

Abstract
Heparanase is an enzyme which cleaves heparan sulfate (HS) polysaccharides of the extracellular matrix. It is a regulator of tumor behavior, plays a key role in kidney related diseases and autoimmune diabetes. We report herein the use of computational studies to extract the natural HS-heparanase interactions as a template for the design of HS mimicking glycopolymers. Upon evaluation, glycopolymer with 12 repeating units was determined to be the most potent inhibitor and to have tight-binding characteristics. This glycopolymer also lacks anticoagulant activity.
Graphical abstract
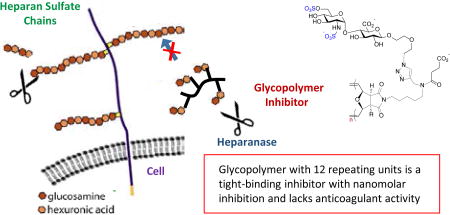
Heparanase is an endo-β-d-glucuronidase that hydrolyzes heparan sulfate (HS) polysaccharide chains in the extracellular matrix (ECM).1 This enzyme is regarded as a regulator of aggressive tumor behavior as recent clinical studies have demonstrated that raised heparanase levels correlated with increased tumor size,1c amplified tumor angiogenesis,1d enhanced metastasis,1e and poor patient prognosis.1f The enzyme cleaves GlcAβ1,4)GlcNS glycosidic bonds (Figure 1) of the HS chains releasing sequestered pools of HS oligosaccharide-binding growth factors for signaling activation to promote angiogenesis.2 Cleavage of HS also degrades the structural integrity of the basement membrane and ECM, permitting malignant cells to enter the blood stream and metastasis.3 Heparanase also plays key a role in Type 1 diabetes as it destroys HS chains within the insulin-producing β cells and causes their death.4 Recently, it has been reported that degradation of HS chains aids in the progression of nephrological diseases5 and releases bound herpes simplex virus-1 for entry into the host.6
Fig. 1.

Heparanase and schematic design of its inhibitors
Heparanase and its main activity has been known for over 30 years.7 A recent major breakthrough was the report of the crystal structure of the enzyme in its apo form as well as when bound to several HS ligands.8 In the crystal structure the ligand was bound inside a 10Å binding cleft with the GlcAβ1,4)GlcNS in the +1 and −1 subsites (Figure 1),9 adjacent to the catalytic residues (Glu225 and Glu343).1 This cleft is flanked by heparin-binding domains (HBD-1 and HBD-2).8 Inhibitors of heparanase are anticancer therapeutics, with carbohydrate mimetics being advanced to clinical trials.1
Although HS mimetics are potent heparanase inhibitors, they are heterogeneous and nonspecific, which could lead to unforeseen adverse effects.10 Heparin inhibits heparanase (IC50=1 µg/mL),12 but its anticoagulant activity has limited its use for cancer treatment due to the risk of bleeding complications.10 Heparin derivatives, SST0001 and M402,13 are known to enhance heparanase inhibition; while SST001 finished Phase I trials in 2016, M402 was halted after Phase I trials. PI-88, a sulfated oligomannan mixture, is heterogeneous in both size and sulfation pattern.14 PI-88’s clinical trials were ended, as patients developed antibody-induced thrombocytopenia.1, 15 While PG545 completed Phase I clincal trials in 2015, it had to switch from subcutaneous injection to IV due to injection site reaction.1a Clearly, there is an urgent need to identify structurally-defined, potent and specific heparanase inhibitors that overcome these limitations.1a,b
To identify novel heparanase inhibitors of known structure, one could consider synthetic HS polysaccharides. However, well-defined polysaccharide synthesis is a formidable challenge. Thus, saccharide- functionalized neo-glycopolymers, which retain the key biological properties of the natural polysaccharides, could be explored.16 HS mimicking neo-glycopolymers have yet been examined as potential inhibitors of heparanase.16 Herein, we describe the design, synthesis, and evaluation of glycopolymers for inhibiting heparanase activity. Our strategy addresses the unmet challenges of heterogeneous sulfation, cross-bioactivity, and HS polysaccharide mimetics.
