΔsPREs is smaller but still sufficient to identify changes in the solvent accessibility caused by complex formation. The fact that 15N R1ρ ΔsPREs appear to be sufficiently sensitive to characterize intermolecular contacts in the GB1:IgG complex is fortunate because the acquisition of high-quality R1ρ sPREs requires much shorter experimental times compared to 15N R1 sPREs (from several days to sometimes less than 24 h; see SI Table 11). The most prominent feature arising from a comparison of 15N R1 and R1ρ ΔsPREs in GB1IgG and 15N R1 ΔsPREs in GB1cryst is that β2 is most protected in both assemblies. This is consistent with the intermolecular hydrogen bonds between GB1 molecules in the crystal and intermolecular hydrogen bonds to the Fab fragment in the GB1:IgG complex. On the other hand, β3, which interacts but does not form hydrogen bonds with the Fc fragment in GB1IgG, is somewhat less protected than in the crystals, where it forms intermolecular hydrogen bonds. Even more interestingly, the N-terminal residues in the helix seem similarly or better protected than β2 as a result of complex formation. The above observations seem to be consistent with creation of the interface between the helix and β3 of GB1 with the Fc part of IgG.
Figure 3.
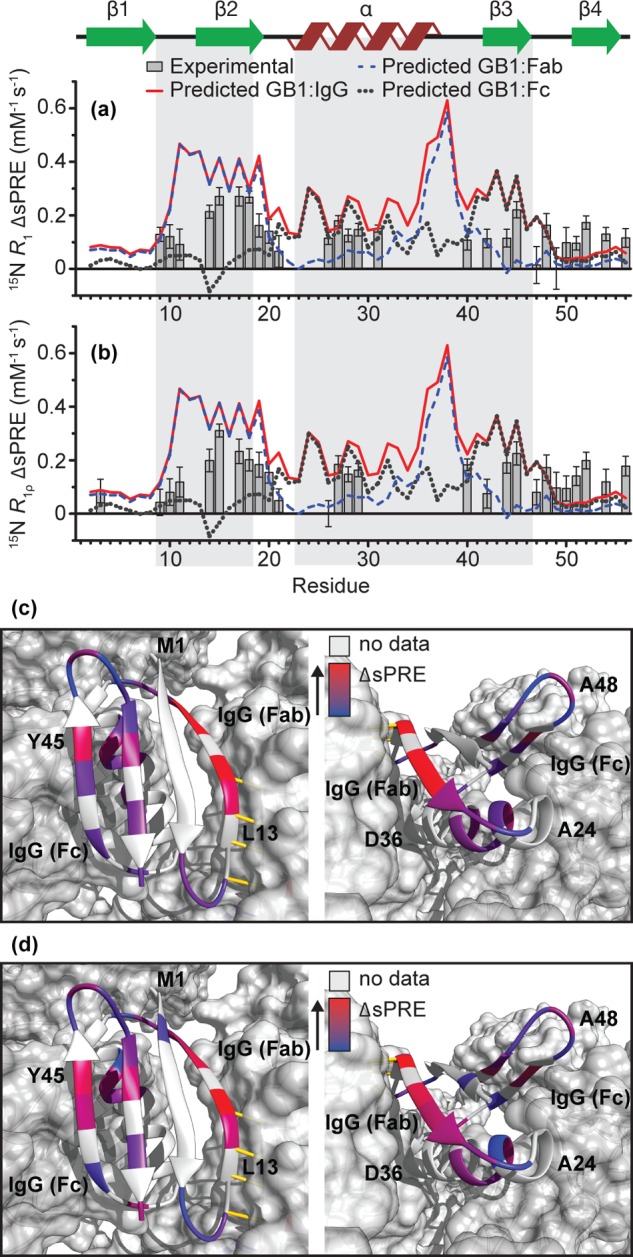
15N ΔsPREs for GB1IgG based on (a) R1 and (b) R1ρ relaxation rates (gray bars). Lines indicate scaled ΔsPREs calculated from back-predicted sPREs: GB1:IgG complex model (red continuous), GB1:Fab complex model (dashed blue), and GB1:Fc complex model (dotted gray). For the comparison, all back-predicted data sets are scaled so that the average of the set is equal to the average of the experimental data. The expected binding sites are highlighted: Fab interface (residues 9–18) and Fc interface (residues 23–46). Experimental 15N R1 (c) and R1ρ (d) ΔsPREs projected onto the structural model of GB1 in a complex with IgG. Red indicates residues with the largest changes in solvent accessibility upon binding and blue the residues with the smallest changes upon binding. Residues for which data are not available either due to severe overlap, missing peak, or insufficient signal-to-noise are indicated in gray.
To further investigate the protein–protein interfaces, we also measured amide 1H solvent PREs. 1H sPREs of the GB1:IgG complex were obtained from 1H R1 measurements (for historical reasons using CuEDTA37 rather than Gd(DTPA-BMA) and are presented in Figure 4b (data in SI Tables 6 and 7, comparison between predicted and experimental sPREs in SI Figure 2). Reference experimental 1H sPREs for isolated GB1 in solution were taken from ref (66).
The overall trend in 1H ΔsPREs for the GB1:IgG complex is similar to that in 15N ΔsPREs: again, as a result of the complex formation, β2 is the most strongly protected; β3 is also protected, but to a lesser extent, with Y45 exhibiting the strongest level of protection. However, relative to the level of protection in β2, the residues in the helix seem to be very slightly less protected than in 15N ΔsPREs.
