

Abstract
Mutations in K-Ras and epidermal growth factor receptor (EGFR) are mutually exclusive, but it is not known how K-Ras activation inactivates EGFR, leading to resistance of cancer cells to anti-EGFR therapy. Here, we report that the K-Ras effector p38γ MAPK confers intrinsic resistance to small molecular tyrosine kinase inhibitors (TKIs) by concurrently stimulating EGFR gene transcription and protein dephosphorylation. We found that p38γ increases EGFR transcription by c-Jun-mediated promoter binding and stimulates EGFR dephosphorylation via activation of protein-tyrosine phosphatase H1 (PTPH1). Silencing the p38γ/c-Jun/PTPH1 signaling network increased sensitivities to TKIs in K-Ras mutant cells in which EGFR knockdown inhibited growth. Similar results were obtained with the p38γ-specific pharmacological inhibitor pirfenidone. These results indicate that in K-Ras mutant cancers, EGFR activity is regulated by the p38γ/c-Jun/PTPH1 signaling network, whose disruption may be a novel strategy to restore the sensitivity to TKIs.
Keywords: epidermal growth factor receptor (EGFR), mitogen-activated protein kinase (MAPK), Ras protein, signal transduction, tumor cell biology
Introduction
Epidermal growth factor receptor (EGFR)3 is a trans-membrane protein-tyrosine kinase and plays a critical role in promoting cell proliferation. In response to the ligand EGF, the receptor dimerizes, leading to autophosphorylation of multiple tyrosine residues on its intracellular domain (1). This in turn activates downstream proliferative pathways, such as Ras/MAPKs and PI3K/AKT (1). EGFR is activated via overexpression and amplification in colon, pancreas, lung, and breast cancers and via mutations in lung and brain tumors. Inhibiting EGFR is therefore considered to be an important anti-cancer strategy (1). EGFR can be inhibited by small molecule tyrosine kinase inhibitors (TKIs) that compete for the ATP-binding site of the catalytic domain (2) and by anti-EGFR antibodies that block the extracellular ligand-binding domain (3). Although anti-EGFR therapy has great potential, resistance remains a major obstacle for its successful clinical application (4).
K-Ras mutations occur in up to 50% of colon cancers, 95% of pancreatic cancers, and 25% of lung cancers (5). Such mutations are invariably associated with the intrinsic resistance to anti-EGFR therapy (4, 6). Recent studies show that mutant K-Ras also causes acquired resistance to anti-EGFR treatment (7). However, the mechanisms by which K-Ras mutation confers the resistance are largely unknown (1, 4, 6). Mutant K-Ras drives malignant initiation and progression through effector pathways downstream of EGFR (1, 5). But K-Ras can also directly regulate EGFR activity and endogenous EGFR is still required for K-Ras tumorigenesis (8–10). These results together suggest an active mechanism by which mutant K-Ras inactivates EGFR therapeutic target activity while still depending on its intrinsic oncogenic potential for K-Ras dependent malignant growth and progression. This potential mechanism could be exploited to develop novel strategies to overcome resistance to anti-EGFR therapy in K-Ras mutant cancers.
p38γ MAPK (gene name: MAPK12), a member of the non-canonical p38 protein family, has a unique PDZ motif on its C terminus (11). p38γ promotes K-Ras invasive and transforming activity in intestinal epithelial cells (12–14). Through its PDZ motif, p38γ binds, phosphorylates, and thus activates protein-tyrosine phosphatase H1 (PTPH1) (13, 15). PTPH1 in turn catalyzes tyrosine dephosphorylation of key signaling molecules, such as EGFR and estrogen receptor (15–17). Moreover, p38γ interacts with the transcription factor c-Jun, through which it is recruited to AP-1 target gene promoters, leading to AP-1–dependent gene expression and malignant progression (14, 18, 19). Importantly, p38γ is overexpressed in K-Ras mutant colon cancer cells (14, 15) and in several types of primary human tumors (13, 18, 20), and increased p38γ predicts a poor clinical prognosis (21, 22). Here, we tested the hypothesis that activated p38γ mediates K-Ras signaling to confer the intrinsic resistance to TKIs through its interaction and activation of c-Jun and PTPH1. Our results show that upon K-Ras mutation, activated p38γ stimulates EGFR gene transcription and protein tyrosine dephosphorylation through increased complex formation with c-Jun and PTPH1. Together, these lead to elevated levels of non-phosphorylated EGFR, which is unresponsive to TKIs but is still proliferative. Further analyses demonstrate that disruption of the p38γ/c-Jun/PTPH1 signaling network can restore the sensitivity to EGFR TKIs.