We designed monomer 2 containing a polymerizable scaffold17 and the [GlcNS(6S)α(1,4)GlcA] disaccharide unit (Figure 2), essential for heparanase recognition.18 Monomer 2 will then undergo ring-opening metathesis polymerization (ROMP) to form glycopolymers that could mimic the multivalent nature of HS polysaccharides.16 The disaccharide unit of 2 lacks iduronic acid (IdA) unit, a key for the anticoagulant properties that could have limited the use of other HS carbohydrate mimetics.16c Since the distance between HBD-1 and HBD-2 is about 25 Å (Figure S10), the [GlcNS(6S)α(1,4)GlcA] moiety was joined to the diethylene glycol-containing azide linker that could interact with heparanase’s catalytic site19 and undergo a “click” reaction with an alkyne unit of a scaffold A (Figure 1).17a We propose that the scaffold A’s carboxylate group could interact with the positively charged residues of heparanase’s HBDs. Monomer 3 serves as a control to determine the effect of the carboxylate group on the binding. Since heparanase hydrolyzes the β-glycosidic bond of GlcAβ1,4)GlcNS, replacement of the GlcNS unit with an aliphatic scaffold as seen in 2 and 3 could potentially disrupt heparanase’s hydrolytic nature.19 The monomers are endowed with a strained bicyclic ring that undergoes ROMP to generate the glycopolymers.17 Incorporation of carbohydrate units onto ROMP polymers was first conceived by Kiessling. 16e ROMP based polymer systems were later explored by the Hsieh-Wilson group to generate [IdoAβ1,4)GlcNS]-functionalized polymers as anticoagulant heparin mimetics.16c
Fig. 2.

Rational design of HS-mimicking monomers
To validate our design, docking studies were explored. Since there are no computational programs that could manage the docking of glycopolymers, the monomeric precursors were investigated in our computational studies. We commenced our docking studies first with tetrasaccharide 1 (Figure 2), a heparanase substrate, into the apo crystal structure of human heparanase (PDB code: 5E8M).8 We chose 1 as a model substrate to determine its location in the binding pocket and as a comparison to our designed monomers. Several salt bridges were formed, with key residues being −2 6-O-sulfate with Lys159 and +1 N-sulfate with Lys231 (Figure S11). The ligand-receptor complex was then subjected to a molecular dynamics (MD) simulation to validate the structure’s stability (Figure S11). During the simulation, a hydrogen bonding network between −2 N-sulfate with Asn64, Gly389, and Tyr391 anchored the GlcA–GlcNS(6S) bond of 1 in close proximity (3.2 – 3.75 Å) to the catalytic residues for cleavage (Figure S11).20 The docking of 2 and 3 was next examined. Analysis of 2 and 3 placed GlcNS(6S)α(1,4)GlcA moiety at the −1/−2 subsite (Figure S11) analogous to 1. Intriguingly, the triazole moiety of these monomers had π-cation and hydrogen bonding interactions with the active site of heparanase. Most importantly, the orientation and distances of the cleavable bond to Glu225 and Glu343 were now farther, and skewed away, making hydrolysis unlikely (Figure S11).20 The attached aliphatic side chain of the scaffold was found in the binding groove forming a network of hydrophobic, π-cation, and hydrogen bonding interactions.19c The strained bicyclic ring of the scaffold tended to stay above the binding groove during the duration of the MD simulation and negates any differences between the linkers in this portion. The difference between monomers 2 and 3 were their interactions with HBD-2. While 3 had little interactions with HBD-2, the carboxylate of 2 interacted with HBD-2 (Figure S11).