The ΔsPREs back-calculated from models of GB1:IgG, GB1:Fab, and GB1:Fc complexes are plotted as lines in Figure 3a,b and Figure 4c. A simple visual inspection is sufficient to see that all three experimental ΔsPRE sets are more compatible with the ΔsPREs back-calculated from a model of the GB1:IgG complex, where both of the binding interfaces are occupied at the same time. In all the cases, ΔsPREs calculated for GB1:Fab grossly underestimate the level of protection for β3 and helix, while ΔsPREs calculated for GB1:Fc grossly underestimate the level of protection for β2 and overestimate the level of protection for β3 acquired upon complex formation.
In order to obtain a more quantitative handle on how well the different structural models reproduce experimental data, we have also performed a series of fits of the experimental ΔsPREs to ΔsPREs back-calculated from the different models (note that scaling of individual back-calculated sPREs is not required before calculation of theoretical ΔsPREs), with a global scaling factor as the only fit parameter. Again, in such data the trends are “cleaned up” from the effects of GB1 conformation simplifying quantification of the contribution from intermolecular interactions. In all the cases, data back-predicted from the GB1:IgG complex, where both Fab and Fc interfaces are occupied, give the lowest χ2, thus identifying it as the best from the considered models of the interaction (see SI Table 12).
Upon closer inspection of the best-fitting theoretical ΔsPRE trends against the experimental ones, we identify one particularly interesting feature where the two types of data differ. According to the experimental data, the protection due to interactions between GB1 and IgG is similar for some residues in β4 to that in β3, suggesting that either the first residues in β3 are less protected than expected or β4 is more protected than expected. Different levels of protection can be explained by either a change of backbone conformation between GB1free and GB1IgG, internal molecular motion, or an additional interaction with IgG.
The Cα secondary chemical shifts for GB1 in complex with IgG are very similar to the ones calculated for GB1 in solution (see SI Figure 5, based on data from ref (7)), with the exception of L6, T11, L12, and K50 (Figure 5a). Consequently, subtle changes in the backbone conformation are unlikely to explain the changes in the solvent accessibility of β4. Internal molecular motions could explain the differences in the ΔsPREs for residues which exhibit large-amplitude backbone motions but can be safely neglected for the rest of the residues, including those in β4.8,61 This means that the observed deviations, if real and not just experimental errors, must arise from changes in the intermolecular interactions: additional interactions for the increased protection and abolished interactions for the decreased protection. Some changes in the solvent accessibility might be expected if GB1 undergoes a small-amplitude anisotropic overall motion in the complex, as we have suggested based on the analysis of relaxation rates measured in the complex.8 To investigate what effect such motion would have on the pattern of solvent accessibility, we generated a series of conformers where the molecule of GB1 was rotated around the axes of the motion by 7°, which corresponds to the approximate amplitude of motion determined in our rather simple analysis8 (the actual amplitude of motion may differ because in the absence of a dipolar order parameter relaxation analysis is not very reliable53,61), followed by translating the molecules, assuming that β2 hydrogen bonded to Fab is the anchoring the point. The distribution of ΔsPREs calculated for five generated conformers is illustrated in Figure 5d,e. In the case of a proposed overall motion, the residues most immune to changes in solvent accessibility as a result of this motion would be located in β1, β4, and the C-terminal half of the helix. Such a motion could, however, contribute to the discrepancies observed for β3, V21, and loop 4 (D47 is the most influenced of all residues).
Figure 5.
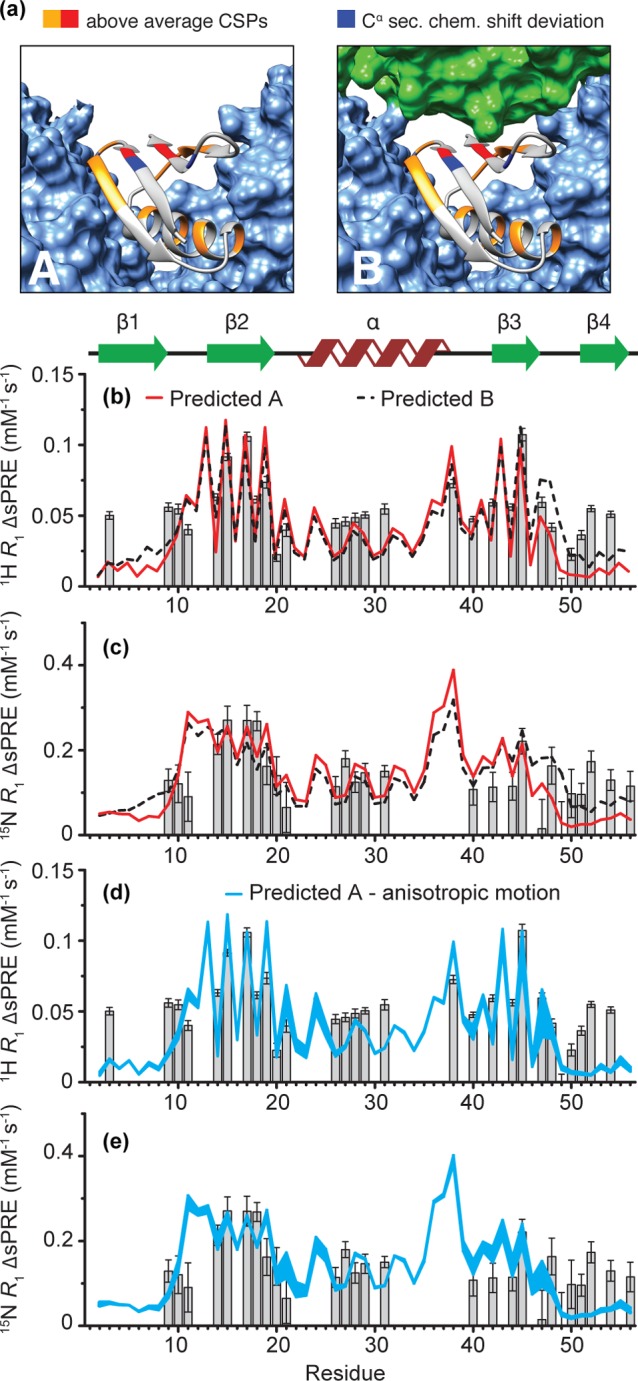
(a) Models of GB1:IgG complex with (A) only Fab and Fc interface (cornflower blue) and (B) Fab, Fc and additional contact with β4 (green). Residues with higher than average chemical shift perturbations (CSPs) are indicated in orange.7 Previously unexplained above average CSPs for residues 7, 53, and 54 are shown in red.7 Sites with deviations of Cα secondary chemical shift compared to solution are indicated in blue.7 (b,c) Experimental ΔsPREs vs ΔsPREs predicted from GB1:IgG models in (a): model A (red line) and model B (black dashed line). (d,e) Back-predicted ΔsPREs for GB1 in complex with IgG assuming small amplitude overall anisotropic motion of GB1 as suggested in ref (8). The lines in (d-e) represent a range of ΔsPREs based on 5 generated conformers.