Results
K-Ras mutation or EGFR dephosphorylation causes intrinsic resistance to TKIs
To determine whether K-Ras mutation causes the unresponsiveness to TKIs as observed clinically (7), colon cancer cells with and without mutated K-Ras were analyzed for growth inhibition by the Food and Drug Administration-approved TKIs lapatinib (Lap) and gefitinib (Gef). EGFR antibody was not included in the analyses because it inhibits EGFR by a distinct mechanism (3). As shown in Fig. 1A and supplemental Fig. S1A, TKIs have no substantial effects on colony formation of HCT116 human colon cancer cells with mutant K-Ras, but they significantly suppress the growth in HCT116 sublines HKe3 and HK2-8, in which the mutant K-Ras allele was selectively disrupted by homologous recombination (23, 24). In a similar manner, doxycycline (Dox)-inducible knockdown of mutant K-Ras in LS174T (15, 25) increases the sensitivity, whereas a forced expression of a oncogenic K-Ras (G12V) in HKe3 cells led to the unresponsiveness (Fig. 1B and supplemental Fig. S1B) (22). These results show that K-Ras mutation causes the unresponsiveness to TKIs in cell culture and suggest that dissecting the signaling events in this system may be able to reveal novel mechanisms that could lead to ways to improve the sensitivity to TKIs.
Figure 1.

Resistance of K-Ras mutated cancer cells to EGFR TKIs couples both with increased p38γ, c-Jun, and EGFR expression and with decreased EGFR phosphorylation. A, the indicated cells were cultured in the presence and absence of TKIs (Lap (2 μm) and Gef (0.125 μm)) for about 2 weeks, and the number of colonies formed was manually counted. Results were normalized to DMSO control (mean ± S.D. (error bars), n = 3). B, cells were cultured with the indicated TKIs in the absence and presence of Dox for colony formation (mean ± S.D., n = 3), with the inset showing mutant K-Ras knockdown after Dox addition overnight. C and D, K-Ras WT and MT cancer cells were analyzed by WB (C, p-EGFR detected with anti-p-EGFR/Tyr-1173 antibody and the same for all other p-EGFR unless specified) and band intensities from these cell lines (C, asterisk indicates results from a separate experiment) were measured by Image Quant software (normalized to β-actin). Quantitative combined results from C are presented in D (mean ± S.D., n = 4). E, EGFR/Y1173F was stably expressed by retrovirus, and the engineered cells were assessed for colony formation. The bar graph (left and middle) is from three separate experiments (mean ± S.D., n = 3), whereas WB shows EGFR expression and phosphorylation (right, EGFR transfection used as a positive control for p-EGFR/Tyr-1173).
Our previous studies demonstrated elevated levels of p38γ and c-Jun protein expression in K-Ras mutant cells (14, 15). EGFR is an AP-1 target gene (26, 27), and its proliferative effects can be inhibited by TKIs through blocking EGFR phosphorylation (2, 28). We next investigated whether the hyperexpression of p38γ/c-Jun in K-Ras mutant cells can impact EGFR expression and phosphorylation. The results (Fig. 1, C and D) show increased p38γ, c-Jun, and EGFR protein levels but decreased EGFR phosphorylation in a panel of K-Ras mutant (MT) cells as compared with those with WT K-Ras. Because TKIs prevent EGFR phosphorylation, subsequently blocking activation of downstream proliferative pathways (29, 30), the unresponsiveness of K-Ras mutant cells to TKIs may be caused by decreased p-EGFR. To directly test this possibility, a non-phosphorable mutant EGFR (Y1173F) (31) was expressed by retroviral infection, and resultant cells were analyzed for growth inhibition by TKIs. Consistent with the decreased p-EGFR and the TKI insensitivity in K-Ras mutant cells, ectopically expressed EGFR/Y1173F leads to TKI resistance in K-Ras wild-type cells without affecting the K-Ras mutant line (Fig. 1E and supplemental Fig. S1C). These results indicate that a non-phosphorylated EGFR is sufficient to confer TKI resistance downstream of mutant K-Ras and that one mechanism for the K-Ras mutation–associated resistance may occur through increased EGFR dephosphorylation.
p38γ stimulates EGFR transcription through c-Jun–mediated binding to the EGFR promoter and c-Jun is required for K-Ras–dependent growth and for TKI resistance
Results (Fig. 1, C and D) also show increased EGFR protein expression in K-Ras mutant cells. Whereas EGFR is frequently overexpressed in human cancers, the responsible mechanisms are largely unknown (1, 32). Having previously demonstrated that p38γ increases AP-1–dependent MMP9 transcription through interacting with and activating c-Jun (14, 19), we determined whether p38γ stimulates EGFR expression in collaboration with c-Jun, which may also contribute to the resistance to TKIs. The results (Fig. 2, A–C) show that p38γ overexpression in normal rat intestinal epithelial cells (IEC-6) (12) stimulates c-Jun and EGFR mRNA and protein expression, whereas p38γ depletion from K-Ras mutant colon cancer HCT116 and SW480 cells has an opposite effect. A forced c-Jun expression in HCT116 cells increases the growth and elevates EGFR protein levels without impacting p38γ expression (Fig. 2D), indicating its oncogenic activity downstream of p38γ and upstream of EGFR. Consistent with this notion, c-Jun depletion in K-Ras mutant cells reduces colony formation and decreases EGFR protein expression without affecting p38γ levels (supplemental Fig. S2, A–C). Of great interest, EGFR knockdown in these cells still inhibits growth without affecting p38γ and c-Jun protein levels (supplemental Fig. S2, A–C). These results indicate that intrinsic EGFR in K-Ras mutant cells is still capable of driving the malignant growth despite the resistance of these cells to TKIs, as observed in patients with K-Ras mutant cancer (1). Together with the ability of p38γ to promote K-Ras oncogenic activity (12–14) and with the increased levels of p38γ, c-Jun, and EGFR in K-Ras mutant cells (Fig. 1, C and D), the data support an essential role of the p38γ/c-Jun/EGFR transcription axis in K-Ras–dependent growth.