To determine if these structural differences corresponded to in vitro changes in inhibition of heparanase, the synthesis of monomers 2 and 3 were then investigated. Due to the 1,2-cis-2-amino linkage nature of the GlcNS(6S)α(1,4)GlcA disaccharide unit, control of its stereoselective formation is quite challenging.21 Capitalizing on our reported nickel-catalyzed 1,2-cis-2-aminoglycoside methodology,22 we investigated the direct coupling of GlcA acceptor 7 with GlcN donors 4 – 6 (Scheme 1) having pivaloyl (Piv), triethylsilyl (TES), and benzyl (Bn) groups, respectively, at O-4. Under our nickel conditions, disaccharides 9–11 were formed in 52–85% yield with exclusive α-selectivity. To explore the reproducibility, 11 was chosen for a large-scale preparation. The product 11, utilized for later preparation of 24 (Figure 4), was isolated in similar yield and α-selectivity (2.1 g, 79% yield, α only).
Scheme 1.

Synthesis of GlcNS(6S)α(1,4)GlcA precursors
Fig. 4.

Tight-binding type graph of glycopolymer 22
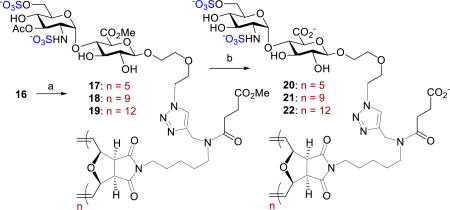
With the ability to produce the GlcNS(6S)α(1,4)GlcA disaccharide precursors in stereoselective control and large quantities via nickel catalysis, we next investigated the suitable protecting groups at O-4 position of GlcN donor and at O-2 and O-3 of GlcA acceptor that has some degree of orthogonality among protecting and functional groups and can be selectively cleaved prior to the ROMP event. To this end, the naphthylmethyl (NAP) group is suitable and could be removed using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ). With these considerations in mind, we conducted the glycosylation of GlcA acceptor 8 with GlcN donor 12 (Scheme 2). Disaccharide 13 was stereoselectively prepared in high yield (79%, α only). We then explored the conditions for sulfation at C(6)-hydroxyl and C(2)-amino groups. We were delighted to find that the C(6)-acetyl group in 13 could be selectively removed using sodium methoxide (1.5 equiv.) in methanol. The N-benzylidene group was removed in less than 5 min with HCl in acetone. Selective sulfation with SO3·Me3N yielded sulfated disaccharide 14 in 65% over three steps. Subsequent DDQ treatment of 14 afforded 15 in 75% yield. A “click” reaction of azide 15 with the alkyne unit of scaffold A (Figure 1) provided 16, which would be utilized for later polymerizations. Global hydrolysis of 16 provided diantennary monomer 2 in 94% yield (Scheme 2).
Scheme 2.

Synthesis of monomer 2. Conditions: a) 15 mol% Ni(OTf)2, 8, CH2Cl2, 35 °C, 12 h, 79%, α only; b) NaOMe, MeOH; c) HCl, acetone; d) SO3.Me3N, Et3N, DMF, 55 °C, 65%, 3 steps; e) DDQ, PBS/CH2Cl2, 75%; f) scaffold A, CuI, DBU, DMF, 50 °C, 64%; g) LiOH, THF/H2O, 94%.