Interestingly, we previously observed large chemical shift perturbations (CSPs) for T53, V54, and L7 upon GB1:IgG complex formation but were unable to completely explain their origin (Figure 5a; L7 could potentially be explained by a small backbone conformation change indicated by change in Cα secondary chemical shift of L6Cα compared to solution data). The presence of these CSPs and the elevated ΔsPREs suggest that there might be an additional interaction between GB1 and IgG, which involves β4 and is not observed in the complexes of protein G domains with IgG fragments. Obviously, with the available data, it is not possible for us to identify the region of IgG responsible for this potential additional interaction. However, we can simulate the effects of proximity of molecular fragments to β4 on the ΔsPRE pattern in GB1IgG. We have generated a model by translating one of the extended IgG loops to make an intermolecular contact with β4 (Figure 5a). This additional contact indeed reduces the anomalous ΔsPRE trend for β4. In spite of complete arbitrariness of this model (the exact structure of the fragment and its position are likely different), it is clear that the presence of a similar additional interaction is consistent with the observed elevated ΔsPREs for β4 and the large CSPs for residues 53 and 54. Interestingly, the existence of such a contact would also help to explain why the complex of GB1 with IgG gives such high-quality NMR spectra that are atypical for a precipitate. With three interfaces, the local environment of GB1 in the complex would be defined almost entirely by the specific interactions with IgG, leaving GB1 largely unaffected by any heterogeneity of the sample.
Conclusions
We introduced 1H and 15N solvent PREs as a general and powerful tool for characterizing intermolecular interfaces in large biomolecular complexes in the solid state. The proposed methods can be applied over the majority of range of spinning frequencies employed in biomolecular solid-state NMR: moderate spinning frequencies (15N R1), intermediate spinning frequencies (15N R1,15N R1ρ), and fast spinning frequencies (15N R1,15N R1ρ,1H R1), which allows to fine-tune this methodology to the specific requirements of different systems and available instrumentation. We demonstrate for 100% back-exchanged protein in the presence of a fully protonated binding partner that 1H–1H spin diffusion is sufficiently slowed down at 100 kHz to allow the use of 1H R1 as a site-specific probe of solvent accessibility. 1H R1 and 15N R1 are the most sensitive probes, enabling accurate measurement of even small PREs. At the same time, 15N R1ρ, even though less sensitive than 1H R1 and 15N R1 as sPRE probes, yields essentially the same picture, with the added benefit of overall shorter experiments, where satisfactory signal-to-noise ratio can be achieved in a reasonable amount of time, even for large biomolecular complexes characterized by low sensitivity. We establish the benefits of comparing sPREs of isolated protein in solution to sPREs of the protein in a complex in the solid state to identify reduced solvent accessibility of regions involved in protein–protein interactions.
Moreover, we demonstrate the utility of solid-state sPREs for determining intermolecular interactions by applying it to characterize intermolecular contacts in GB1 crystal and protein–protein interfaces in GB1 in a complex with full-length IgG. The experimental sPREs are in very good agreement with predicted sPREs based on crystal structures. All three sPRE probes—15N R1, 15N R1ρ, and 1H R1—yield a highly consistent view of the GB1 interactions with IgG. Based on the local deviations of ΔsPRE trends and CSPs, we suggest that the extraordinary GB1:IgG binding interface might involve three different regions, painting a more complex picture than what can be deduced from the structures of protein G with IgG fragments, emphasizing the importance of using full-length proteins in interaction studies if at all possible.
We envision the proposed approach to be widely applicable for characterization of intermolecular interfaces in large protein complexes and especially the ones that are not accessible to other high-resolution techniques, as is the case for the precipitated complex of GB1:IgG.