Figure 2.

p38γ and c-Jun cooperate to stimulate EGFR RNA/protein expression. A, IEC-6 cells with tetracycline-inducible p38γ expression (Tet-on) were cultured in the absence or presence of Tet for the indicated times (left and middle); alternatively, cells were infected for 24 h with adenovirus expressing p38γ (Ad-p38γ) or β-galactosidase (Ad-β-Gal) (right). Protein and RNA samples were analyzed by WB and qRT-PCR, respectively (bar graph, mean ± S.D. (error bars), n = 3). B and C, p38γ was stably depleted by shRNAs through lentiviral infection of the indicated K-Ras mutant cells, and the resultant cells were analyzed for protein levels by WB (left) and for mRNA expression by qRT-PCR (right, mean ± S.D., n = 3) (14). D, c-Jun was stably expressed, and engineered cells were assessed for protein expression by WB and for colony formation (mean ± S.D., n = 3).
Previous studies showed that p38γ is both cytoplasmic and nuclear, whereas its phosphorylated form is predominantly localized in the nucleus (33–35). Because phosphorylated p38γ protein is up-regulated in K-Ras mutant cells (15), we examined whether K-Ras mutation triggers p38γ nuclear translocation, thus increasing its interaction with c-Jun. Cell fractionation analyses show that there is increased p38γ nuclear accumulation in K-Ras mutant cells relative to their K-Ras wild-type counterparts (Fig. 3A). These increases in p38γ also correlate with elevated c-Jun in the nucleus, as revealed by cell fractionation and co-localization analyses (Fig. 3A and supplemental Fig. S3A). These results indicate that p38γ and c-Jun may collaborate to promote K-Ras oncogenesis through their enhanced nuclear activities.
Figure 3.
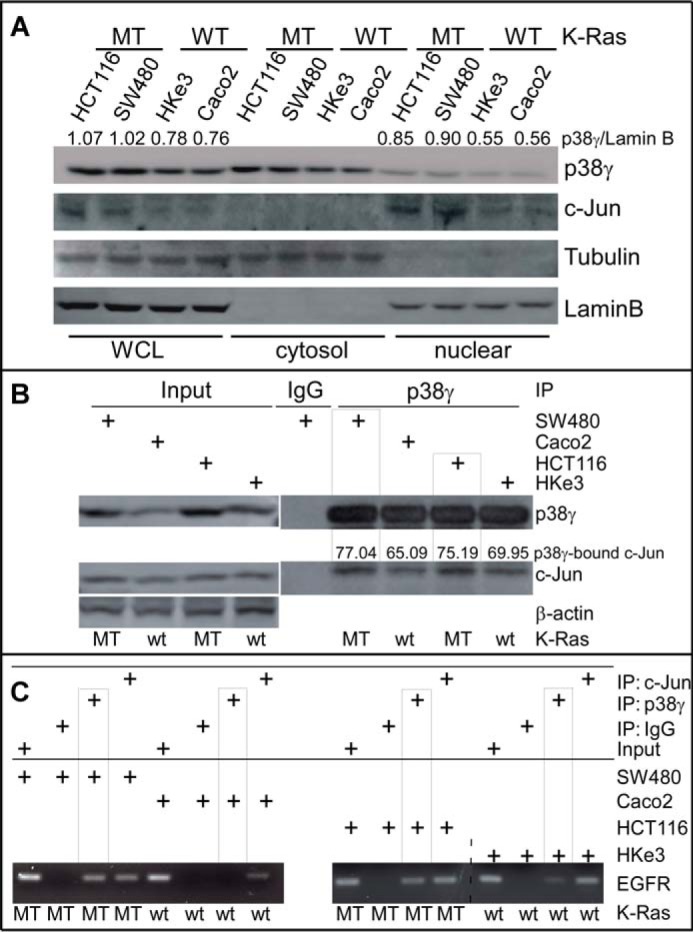
K-Ras mutation induces p38γ nuclear translocation and confers constitutive p38γ binding to the EGFR promoter through interaction with c-Jun. A, representative Western blots of equal protein loading of whole-cell lysates (WCL) and cytosolic or nuclear fractions of cells with either MT or WT K-Ras (the number indicates normalized p38γ over Lamin B). B and C, the indicated cells were processed for IP/WB (B) and ChIP (C) analysis using the indicated specific antibodies as described (14, 19), and p38γ-bound c-Jun was measured from p38γ precipitates (B). Similar results were obtained in a separate experiment.
