
Figure 10.
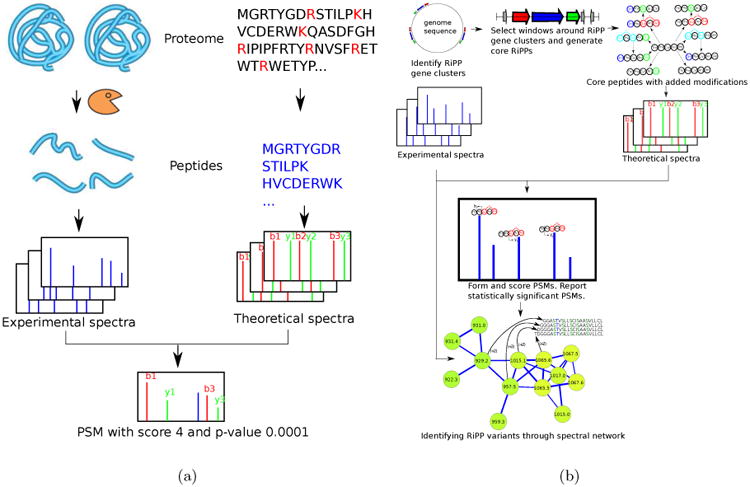
(a) Standard proteomic database search tools (e.g., Sequest [9]) are based on digesting the proteins by an enzyme, and collecting tandem spectra of the resulting peptides. Each spectrum is matched against theoretical spectra of all peptides in a protein database (with mass equal to the the precursor mass of the spectrum) and PSMs with highest scores/lowest p-values are reported. (b) RiPPquest pipeline starts with genome mining for RiPP biosynthetic gene clusters. Genome mining identifies short windows where genes encoding RiPPs are located. Short ORFs are identified in the six frame translation of the selected windows, and putative core RiPPs are selected from these ORFs. Various combinations of modifications are applied to core RiPPs, and the resulting mature RiPPs are scored against the spectral dataset by analyzing all possible PSMs using spectral alignment. Spectral network analysis helps in discovery of novel variants of RiPPs with various modifications.