Experimental Section
Sample Preparation
Isotope-labeled GB1 2Q6I was expressed using pGEV2 in BL21(DE3).67 [U-13C,15N]GB1 was purified from cultures grown in M9 supplemented with [U-13C]glucose and 15NH4Cl. [U-2H,13C,15N]GB1 was expressed in M9 prepared in D2O with deuterated [U-13C]glucose and 15NH4Cl. Cells were grown to an OD600 > 1.0 in 2 L of LB medium for each liter of M9 and washed once with PBS before resuspension in M9. Expression was induced with 0.5 mM IPTG after 1 h incubation at 37 °C. After expression for 4 h at 37 °C, the cells were pelleted (4000g for 20 min at 16 °C) and lysed by sonication in buffer (50 mM potassium phosphate; 200 mM NaCl; 1 mg/mL lysozyme; pH 7.0). The lysate was then incubated at 75 °C for 10 min and cleared by centrifugation (12000g for 50 min). After precipitation overnight with 80% ammonium sulfate, GB1 was pelleted (15000g for 50 min), redissolved in buffer (50 mM potassium phosphate; 200 mM NaCl; pH 7.0), and purified on a 16/600 Sephadex pg75 (GE Healthcare) gel filtration column. Fractions containing GB1 were collected, desalted, freeze-dried, and stored at −20 °C.
Freeze-dried [U-2H,13C,15N]GB1 was dissolved in buffer (50 mM sodium phosphate buffer pH 5.5) to obtain a protein concentration of 10 mg/mL and crystallized with the aid of 2:1 2-methyl-2,4-pentanediol (MPD):isopropanol.68 Lyophilized IgG from human serum was purchased from Sigma-Aldrich. GB1:IgG complex was formed by mixing GB1 and IgG solutions in 2:1 molar ratio.7 Crystalline GB1 and precipitated GB1:IgG complex were packed into NMR rotors using the following procedure: The crystals/precipitate were spun down by centrifugation (1 min at 20000g using a benchtop centrifuge) and resuspended in a small volume of the supernatant containing 2% DSS and Gd(DTPA-BMA) at the desired concentration. The 1.3 mm rotors were packed by centrifugation (20000g) and the rotor caps sealed with a silicone-based glue to prevent leakage. The smaller 0.81 mm rotors were filled manually using microspatulas.
The solution NMR sample was prepared in a 3 mm tube containing 200 μL of 1 mM [U-13C,15N]GB1 in 50 mM sodium phosphate, pH 5.5, 10% D2O, and 30 μM DSS.
Solution NMR
All solution NMR data were recorded at 298 K on a 700 MHz Bruker Avance spectrometer equipped with a cryogenically cooled probehead. 15N longitudinal relaxation rates (R1) were measured with a 15N-HSQC-based standard Bruker pseudo-3D (hsqct1etf3gpsi3d.2), with 8–10 points, using delays between 0.05 and 2.0 s (details are listed in SI Table 11). Spectral widths were 8400 Hz for 1H and 2700 Hz for 15N, and FIDs had 2048 and 256 points, respectively. The recycle delay was 3.5 s. To obtain the sPREs, the sample was titrated with Gd(DTPA-BMA) (Omniscan; stock 20 mM) up to 2.5 mM (details in SI Table 11).
All spectra were processed in TopSpin 3.2 and CCPNMR,69 and MatLab R2014a was used to analyze the relaxation data.
Solid-State NMR
Solid-state NMR spectra were recorded at 600 MHz Bruker Avance II+, 700 MHz Bruker Avance III HD, and 850 MHz Bruker Avance III spectrometers, using Bruker 1.3 mm triple-resonance probes (at MAS frequencies of 50–60 kHz) or a volume-optimized 0.81 mm double-resonance probe from Samoson laboratory (for experiments at ∼100 kHz MAS). A Bruker BCU-X cooling unit was used to regulate the internal sample temperature to 27 ± 1 °C (measured from the chemical shift of water with respect to DSS). For experiments recorded at 700 MHz 1H Larmor frequency, 10% D2O was added to the sample buffer before packing the rotors, and deuterium locking was used in the same way as in solution NMR. 15N–1H 2D correlation spectra were recorded using a proton-detected heteronuclear correlation sequence. Double-quantum cross-polarization (CP) contact times were between 0.5 and 1.5 ms and individually optimized for each sample. Recycle delays between 0.2 and 2.5 s were used, depending on the amount of paramagnetic agent and magnetic field.
The maximum employed concentrations were chosen so that the paramagnetic effect does not lead to significant line broadening and are thus different for different samples; e.g., larger concentrations could be used in GB1:IgG complex than in GB1 crystal to obtain similar line widths.
In all solid-state experiments, hard pulses were applied at nutation frequencies of 100 kHz (1.3 mm probe) or 125 kHz (0.81 mm probe) for 1H and 83.3 kHz for 15N. WALTZ-16 decoupling at 10 kHz was applied on protons during 15N evolution and on 15N channel during direct 1H acquisition, while quadrature detection was achieved using the States-TPPI method. Suppression of the water signal was achieved by saturation, with 50–200 ms of slpTPPM 1H decoupling applied at an amplitude of one-fourth of the MAS frequency38 or with 100–140 ms of MISSISSIPPI70 at an amplitude of half the MAS frequency on resonance with the water signal.
R1 and R1ρ relaxation curves were sampled using 8–11 points for all experiments except the diamagnetic 15N R1 in the complex, where only 2 points were used (SI Tables 10 and 11). Error estimates for the integrals were achieved by duplicating one of the relaxation delays (R1) or spin-lock lengths (R1ρ). A 10 kHz nutation frequency, measured by a nutation experiment, was used for the spin-lock field in the R1ρ experiments.
All spectra were processed using TopSpin 3.2. GB1 resonances in the complex with IgG were previously assigned on the basis of 3D H(H)NH, CONH, CO(CA)NH, and CANH experiments.7 Peak integrals were calculated in TopSpin 3.2. OriginPro 2016 and MatLab R2014a were used to analyze the relaxation data.