We next used ChIP assays (14) to determine whether p38γ and c-Jun cooperate to stimulate EGFR transcription through their binding to the EGFR promoter. Of great interest, p38γ is only significantly recruited to the EGFR promoter at an AP-1 site in K-Ras mutant, but not in K-Ras wild-type, cells, whereas c-Jun binds the same location independent of K-Ras mutation status (Fig. 3, B and C). Consistent with a mediating role for c-Jun in the p38γ/AP-1 promoter DNA binding (14), p38γ immunoprecipitates from K-Ras mutant cells consistently contain higher levels of c-Jun (Fig. 3B). These results indicate that K-Ras mutation results in constitutive binding of p38γ to the EGFR promoter through its increased interaction with c-Jun. Furthermore, c-Jun depletion from K-Ras mutant cells not only decreases the growth and down-regulates EGFR expression, but also increases the growth-inhibitory activity of TKIs (supplemental Figs. S2 (A and C) and S3B). These results together indicate that K-Ras mutation can directly trigger p38γ binding to the EGFR promoter through its increased nuclear translocation in a c-Jun-dependent manner and that increased c-Jun in K-Ras mutant colon cancer cells is, at least in part, responsible for elevated EGFR expression and the unresponsiveness to TKIs.
K-Ras mutation increases the binding of EGFR with both p38γ and PTPH1, which promotes EGFR dephosphorylation and TKI resistance
EGFR can be dephosphorylated by several phosphatases, including protein-tyrosine phosphatase TCPTP (36) and receptor-type protein-tyrosine phosphatase-κ (RRTO) (37). The consequences of such EGFR dephosphorylation for sensitivities to TKIs, however, have not been explored. We previously demonstrated that PTPH1 specifically decreases EGFR phosphorylation at Tyr-1173, but not Tyr-1068 and that this increases breast cancer sensitivity to TKIs through disruption of an EGFR inhibitory complex with the estrogen receptor (17). In this study, the growth-inhibitory activity of TKIs was positively correlated with PTPH1 protein levels in EGFR immunoprecipitates (17). Because the EGFR dephosphorylating activity of PTPH1 requires its interaction with and phosphorylation by p38γ (15), we next determined whether K-Ras mutation alters the binding of EGFR with PTPH1 and its activator p38γ. The results (Fig. 4A) show that EGFR immunoprecipitates contain higher levels of both PTPH1 and p38γ proteins in K-Ras mutant cells as compared with those in K-Ras wild-type cells. Further analyses (Fig. 4B) show that ectopically expressed EGFR binds PTPH1 and p38γ, but not the C-terminal deleted p38γΔ4 or the non-phosphorable p38γ/AGF mutant. These results indicate that EGFR binds PTPH1 and p38γ by a mechanism that depends on both the p38γ C terminus and p38γ phosphorylation. An enhanced binding of EGFR to the p38γ–PTPH1 complex may play an important role in decreasing the levels of p-EGFR in K-Ras mutant cells (Fig. 1, C and D).
Figure 4.
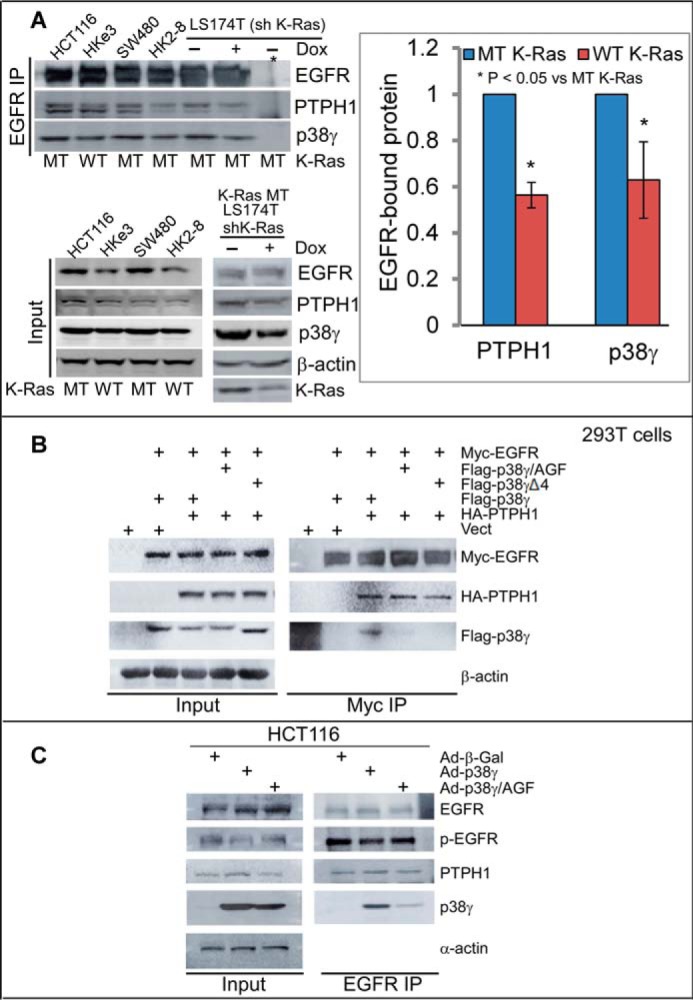
There is an increased complex formation of EGFR with PTPH1 and p38γ in K-Ras mutant cells. A, equal protein amounts from K-Ras WT and MT cell lysates were precipitated with a specific EGFR antibody, and the precipitates were analyzed by WB for PTPH1 and p38γ (top left, *, IP with control IgG). PTPH1 and p38γ proteins in EGFR precipitates from K-Ras WT cells were measured and normalized by those from K-Ras MT cells (right panel, mean ± S.D. (error bars), n = 3, from top left). Direct Western blots of the inputs are shown (bottom, left). B, the indicated constructs were transiently expressed in 293T cells, and Myc precipitates were analyzed by WB for EGFR binding to HA-PTPH1 and/or FLAG-p38γ. C, cells were transduced with the indicated adenovirus and EGFR precipitates were analyzed by WB after a 24-h incubation.