On the basis of docking studies (Figure S11) and the heparanase inhibition activities (vide infra, Figure 3), we investigated a series conditions for ROMP of monomer 16 with Grubbs’ III catalyst 23 (Table 1). Use of weakly coordinating 2,2,2-trifluoro-ethanol (TFE) as part of the co-solvent system, [2.5:1, (CH2Cl)2/TFE], prevented catalyst deactivation and allowed ROMP to proceed to completion within 1 h. Varying the amount of catalyst (9 – 20 mol%) allowed for precise control over the degrees of polymerization, (DP = 5 – 12). Increasing DP is known to increase binding,16c unfortunately under these conditions lowering catalyst below 9 mol% resulted in low conversion. After polymerization, both DP and molecular weights (Mn) were determined by 1H-NMR end group analysis (Table 1).23 Polymers were then hydrolyzed and purified by dialysis to remove impurities, affording polymers 20–22 (Table 1). The turbidimetric assay was also conducted to quantify sulfate content in polymers, and sulfate content is equal to 1.86 per disaccharide unit.24 Due to their amphiphilic nature, these glycopolymers aggregate to form micelles. Using transmission electron microscopy (TEM) and dynamic light scattering (DLS), the shape and size of these polymers 20–22 were determined to be spherical with uniform Z-average radii in the range of 70 – 90 nm (PDI = 14.7–20.1%) (Figures S4 and S5). Fig. 3 Inhibition of heparanase by HS mimicking monomers and neo-glycopolymers using TR-FRET assay.
Fig. 3.

Inhibition of heparanase by HS mimicking monomers and neo-glycopolymers using TR-FRET assay
Table 1.
Synthesis of HS mimicking glycopolymers. Conditions: a) (H2IMes)(3-BrPy)2(Cl)2Ru=CHPh (23), DCE/TFE (2.5:1, 55 °C, 1 h. b) LiOH, H2O/THF, H2O, 20 (80%), 21 (83%), 22 (89%).
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Polymers | DPn | 23 Mol% |
DCE:TFE | Conv. (%) |
Yield (%) |
Mn (Thereotical) |
Mn (NMR) |
| 1 | 17 | 5 | 20 | 2.5:1 | 100 | 81 | 5,425 | 5,425 |
| 2 | 18 | 9 | 11.5 | 2.5:1 | 100 | 97 | 9,200 | 9,765 |
| 3 | 19 | 12 | 9 | 2.5:1 | 100 | 89 | 12,075 | 13,030 |
The heparanase inhibition activities of monomers 2 and 3 (see Scheme S1) as well as polymers 20–22 were then assessed by in vitro TR-FRET assay against fluorescent-tagged heparan sulfate.25 For the monomer series (Figure 3), monoantennary monomer 3 exhibited no increase in binding (49.44 ± 1.56 µM) when compared to that of control disaccharide 24 with no attached scaffold (IC50 = 50.07 ± 3.46 µM). The results suggest that the strained oxanorbornene system and attached linker had no influence on binding, as seen in the MD simulation. Diantennary monomer 2 is the most potent compound with IC50 value of 11.59 ± 0.95 µM. Comparing monomers 2 and 3, it was deemed that the carboxylate of the diantennary scaffold is of importance as it enhances the binding to heparanase (R&D Systems). The increase in binding was apparent in the inhibition of heparanase with glycopolymers 20 – 22, as IC50 values decreased by 1000-fold to the nanomolar range (Figure 3) even at a low DP of five (20, 7.49 ± 0.48 nM). Polymer 21 (DP = 9) was slightly more effective with an IC50 value of 4.43 ± 0.13 nM. Polymer 22 (n=12) was the most potent inhibitor. We also measured the inhibitory activity of heparin (MW = 18kDa) against heparanase (IC50 = 0.54 ± 0.028 nM). Interestingly, when fitting the saturation curve for inhibition of 22 to the standard hyperbolic function, a high degree of variation was observed. When applying the standard hyperbolic fit, the IC50 for 22 was comparable to the enzyme concentration.26 As such, refitting the inhibition of 22 (Figure 4) to a tight-binding equation (see SI) resulted in a fit with little variation from the data and extrapolated an IC50 value of 0.10 ± 0.036 nM.27 Importantly, critical micelle concentration (CMC) value of 22 (3.3 µM, see Figure S2)28 is significantly above its IC50 value, suggesting that heparanase is inhibited by unicellular glycopolymer strands rather than micelles.29 We also performed CMC of 22 at different concentrations below its measured CMC using the same FRET assay conditions. We observed that there was no change in the fluorescence ratio of fluorogenic probe pyrene, suggesting that the inhibitory activity is not due to aggregation assisted by heparanase.