Error Estimates
Peak integrals from TopSpin or peak volumes from CCPNMR were exported to MatLab, where an exponential function was used with the fminsearchbnd function to fit the relaxation data. Errors were calculated by Monte Carlo error estimations for R1 and R1ρ exponential fits. A random number between 0 and 1 was multiplied with the integral error and added to the recalculated integrals or volumes. The fitting was then repeated 2000 times with a new random number between 0 and 1 generated each time. Values of 2 times the standard deviations of the R1 or R1ρ values received from the fits for each residue were used as errors. Errors for sPREs were obtained in the same way but with linear fits instead of exponential. Error propagations for ΔPREs and PRE ratios were calculated using standard formulas for error propagation.
PRE Predictions
Predicted sPRE data were computed using a previously published grid-based approach.30,33,56 To this end, the structural model was placed in a grid with equally spaced grid points. The grid point to grid point distance was set to 0.2 Å, and the distance between the outer atoms of the protein and the edges of the grid box was set to 20 Å. Next, all grid points that were positioned within a radius rclash around an atom of the protein were removed. The radius rclash was set to rclash = rvdW,i + rGd, where rvdW,i is the van der Waals radius of atom i and rGd is the radius of the paramagnetic compound and was set to 3.5 Å. Next, the sPRE value of atom i of the protein was computed by summing up the contributions of all remaining grid points according to
where i is the index of the protein atom, the index j iterates over all remaining grid points, and dij is the distance between the atom i and grid point j.
Whenever two sPREs data sets of different origin (e.g., theoretical and experimental sPREs or experimental sPREs derived from different relaxation measurements) were compared directly, one of the data sets is scaled by the ratio of the averages of the sPREs in each data set. Only data points for residues present in both data sets were used to calculate the average.
C++ code for calculating sPREs is available from the authors upon request. For published structures that contained several models (like solution NMR structure of GB1), the error of the sPRE prediction was estimated using the standard deviation of the sPRE values of the different models. For calculation of sPREs for GB1free, a solution structure (PDB ID: 3gb1;58 our construct is a T2Q mutant, but comparison of sPREs calculated from isolated GB1 from several different solution and X-ray structures suggested that, in the case of amide sPREs, differences are very small) was used, with the sPRE reported as an average of the sPREs calculated for each conformer in the bundle. For calculation of sPREs for GB1cryst, an X-ray structure (PDB ID: 2qmt)59 was used, with a molecule in the middle of a 3×3 unit cell crystal lattice generated in Chimera71 using the Multiscale Models tool. For sPREs for GB1:Fab complex, a model obtained by structural alignment of the X-ray structure of GB1 (PDB ID: 2qmt) with GB3 in the X-ray structure of GB3:Fab (PDB ID: 1igc)72 was used. For sPREs for GB1:Fc complex, a model obtained by structural alignment of the X-ray structure of GB1 (PDB ID: 2qmt) with GB3 in the X-ray structure of GB2:Fc (PDB ID: 1fcc)65 was used. For sPREs for GB1:IgG complex, a model obtained by the alignment of the above two complexes on GB1 (PDB IDs: 1igc and 1fcc) was used. Before sPRE calculation, protons were added to the models using the default tool in Chimera.
ΔsPREs were calculated as a difference between sPREs for isolated molecules in solution and sPREs in the assembly. Both experimental and theoretical ΔsPREs were calculated. sPREs were scaled by the ratio of averages before calculating ΔsPREs to minimize bias from any particular data set. Here we typically scaled up the sPREs from the assembly to solution sPREs. Note that comparison of the two different theoretical sPRE data sets does not require scaling.
Fitting of the experimental ΔsPREs to ΔsPREs back-predicted from various models was performed in Matlab. The best fit was determined by minimizing the χ2 target function:
where ΔsPREexp,i is experimental ΔsPRE for residue i, ΔsPREcalc,i is ΔsPRE for residue i calculated from a given model, σΔsPREexp,i2 is error for experimental ΔsPRE for residue i, and A is a constant and the only fit parameter.
Acknowledgments
The research leading to these results has received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement 639907. J.R.L. also acknowledges funding from Royal Society Grant RG130022, EPSRC Grant EP/L025906/1, BBSRC Grant BB/L022761/1, and Gates Foundation OPP1160394. C.Ö. acknowledges funding from the European Union under a Marie Curie Initial Training Network FP7-PEOPLE-2012-ITN Grant Agreement Number 316630 CAS-IDP. S.K. acknowledges support from BBSRC Grant BB/L022761/1. J.M.L. acknowledges support from EPSRC DTG. T.M. is supported by the OmicsCenter Graz, the Bavarian Ministry of Sciences, Research and the Arts (Bavarian Molecular Biosystems Research Network, to T.M.), the President’s International Fellowship Initiative of CAS (No. 2015VBB045), the National Natural Science Foundation of China (No. 31450110423), the Austrian Science Fund (FWF: P28854, W1226-B18), and the Deutsche Forschungsgemeinschaft (DFG, MA5703/1-1), Integrative Metabolism Research Center Graz, the Austrian infrastructure program 2016/2017, BioTechMed/Graz. The UK 850 MHz solid-state NMR Facility used in this research was funded by EPSRC and BBSRC (contract reference PR140003), as well as the University of Warwick, including via partial funding through Birmingham Science City Advanced Materials Projects 1 and 2 supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). The Tallinn group was supported by PUT 1534 grant of ETAG. Figures 1, 3, 4, 5, and SI Figure 4 were produced using the UCSF Chimera package, which was developed by the RBVI at the University of California, San Francisco (supported by NIGMS P41-GM103311). All data supporting this study are provided as Supplementary Information accompanying this paper with exception of the raw NMR data, which can be downloaded from http://wrap.warwick.ac.uk/91300/1/All_data_sPRE.zip.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b03875.