Consistent with this premise, overexpression of p38γ, but not its p38γ/AGF mutant, stimulates both PTPH1 expression and EGFR dephosphorylation in association with an enhanced EGFR–PTPH1–p38γ complex formation (Fig. 4C). To demonstrate whether K-Ras mutation alters PTPH1-mediated EGFR dephosphorylation, p-EGFR proteins were measured using a p-EGFR/Tyr-1173–specific antibody after endogenous PTPH1 was knocked down. Of great interest, PTPH1 depletion significantly increases p-EGFR levels in K-Ras mutant HCT116 cells, but not in the K-Ras-disrupted HKe3 subline (Fig. 5A), indicating that endogenous PTPH is responsible for decreased levels of p-EGFR in K-Ras mutant cells. These results, together with those in Fig. 4 (A and B), indicate that decreased p-EGFR in K-Ras mutant cells (Fig. 1, C and D) is probably caused by enhanced EGFR dephosphorylation by PTPH1 through its increased complex formation with PTPH1 and p38γ. Furthermore, knockdown of p38γ or PTPH1 increases the growth-inhibitory activity of TKIs in K-Ras mutant cells (supplemental Fig. S4, A and B), whereas PTPH1 depletion has no consistent effect in K-Ras wild-type cells (supplemental Fig. S5, A and B). Together, these results demonstrate a causative role of the p38γ–PTPH1 signaling nodule in dephosphorylating EGFR and in resistance of K-Ras mutant cells to TKIs.
Figure 5.

PTPH1 is only active in decreasing EGFR tyrosine phosphorylation in K-Ras mutant cells and cooperates with mutant K-Ras and p38γ to promote EGFR dephosphorylation. A, control cells and cells stably depleted of PTPH1 were analyzed by WB for EGFR expression and phosphorylation (EGFR/Tyr-1173); relative levels of p-EGFR in shPTPH1 versus shLuc cells were normalized to EGFR (55). B, the indicated constructs were transiently transfected into 293T cells, and the resultant cells were analyzed by WB. C, Myc-EGFR protein isolated from transiently transfected 293T cells was incubated in vitro with the indicated proteins, and the mixtures were then analyzed by WB for p-EGFR levels. D, K-Ras mutation in situ activates the p38γ/c-Jun/PTPH1 signaling network, leading to both increased EGFR protein expression and decreased EGFR/Tyr-1173 phosphorylation. Cells were cultured with Dox for 72 h to silence the MT K-Ras. Protein lysates were prepared at the indicated time after Dox removal for K-Ras re-expression and analyzed by WB. The number indicates relative protein amounts over β-actin at each time point (normalized to 0 min), and similar results were obtained in a separate experiment.
To further determine the cooperative role of p38γ and PTPH1 in EGFR dephosphorylation, a reconstitution experiment was performed in 293T cells by transient co-transfection. The results (Fig. 5B) show that FLAG-p38γ cooperates with HA-PTPH1 to regulate EGFR phosphorylation and expression in association with increased HA-PTPH1 phosphorylation. Furthermore, the addition of p38γ protein to Myc-isolated EGFR precipitates in vitro failed to decrease p-EGFR levels as compared with the positive control of HA-PTPH1 (Fig. 5C), indicating that p38γ is not able to directly dephosphorylate EGFR. These results, together with those in Figs. 1–4, suggest that p38γ MAPK increases non-phosphorylated EGFR by concurrently enhancing PTPH1-mediated EGFR dephosphorylation and stimulating c-Jun–dependent EGFR trans-activation. To determine whether an in situ K-Ras mutation activates this dynamic signaling network, mutant K-Ras was re-expressed by Dox removal following its initial knockdown by inducible shRNA (15, 25), and cells were then analyzed for protein expression and phosphorylation. Results (Fig. 5D) show that an increase in mutant K-Ras protein stimulates p38γ expression in <5 min, leading to decreased p-EGFR and increased total EGFR protein for up to 60 min. These results further indicate that the in situ K-Ras mutation can indeed trigger increased EGFR expression and dephosphorylation through endogenous p38γ activity.