We also assessed anticoagulant activity of 22 in comparison to heparin (MW = 18 kDa) and LMWH (MW = 3 kDa).16c Polymer 22 (IC50 > 4500 nM) did not inhibit either FXa or FIIa (see SI). To compare, LMWH exhibited a much higher activity against FXa (266 ± 11.5 nM) and heparin for FIIa (4.63 ± 0.22 nM) (Figures S6 and S7). These results illustrate 22 is as potent as heparin at inhibiting heparanase activity. In contrast to heparin, 22 has no cross-anticoagulant activity.
A colorimetric assay was next performed to determine if cleavage of the disaccharide unit from the polymer scaffold of 22 is possible using a tetrazolium sodium salt (WST-1), which reacts with a newly formed anomeric hemiacetal (Figure 5).30 Over 21 h, there was no change in absorbance for the sample containing a mixture of 22 and heparanase relative to the control. This result suggests that 22 is not hydrolyzed by heparanase. In stark contrast, Fondaparinux, whose structure incorporates a scissible disaccharide GlcAβ1,4)GlcNS(6S) unit, was readily cleaved by heparanase (Figure 5).
Fig. 5.

The hydrolytic potential over time of heparanase on 22 and Fondaparinux.
Rational design allows access synthetically well-defined HS-mimicking neo-glycopolymers, with control and predictable degree of polymerization, which are highly potent against heparanase and exhibit no anticoagulant activity. Several key biological assays are under investigation to illustrate that the glycopolymers are viable for cancer therapeutics and will be reported in due course.
Financial support from the NIH (GM098285) and the University of Iowa (computational resources) are acknowledged. The authors also acknowledge Dr. Sai Ramadugu for assistance in computational studies, the Sebag group for assistance in TR-FRET assay as well as Professors Dan Quinn and Todd Lowary for helpful discussions.
Supplementary Material
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures, computational data, and NMR spectra. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.(a) Rivara S, Milazzo FM, Giannini G. Future Med. Chem. 2016;8:647. doi: 10.4155/fmc-2016-0012. [DOI] [PubMed] [Google Scholar]; (b) Vlodavsky I, Singh P, Boyango I, Gutter-Kapon L, Elkin M, Sanderson RD, Ilan N. Drug Resist. Updates. 2016;29:54. doi: 10.1016/j.drup.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Maxhimer JB, Quiros RM, Stewart R, Dowlatshahi K, Gattuso P, Fan M, Prinz RA, Xu X. Surgery. 2002;132:326. doi: 10.1067/msy.2002.125719. [DOI] [PubMed] [Google Scholar]; (d) Elkin M, Ilan N, Ishai-Michaeli R, Friedmann Y, Papo O, Pecker I, Vlodavsky I. FASEB J. 2001;15:1661. doi: 10.1096/fj.00-0895fje. [DOI] [PubMed] [Google Scholar]; (e) Cohen E, Doweck I, Naroditsky I, Ben-Izhak O, Kremer R, Best LA, Vlodavsky I, Ilan N. Cancer. 2008;113:1004. doi: 10.1002/cncr.23680. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ramani VC, Vlodavski I, Ng M, Zhang Y, Barbieri P, Noseda A, Sanderson RD. Matrix Biol. 2016;55:22. doi: 10.1016/j.matbio.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Vlodavsky I, Friedmann Y. J Clin. Invest. 2001;108:341. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Iozzo RV, San Antonio JD. J. Clin. Invest. 2001;108:349. doi: 10.1172/JCI13738. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Reiland J, Kempf D, Roy M, Denkins Y, Marchetti D. Neoplasia. 2006;8:596. doi: 10.1593/neo.06244. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Barker HE, Paget JTE, Khan AA, Harrington KJ. Nat. Rev. Cancer. 2015;15:409. doi: 10.1038/nrc3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanderson RD. Semin. Cell Dev. Biol. 2001;12:89. doi: 10.1006/scdb.2000.0241. [DOI] [PubMed] [Google Scholar]