Comparison between experimental and predicted sPREs, model of GB1:IgG complex used for sPRE predictions, examples of linear fits for sPRE calculations, tables with all relaxation rates and sPREs, and tables with relaxation delays and spin-lock lengths used for relaxation measurements (PDF)
Author Contributions
‡ C.Ö. and S.K. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Gardiennet C.; Schütz A. K.; Hunkeler A.; Kunert B.; Terradot L.; Böckmann A.; Meier B. H. Angew. Chem., Int. Ed. 2012, 51 (31), 7855. 10.1002/anie.201200779. [DOI] [PubMed] [Google Scholar]
- Loquet A.; Sgourakis N. G.; Gupta R.; Giller K.; Riedel D.; Goosmann C.; Griesinger C.; Kolbe M.; Baker D.; Becker S.; Lange A. Nature 2012, 486 (7402), 276. 10.1038/nature11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M.; Hou G.; Zhang H.; Suiter C. L.; Ahn J.; Byeon I.-J. L.; Perilla J. R.; Langmead C. J.; Hung I.; Gor’kov P. L.; Gan Z.; Brey W.; Aiken C.; Zhang P.; Schulten K.; Gronenborn A. M.; Polenova T. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (47), 14617. 10.1073/pnas.1516920112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainz A.; Religa T. L.; Sprangers R.; Linser R.; Kay L. E.; Reif B. Angew. Chem., Int. Ed. 2013, 52 (33), 8746. 10.1002/anie.201301215. [DOI] [PubMed] [Google Scholar]
- Barbet-Massin E.; Huang C.-T.; Daebel V.; Hsu S.-T. D.; Reif B. Angew. Chem., Int. Ed. 2015, 54 (14), 4367. 10.1002/anie.201409393. [DOI] [PubMed] [Google Scholar]
- Jehle S.; Rajagopal P.; Bardiaux B.; Markovic S.; Kühne R.; Stout J. R.; Higman V. A.; Klevit R. E.; van Rossum B.-J.; Oschkinat H. Nat. Struct. Mol. Biol. 2010, 17 (99), 1037. 10.1038/nsmb.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamley J. M.; Iuga D.; Öster C.; Sass H.-J.; Rogowski M.; Oss A.; Past J.; Reinhold A.; Grzesiek S.; Samoson A.; Lewandowski J. R. J. Am. Chem. Soc. 2014, 136 (48), 16800. 10.1021/ja5069992. [DOI] [PubMed] [Google Scholar]
- Lamley J. M.; Öster C.; Stevens R. A.; Lewandowski J. R. Angew. Chem., Int. Ed. 2015, 54 (51), 15374. 10.1002/anie.201509168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoop C. L.; Lin H.-K.; Kar K.; Magyarfalvi G.; Lamley J. M.; Boatz J. C.; Mandal A.; Lewandowski J. R.; Wetzel R.; van der Wel P. C. A. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (6), 1546. 10.1073/pnas.1521933113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good D. B.; Wang S.; Ward M. E. M. E.; Struppe J.; Brown L. S. L. S.; Lewandowski J. R.; Ladizhansky V. J. Am. Chem. Soc. 2014, 136 (7), 2833. 10.1021/ja411633w. [DOI] [PubMed] [Google Scholar]
- Jaroniec C. P. J. Magn. Reson. 2015, 253, 50. 10.1016/j.jmr.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight M. J.; Pell A. J.; Bertini I.; Felli I. C.; Gonnelli L.; Pierattelli R.; Herrmann T.; Emsley L.; Pintacuda G. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (28), 11095. 10.1073/pnas.1204515109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe N. P.; Kotecha M.; Samoson A.; Past J.; Ishii Y. J. Magn. Reson. 2007, 184 (2), 350. 10.1016/j.jmr.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadaud P. S.; Helmus J. J.; Sengupta I.; Jaroniec C. P. J. Am. Chem. Soc. 2010, 132 (28), 9561. 10.1021/ja103545e. [DOI] [PubMed] [Google Scholar]
- Ward M. E.; Wang S.; Krishnamurthy S.; Hutchins H.; Fey M.; Brown L. S.; Ladizhansky V. J. Biomol. NMR 2014, 58 (1), 37. 10.1007/s10858-013-9802-2. [DOI] [PubMed] [Google Scholar]
- Balayssac S.; Bertini I.; Bhaumik A.; Lelli M.; Luchinat C. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (45), 17284. 10.1073/pnas.0708460105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linser R.; Fink U.; Reif B. J. Am. Chem. Soc. 2009, 131 (38), 13703. 10.1021/ja903892j. [DOI] [PubMed] [Google Scholar]
- Sengupta I.; Nadaud P. S.; Jaroniec C. P. Acc. Chem. Res. 2013, 46 (9), 2117. 10.1021/ar300360q. [DOI] [PubMed] [Google Scholar]
- Ullrich S. J.; Hölper S.; Glaubitz C. J. Biomol. NMR 2014, 58 (1), 27. 10.1007/s10858-013-9800-4. [DOI] [PubMed] [Google Scholar]
- Nadaud P. S.; Helmus J. J.; Kall S. L.; Jaroniec C. P. J. Am. Chem. Soc. 2009, 131 (23), 8108. 10.1021/ja900224z. [DOI] [PubMed] [Google Scholar]