The p38γ inhibitor pirfenidone disrupts the EGFR–p38γ–PTPH1 complex, increases EGFR tyrosine phosphorylation, and restores sensitivity of K-Ras mutant cancer cells to TKIs
p38γ depends on its phosphorylation status to activate both c-Jun and PTPH1 (13–15), to interact with EGFR (Fig. 4B), and to trigger decreased p-EGFR expression (Fig. 4C). We next explored whether inhibition of p38γ activity by its pharmacological inhibitor pirfenidone (PFD) (38, 39) impacts EGFR phosphorylation and TKI sensitivity in K-Ras mutant cancer cells. PFD more significantly inhibits p38γ in vitro than its family member p38α and p38β (40). Because of its strong anti-fibrotic effect and relatively non-toxic properties, PFD is approved for the treatment of lung fibrosis (39, 41). Treatment of K-Ras mutant cells (colon cancer: HCT116 and SW480; pancreatic cancer: Panc-1 and Mia2PaCa2; and lung cancer: A549) with TKIs alone has no substantial effects on their colony formation. The PFD addition (at a concentration for which PFD was non-inhibitory itself) significantly enhanced the growth inhibition by Lap and Gef (Fig. 6A and supplemental Fig. S6A). Further, PFD treatment completely abolishes the EGFR binding of p38γ in two K-Ras mutant cell lines, leading to decreased p-PTPH1 and increased p-EGFR (Fig. 6B, Input). These results demonstrate a required role for p38γ activity in PTPH1-mediated EGFR dephosphorylation through a complex formation and suggest that the sensitivity to TKIs in K-Ras mutant cells can be restored by PFD-induced p38γ depletion from the EGFR–PTPH1 complex. Consistent with this notion, the sensitization effect was further demonstrated in HCT116 xenografts in which systemic PFD administration disrupts the binding of EGFR with both p38γ and PTPH1, alone and in combination with Lap (Fig. 6C and supplemental Fig. S6B). The application of Lap, instead of Gef, for the in vivo combination with PFD is due to the fact that Lap was previously tested in colon cancer patients in combination studies (42). These results together demonstrate that PFD can sensitize K-Ras mutant cancers to TKIs by disrupting the EGFR–p38γ–PTPH1 signaling complex.
Figure 6.

The p38γ inhibitor PFD restores the therapeutic response of K-Ras mutated cancers to TKIs by depleting p38γ and/or PTPH1 from the EGFR complexes. A, colony formation of various cells was assessed in the absence and presence of TKIs with or without PFD (Lap, 2 μm; Gef, 0.125 μm; PFD, 30 μg/ml) (mean ± S.D. (error bars), n = 3). B, HCT116 and SW480 cells were treated with PFD (50 μg/ml) or DMSO for 24 h and then were analyzed by EGFR IP/WB. C, Lap (40 mg/kg) or solvent (DMSO) solution (in 50 μl) was administered i.p. to tumor-bearing nude mice twice a week (51), whereas PFD (200 mg/kg in 100 μl of water) was given by oral gavage daily for 2 weeks. Changes in tumor volume were monitored every other day (results are means of five tumors ± S.E.). D, an experimental model indicates that K-Ras mutation may confer TKI resistance through p38γ MAPK-induced concurrent activation of c-Jun-dependent EGFR gene transcription and of PTPH1-induced EGFR dephosphorylation leading to elevated non-phosphorylated EGFR, which may contribute to both K-Ras-dependent growth and unresponsiveness to TKIs.
Discussion
K-Ras mutation has been long known to be associated with resistance to TKIs, and there is an urgent need to identify novel signaling mechanisms that can be used to restore TKI sensitivity (43–45). p38γ MAPK promotes K-Ras oncogenesis through activating its signaling network c-Jun and PTPH1 by complex formation (11–15). Here, our results provide several key pieces of evidence that, together with the previous findings, indicate that the K-Ras effector p38γ confers resistance to TKIs through its concurrent stimulation of c-Jun-dependent EGFR transcription and PTPH1-catalyzed EGFR dephosphorylation (Fig. 6D). Because expression of EGFR/Y1173F increases the sensitivity to TKIs in K-Ras wild-type but not K-Ras mutant cells (Fig. 1E), in which EGFR silencing still decreases the colony formation (supplemental Fig. 2), these results suggest that endogenous EGFR in K-Ras mutant cells may drive the growth independent of kinase activity. Importantly, inhibiting p38γ with its pharmacological inhibitor PFD restores the TKI sensitivity of K-Ras mutant cells in vitro and in vivo (Fig. 6 and supplemental Fig. S6). Together, these results demonstrate that p38γ converts K-Ras oncogene signaling to TKI resistance through its dual stimulating activity on EGFR gene expression and protein dephosphorylation (Fig. 6D).
An autocrine mechanism is believed to be responsible for the signaling cross-talk between Ras and EGFR; however, the factors involved have been mostly unidentified (46–49). Our results suggest that the K-Ras mutation itself simultaneously stimulates EGFR transcription and dephosphorylation by activating the p38γ signaling network through regulating dynamic protein–protein and protein–DNA interactions. This is supported by enhanced p38γ binding to the EGFR promoter DNA through interaction with c-Jun and by increased complex formation of EGFR with p38γ and PTPH1 proteins in K-Ras mutant cells (Figs. 3 and 4). The functional role of this complex is suggested by the fact that the p38γ inhibitor PFD disrupts the p38γ interaction with c-Jun/AP-1 DNA (19), suppresses the EGFR binding with p38γ and PTPH1, and increases the sensitivity of K-Ras mutant cells to TKIs (Fig. 6 and supplemental Fig. S6). Moreover, depletion of PTPH1 from the ternary EGFR–PTPH1–p38γ complex increases p-EGFR levels and enhances the sensitivity of K-Ras mutant cells to TKIs (Fig. 5 and supplemental Fig. S4B). In addition to elevated p38γ, hyperexpression of c-Jun and EGFR (Fig. 1, C and D) increased p38γ-induced PTPH1 phosphorylation in K-Ras mutant cells, and a phosphorylation-dependent p38γ interaction with c-Jun and PTPH1 (13–15) may also contribute to the distinct complex formation. These results together reveal a novel mechanism by which the K-Ras oncogene may inactivate EGFR by a p38γ-activated signaling network through its increased interaction with c-Jun/PTPH1/EGFR proteins as well as with the EGFR promoter DNA.