- 4.Ziolkowski AF, Popp SK, Freeman C, Parish CR, Simeonovic CJ. J. Clin. Invest. 2012;122:132. doi: 10.1172/JCI46177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van den Hoven MJ, Rops AL, Vlodavsky I, Levidiotis V, Berden JH, van der Vlag J. Kidney Int. 2007;72:543. doi: 10.1038/sj.ki.5002337. [DOI] [PubMed] [Google Scholar]
- 6.Hadigal SR, Agelidis AM, Karasneh GA, Antoine TE, Yakoub AM, Ramini VC, Djalilian AR, Sanderson RD, Shukla D. Nat. Comm. 2015;6:6985. doi: 10.1038/ncomms7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Vlodavsky I, Friedmann Y, Elkin M, Aingorn H, Atzmon R, Ishai-Michaeli R, Bitan M, Pappo O, Peretz T, Michal I, Spector L, Pecker I. Nat. Med. 1999;5:793. doi: 10.1038/10518. [DOI] [PubMed] [Google Scholar]; (b) Nakajima N, Irimura T, Di Ferrante N, Nicolson GL. J. Biol. Chem. 1984;259:2283. [PubMed] [Google Scholar]
- 8.Wu L, Viola CM, Brzozowski AM, Davies GJ. Nat. Struct. Mol. Biol. 2015;22:1016. doi: 10.1038/nsmb.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davies GJ, Wilson KS, Henrissat B. Biochem. J. 1997;321:557. doi: 10.1042/bj3210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Guerrini M, Beccati D, et al. Nat. Biotechnol. 2008;26:669. doi: 10.1038/nbt1407. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Warkentin TE, Levine MN, Hirsh J, Horsewood P, Roberts RS, Gent M, Kelton JG. N. Engl. J. Med. 1995;332:1330. doi: 10.1056/NEJM199505183322003. [DOI] [PubMed] [Google Scholar]
- 11.Sarrazin S, Lamanna WC, Esko JD. Cold Spring Harb. Perspect. Biol. 2011;3:a004952. doi: 10.1101/cshperspect.a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Bar-Ner M, Eldor A, Wasserman L, Matzner Y, Cohen IR, Fuks Z, Vlodavsky I. Blood. 1987;70:551. [PubMed] [Google Scholar]; (b) Parish CR, Freeman C, Brown KJ, Francis DJ, Cowden WB. Cancer Res. 1999;59:3433–3441. [PubMed] [Google Scholar]
- 13.(a) Naggi A, Casu B, Perez M, Torri G, Cassinelli G, Penco S, Pisano C, Giannini G, Ishai-Michaeli R, Vlodavsky I. J. Biol. Chem. 2005;280:12103. doi: 10.1074/jbc.M414217200. [DOI] [PubMed] [Google Scholar]; (b) Zhou H, Roy S, Cochran E, et al. PLoS ONE. 2011;6:e21106. doi: 10.1371/journal.pone.0021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferro V, Fewings K, Palermo M, Li C. Carbohydr. Res. 2001;332:183. doi: 10.1016/s0008-6215(01)00061-1. [DOI] [PubMed] [Google Scholar]
- 15.Kudchadkar R, Gonzalez R, Lewis KD. Expert Opin. Invest. Drugs. 2008;17:1769. doi: 10.1517/13543784.17.11.1769. [DOI] [PubMed] [Google Scholar]