- Wang S.; Parthasarathy S.; Xiao Y.; Nishiyama Y.; Long F.; Matsuda I.; Endo Y.; Nemoto T.; Yamauchi K.; Asakura T.; Takeda M.; Terauchi T.; Kainosho M.; Ishii Y. Chem. Commun. 2015, 51 (81), 15055. 10.1039/C5CC04618A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadaud P. S.; Helmus J. J.; Höfer N.; Jaroniec C. P. J. Am. Chem. Soc. 2007, 129 (24), 7502. 10.1021/ja072349t. [DOI] [PubMed] [Google Scholar]
- Pintacuda G.; Giraud N.; Pierattelli R.; Böckmann A.; Bertini I.; Emsley L. Angew. Chem., Int. Ed. 2007, 46 (7), 1079. 10.1002/anie.200603093. [DOI] [PubMed] [Google Scholar]
- Jaroniec C. P. Solid State Nucl. Magn. Reson. 2012, 43–44, 1. 10.1016/j.ssnmr.2012.02.007. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Spiller M.; Caravan P. Magn. Reson. Med. 2010, 63 (3), 609. 10.1002/mrm.22214. [DOI] [PubMed] [Google Scholar]
- Hocking H. G.; Zangger K.; Madl T. ChemPhysChem 2013, 14 (13), 3082. 10.1002/cphc.201300219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petros A. M.; Mueller L.; Kopple K. D. Biochemistry 1990, 29 (43), 10041. 10.1021/bi00495a005. [DOI] [PubMed] [Google Scholar]
- Sakakura M.; Noba S.; Luchette P. A.; Shimada I.; Prosser R. S. J. Am. Chem. Soc. 2005, 127 (16), 5826. 10.1021/ja047825j. [DOI] [PubMed] [Google Scholar]
- Bernini A.; Venditti V.; Spiga O.; Niccolai N.. Progress in Nuclear Magnetic Resonance Spectroscopy; Elsevier B.V.: Amsterdam, 2009; pp 278–289. [Google Scholar]
- Pintacuda G.; Otting G. J. Am. Chem. Soc. 2002, 124 (3), 372. 10.1021/ja016985h. [DOI] [PubMed] [Google Scholar]
- Madl T.; Bermel W.; Zangger K. Angew. Chem., Int. Ed. 2009, 48 (44), 8259. 10.1002/anie.200902561. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Schwieters C. D.; Tjandra N. J. Magn. Reson. 2012, 221, 76. 10.1016/j.jmr.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlmüller C.; Göbl C.; Madl T. Angew. Chem., Int. Ed. 2016, 55 (39), 11970. 10.1002/anie.201604788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson J. H.; Thompson G. S.; Kalverda A. P.; Zhuravleva A.; O’Neill A. J. Sci. Rep. 2016, 6 (1), 19524. 10.1038/srep19524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madl T.; Güttler T.; Görlich D.; Sattler M. Angew. Chem., Int. Ed. 2011, 50 (17), 3993. 10.1002/anie.201007168. [DOI] [PubMed] [Google Scholar]
- Parthasarathy S.; Nishiyama Y.; Ishii Y. Acc. Chem. Res. 2013, 46 (9), 2127. 10.1021/ar4000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe N. P.; Parthasarathy S.; Jones C. R.; Bhardwaj C.; Long F.; Kotecha M.; Mehboob S.; Fung L. W.-M.; Past J.; Samoson A.; Ishii Y. Nat. Methods 2009, 6 (3), 215. 10.1038/nmeth.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski J. R.; Sein J.; Sass H. J.; Grzesiek S.; Blackledge M.; Emsley L. J. Am. Chem. Soc. 2010, 132 (24), 8252. 10.1021/ja102744b. [DOI] [PubMed] [Google Scholar]
- Lewandowski J. R. Acc. Chem. Res. 2013, 46 (9), 2018. 10.1021/ar300334g. [DOI] [PubMed] [Google Scholar]
- Björck L.; Kronvall G. J. Immunol. 1984, 133 (2), 969. [PubMed] [Google Scholar]
- Nordenfelt P.; Waldemarson S.; Linder A.; Mörgelin M.; Karlsson C.; Malmström J.; Björck L. J. Exp. Med. 2012, 209 (13), 2367. 10.1084/jem.20120325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S.; Seu C.; Kovacs Z.; Sherry A. D.; Chen Y. J. Am. Chem. Soc. 2006, 128 (41), 13474. 10.1021/ja0634526. [DOI] [PubMed] [Google Scholar]
- Hocking H. G.; Zangger K.; Madl T. ChemPhysChem 2013, 14 (13), 3082. 10.1002/cphc.201300219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich S. J.; Hölper S.; Glaubitz C. J. Biomol. NMR 2014, 58 (1), 27. 10.1007/s10858-013-9800-4. [DOI] [PubMed] [Google Scholar]
- Lewandowski J. R.; Dumez J.-N.; Akbey Ü.; Lange S.; Emsley L.; Oschkinat H. J. Phys. Chem. Lett. 2011, 2 (17), 2205. 10.1021/jz200844n. [DOI] [Google Scholar]
- Andreas L. B.; Jaudzems K.; Stanek J.; Lalli D.; Bertarello A.; Le Marchand T.; Cala-De Paepe D.; Kotelovica S.; Akopjana I.; Knott B.; Wegner S.; Engelke F.; Lesage A.; Emsley L.; Tars K.; Herrmann T.; Pintacuda G. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (33), 9187. 10.1073/pnas.1602248113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann J. J.; Agarwal V.; Hellwagner J.; Lends A.; Cadalbert R.; Meier B. H.; Ernst M. J. Biomol. NMR 2016, 66 (4), 233. 10.1007/s10858-016-0071-8. [DOI] [PubMed] [Google Scholar]