EGFR is a well-established target for cancer therapy (50, 51), and our results indicate that levels of EGFR expression and phosphorylation are both important for its therapeutic target activity. The previous studies showed that knock-out of mutant K-Ras by shRNA restores sensitivity to TKIs, but the application potential of this strategy was uncertain (52). Our results show that K-Ras mutation confers unresponsiveness to TKIs through p38γ-induced EGFR gene transcription and EGFR protein dephosphorylation, respectively, via the transcription factor c-Jun and the phosphatase PTPH1 (11–15) (Fig. 6D). Knocking down each member of the p38γ/c-Jun/PTPH1 network increases the sensitivity of K-Ras mutant cancer cells to TKIs (supplemental Fig. S3B and Fig. 4 (A and B)). Most importantly, the non-toxic p38γ inhibitor PFD restores the sensitivity of K-Ras mutant cancers in vitro and in vivo (Fig. 6 and supplemental Fig. S6). These results, together with its inhibitory effects on the p38γ interaction with c-Jun (19) and on the EGFR association with p38γ and PTPH1 (supplemental Fig. S6B), further indicate that disruption of the p38γ-activated signaling network (such as by using PFD) may have great application potential to restore the therapeutic response of K-Ras mutant cancers to TKIs.
Experimental procedures
Cell lines, constructs, and cell culture
Human colon cell lines were purchased from ATCC and have been maintained and used as described in our previous publications (13–15, 22). The tetracycline-inducible system (Tet-on) for p38γ expression in intestinal epithelial cells (IEC-6) was reported earlier (14). Human colon cancer LS174T cells with Dox-inducible shRNA to knockdown mutant K-Ras were kindly provided by Dr. Gambacorti-Passerini (25). HCT-116 and its sublines in which mutant K-Ras was disrupted (HKe3 and HK2-8) were provided by Dr. Shirasawa (23). Both of these engineered cell lines have been used previously in our laboratory (14, 15, 22, 24). The pLenti6/Block-iT system was used to clone sequences for shRNAs against luciferase (shLuc), PTPH1 (shPTPH1), EGFR (shEGFR), p38γ (shp38γ), and c-Jun (shc-Jun) as described (13, 15, 53) (supplemental Experimental procedures). Human EGFR cDNA and its Y1173F mutant were provided by Dr. Mien-Chie Hung (31) and were subcloned into pLHCX retroviral vector as described previously (12). Other constructs for p38γ, PTPH1, and their mutants were described previously (13–15, 22). Cell culture materials were supplied by Invitrogen.
RNA preparation, quantitative RT-PCR (qRT-PCR), and ChIP
qRT-PCR was carried out as described previously (14). Total RNA was prepared using the TRIzol extraction kit, and qRT-PCR was performed using the Express One-Step SYBER GreenER qPCR kit (Invitrogen). Samples were analyzed by the ΔΔCt method for -fold changes in expression, and the ratios of the individual genes relative to β-actin were expressed relative to the respective controls (14, 19). For ChIP assays, cells were incubated with 5% formaldehyde, and lysates were then subjected to immunoprecipitation with the indicated antibodies. These precipitated DNAs were used as a template for PCR analysis using primers covering the AP-1 site (14, 19, 26). An aliquot of each DNA before PCR was included as an input control on the same gel as the PCR products. Other procedures were the same as described (14, 19).
Colony formation and animal studies
For colony formation, 200 cells were plated in duplicate in 6-well plates and incubated with TKIs and/or PFD for about 2 weeks. The colonies formed were stained and counted as described (17, 22, 54). Animal studies were conducted according to the protocol approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee. Briefly, HCT116 cells (2 × 106 in 0.1 ml of PBS) were subcutaneously injected into male BALB/c nude mice (Charles River), and therapy with Lapatinib and/or PFD (or DMSO control) was initiated when tumors became palpable. Tumor volume was measured every 2–3 days (22), with representative tumors at sacrifice photographed and shown as the inset (Fig. 6C). Moreover, protein lysates were prepared from tumors and analyzed by WB/IP (19, 22).
Statistical analysis
The results were analyzed by Student's t test unless otherwise specified.
Author contributions
N. Y., A. L., Y. J., X.-M. Q., and G. C. conceived and designed the study. N. Y., A. L., Y. J., S. H., X.-M. Q., and C. R. M. conducted experiments and provided reagents. N. Y., A. L., M. M., X.-M. Q., and G. C. analyzed and interpretated data. N. Y., A. L., C. R. M., and G. C. wrote, reviewed, and revised the manuscript. G. C. supervised the study.
Supplementary Material
Acknowledgments
We thank Drs. Mien-Chie Hung, Gambacorti-Passerini, and Senji Shirasawa for reagents that made this study possible.