- 16.(a) Rabuka D, Forstner MB, Groves JT, Bertozzi CR. J. Am. Chem. Soc. 2008;130:5947. doi: 10.1021/ja710644g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kiessling LL, Grim JC. Chem. Soc. Rev. 2013;42:4476. doi: 10.1039/c3cs60097a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Oh YI, Sheng GJ, Chang S-K, Hsieh-Wilson LC. Angew. Chem. Int. Ed. 2013;52:11796. doi: 10.1002/anie.201306968. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sheng GJ, Oh YI, Chang S-K, Hsieh-Wilson LC. J. Am. Chem. Soc. 2013;135:10898. doi: 10.1021/ja4027727. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Spaltenstein A, Whitesides GM. J. Am. Chem. Soc. 1991;113:686. [Google Scholar]; (f) Mortell KH, Gingras M, Kiessling LL. J. Am. Chem. Soc. 1994;116:12053–12054. For glycopolymer GAGs mimic review: [Google Scholar]; (g) Paluck SJ, Nguyen TH, Maynard HD. Biomacromolecules. 2016;17:3417. doi: 10.1021/acs.biomac.6b01147. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Miura Y, Fukuda T, Seto H, Hoshino Y. Polym. J. 2016;48:229. [Google Scholar]
- 17.(a) Johnson JA, Lu YY, Burts AO, Xia Y, Durrell AC, Tirrell DA, Grubbs RH. Macromolecules. 2010;43:10326. doi: 10.1021/ma1021506. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Loka RS, McConnell MS, Nguyen HM. Biomacromolecules. 2015;16:4013. doi: 10.1021/acs.biomac.5b01380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peterson SB, Liu J. Matrix Biol. 2013;32:223. doi: 10.1016/j.matbio.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 19.(a) Pearson AG, Kiefel MJ, Ferro V, von Itzstein M. Org. Biomol. Chem. 2011;9:4614. doi: 10.1039/c1ob05165b. [DOI] [PubMed] [Google Scholar]; (b) Ferro V, Liu L, et al. J. Med. Chem. 2012;55:3804. doi: 10.1021/jm201708h. [DOI] [PubMed] [Google Scholar]; (c) Spaltenstein A, Whitesides GM. J. Am. Chem. Soc. 1991;113:686. [Google Scholar]
- 20.Gandhi NS, Freeman C, Parish CR, Mancera RL. Glycobiology. 2012;22:35. doi: 10.1093/glycob/cwr095. [DOI] [PubMed] [Google Scholar]
- 21.Dulaney SB, Huang X. Adv. Carbohydr Chem. Biochem. 2012;67:95. doi: 10.1016/B978-0-12-396527-1.00003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sletten ET, Ramadugu SK, Nguyen HM. Carbohydr. Res. 2016;435:195. doi: 10.1016/j.carres.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radzinski SC, Foster JC, Chapleski RC, Troya D, Matson JB. J. Am. Chem. Soc. 2016;138:6998. doi: 10.1021/jacs.5b13317. [DOI] [PubMed] [Google Scholar]
- 24.Terho TT, Hartiala K. Anal. Biochem. 1971;41:471–476. doi: 10.1016/0003-2697(71)90167-9. [DOI] [PubMed] [Google Scholar]
- 25.Roy S, El Hadri A, Richard S, Denis F, Holte K, Duffner J, Yu F, Galcheva-Gargova Z, Capila I, Schultes B, Petitou M, Kaundinya GV. J. Med. Chem. 2014;57:4511. doi: 10.1021/jm4016069. [DOI] [PubMed] [Google Scholar]
- 26.Henderson PJF. Biochem. J. 1972;127:321. doi: 10.1042/bj1270321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The assay was repeated with different batch of 22 to enure the result is reproducible (IC50 of 22 = 0.11 ± 0.05 nM, see SI).
- 28.Kalyanasundaram K, Thomas JK. J. Am. Chem. Soc. 1977;99:2039. [Google Scholar]
- 29.We also conducted a control heparanase assay by replacing ionic CHAPS with non-ionic tween-20 detergent (IC50 of 22 = 0.080 ± 0.04 nM). This result suggests that aggregation is not responsible for the inhibition.
- 30.Hammond E, Li CP, Ferro V. Anal. Biochem. 2010;396:112. doi: 10.1016/j.ab.2009.09.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.