- Ye Y. Q.; Malon M.; Martineau C.; Taulelle F.; Nishiyama Y. J. Magn. Reson. 2014, 239, 75. 10.1016/j.jmr.2013.12.010. [DOI] [PubMed] [Google Scholar]
- Chevelkov V.; Diehl A.; Reif B. J. Chem. Phys. 2008, 128 (5), 052316. 10.1063/1.2819311. [DOI] [PubMed] [Google Scholar]
- Giraud N.; Blackledge M.; Goldman M.; Böckmann A.; Lesage A.; Penin F.; Emsley L. J. Am. Chem. Soc. 2005, 127 (51), 18190. 10.1021/ja055182h. [DOI] [PubMed] [Google Scholar]
- Krushelnitsky A.; Bräuniger T.; Reichert D. J. Magn. Reson. 2006, 182 (2), 339. 10.1016/j.jmr.2006.06.028. [DOI] [PubMed] [Google Scholar]
- Smith A. A.; Testori E.; Cadalbert R.; Meier B. H.; Ernst M. J. Biomol. NMR 2016, 65 (3), 171. 10.1007/s10858-016-0047-8. [DOI] [PubMed] [Google Scholar]
- Haller J. D.; Schanda P. J. Biomol. NMR 2013, 57 (3), 263. 10.1007/s10858-013-9787-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krushelnitsky A.; Zinkevich T.; Reichert D.; Chevelkov V.; Reif B. J. Am. Chem. Soc. 2010, 132 (34), 11850. 10.1021/ja103582n. [DOI] [PubMed] [Google Scholar]
- Lewandowski J. R.; Sass H. J. J.; Grzesiek S.; Blackledge M.; Emsley L. J. Am. Chem. Soc. 2011, 133 (42), 16762. 10.1021/ja206815h. [DOI] [PubMed] [Google Scholar]
- Hartlmüller C.; Günther J. C.; Wolter A. C.; Wöhnert J.; Sattler M.; Madl T. Sci. Rep. 2017, 7 (1), 5393. 10.1038/s41598-017-05821-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvignies G.; Bernadó P.; Meier S.; Cho K.; Grzesiek S.; Brüschweiler R.; Blackledge M. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (39), 13885. 10.1073/pnas.0505129102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuszewski J.; Gronenborn A. M.; Clore G. M. J. Am. Chem. Soc. 1999, 121 (10), 2337. 10.1021/ja9843730. [DOI] [Google Scholar]
- Frericks Schmidt H. L.; Sperling L. J.; Gao Y. G.; Wylie B. J.; Boettcher J. M.; Wilson S. R.; Rienstra C. M. J. Phys. Chem. B 2007, 111 (51), 14362. 10.1021/jp075531p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher T.; Alexander P.; Bryan P.; Gilliland G. L. Biochemistry 1994, 33, 4721. 10.1021/bi00181a032. [DOI] [PubMed] [Google Scholar]
- Lamley J. M.; Lougher M. J.; Sass H. J.; Rogowski M.; Grzesiek S.; Lewandowski J. R. Phys. Chem. Chem. Phys. 2015, 17 (34), 21997. 10.1039/C5CP03484A. [DOI] [PubMed] [Google Scholar]
- Derrick J. P.; Wigley D. B. Nature 1992, 359, 752. 10.1038/359752a0. [DOI] [PubMed] [Google Scholar]
- Lian L.-Y.; Barsukov I. L. L.; Derrick J. P. P.; Roberts G. C. K. C. K. Nat. Struct. Mol. Biol. 1994, 1 (6), 355. 10.1038/nsb0694-355. [DOI] [PubMed] [Google Scholar]
- Gronenborn A. M.; Clore G. M. J. Mol. Biol. 1993, 233, 331. 10.1006/jmbi.1993.1514. [DOI] [PubMed] [Google Scholar]
- Sauer-Eriksson A. E.; Kleywegt G. J.; Uhlén M.; Jones T. A. Structure 1995, 3 (3), 265. 10.1016/S0969-2126(01)00157-5. [DOI] [PubMed] [Google Scholar]
- Gu X.-H.; Gong Z.; Guo D.-C.; Zhang W.-P.; Tang C. J. Biomol. NMR 2014, 58 (3), 149. 10.1007/s10858-014-9817-3. [DOI] [PubMed] [Google Scholar]
- Vajpai N.; Gentner M.; Huang J.-R.; Blackledge M.; Grzesiek S. J. Am. Chem. Soc. 2010, 132 (9), 3196. 10.1021/ja910331t. [DOI] [PubMed] [Google Scholar]
- Franks W. T.; Zhou D. H.; Wylie B. J.; Money B. G.; Graesser D. T.; Frericks H. L.; Sahota G.; Rienstra C. M. J. Am. Chem. Soc. 2005, 127 (35), 12291. 10.1021/ja044497e. [DOI] [PubMed] [Google Scholar]
- Fogh R. H.; Ionides J.; Ulrich E. L.; Boucher W.; Vranken W. F.; Linge J. P.; Habeck M.; Rieping W.; Bhat T. N.; Westbrook J.; Henrick K.; Gilliland G.; Berman H.; Thornton J.; Nilges M.; Markley J.; Laue E. Nat. Struct. Biol. 2002, 9 (6), 416. 10.1038/nsb0602-416. [DOI] [PubMed] [Google Scholar]
- Zhou D. H.; Rienstra C. M. J. Magn. Reson. 2008, 192 (1), 167. 10.1016/j.jmr.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. J. Comput. Chem. 2004, 25 (13), 1605. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Derrick J. P.; Wigley D. B. J. Mol. Biol. 1994, 243 (5), 906. 10.1006/jmbi.1994.1691. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.