Note added in proof
In the version of this article that was published as a Paper in Press on July 24, 2017, Fig. 3C did not indicate the border between different sections of a gel. This error has now been corrected and does not affect the results or conclusions of this work.
This study was supported by Department of Veterans Affairs Merit Review Grant 1I01BX002883, Department of Defense Grant BC141898, and the Cancer Center of the Medical College of Wisconsin (to G. C.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Experimental procedures and Figs. S1–S6.
- EGFR
- epidermal growth factor receptor
- p-EGFR
- phosphorylated EGFR
- TKI
- tyrosine kinase inhibitor
- PTPH1
- protein-tyrosine phosphatase H1
- Lap
- lapatinib
- Gef
- gefitinib
- MT
- mutant
- PFD
- pirfenidone
- Dox
- doxycycline
- qRT-PCR
- quantitative RT-PCR
- WB
- Western blotting
- IP
- immunoprecipitation.
References
- 1. Avraham R., and Yarden Y. (2011) Feedback regulation of EGFR signaling: decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 12, 104–117 [DOI] [PubMed] [Google Scholar]
- 2. Arora A., and Scholar E. M. (2005) Role of tyrosine kinase inhibitors in cancer therapy. J. Pharmacol. Exp. Ther. 315, 971–979 [DOI] [PubMed] [Google Scholar]
- 3. Wheeler D. L., Dunn E. F., and Harari P. M. (2010) Understanding resistance to EGFR inhibitors: impact on future treatment strategies. Nat. Rev. Clin. Oncol. 7, 493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shaib W., Mahajan R., and El-Rayes B. (2013) Markers of resistance to anti-EGFR therapy in colorectal cancer. J. Gastrointest. Oncol. 4, 308–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prior I. A., Lewis P. D., and Mattos C. (2012) A comprehensive survey of ras mutations in cancer. Cancer Res. 72, 2457–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Normanno N., Tejpar S., Morgillo F., De Luca A., Van Cutsem E., and Ciardiello F. (2009) Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat. Rev. Clin. Oncol. 6, 519–527 [DOI] [PubMed] [Google Scholar]
- 7. Misale S., Di Nicolantonio F., Sartore-Bianchi A., Siena S., and Bardelli A. (2014) Resistance to anti-EGFR therapy in colorectal cancer: from heterogeneity to convergent evolution. Cancer Discov. 4, 1269–1280 [DOI] [PubMed] [Google Scholar]
- 8. Zhao S., Wang Y., Cao L., Ouellette M. M., and Freeman J. W. (2010) Expression of oncogenic K-Ras and loss of Smad4 cooperate to induce the expression of EGFR and to promote invasion of immortalized human pancreas ductal cells. Int. J. Cancer 127, 2076–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Young A., Lou D., and McCormick F. (2013) Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 3, 112–123 [DOI] [PubMed] [Google Scholar]
- 10. Navas C., Hernández-Porras I., Schuhmacher A. J., Sibilia M., Guerra C., and Barbacid M. (2012) EGF receptor signaling is essential for K-Ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell 22, 318–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Loesch M., and Chen G. (2008) The p38 MAPK stress pathway as a tumor suppressor or more? Front. Biosci. 13, 3581–3593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tang J., Qi X., Mercola D., Han J., and Chen G. (2005) Essential role of p38γ in K-Ras transformation independent of phosphorylation. J. Biol. Chem. 280, 23910–23917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hou S. W., Zhi H. Y., Pohl N., Loesch M., Qi X. M., Li R. S., Basir Z., and Chen G. (2010) PTPH1 dephosphorylates and cooperates with p38γ MAPK to increases Ras oncogenesis through PDZ-mediated interaction. Cancer Res. 70, 2901–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Loesch M., Zhi H. Y., Hou S. W., Qi X. M., Li R. S., Basir Z., Iftner T., Cuenda A., and Chen G. (2010) p38γ MAPK cooperates with c-Jun in trans-activating matrix metalloproteinase 9. J. Biol. Chem. 285, 15149–15158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hou S., Suresh P. S., Qi X., Lepp A., Mirza S. P., and Chen G. (2012) p38g MAPK signals through phosphorylating its phosphatase PTPH1 in regulating Ras oncogenesis and stress response. J. Biol. Chem. 287, 27895–27905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Suresh P. S., Ma S., Migliaccio A., and Chen G. (2014) Protein-tyrosine phosphatase H1 increases breast cancer sensitivity to antiestrogens by dephosphorylating estrogen receptor at tyr537. Mol. Cancer Ther. 13, 230–238 [DOI] [PubMed] [Google Scholar]
- 17. Ma S., Yin N., Qi X., Pfister S. L., Zhang M. J., Ma R., and Chen G. (2015) Tyrosine dephosphorylation enhances the therapeutic target activity of epidermal growth factor receptor (EGFR) by disrupting its interaction with estrogen receptor (ER). Oncotarget 6, 13320–13333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qi X., Zhi H., Lepp A., Wang P., Huang J., and Basir Z., Chitambar C. R., Myers C. R., and Chen G. (2012) p38γ mitogen-activated protein kinase (MAPK) confers breast cancer hormone sensitivity by switching estrogen receptor (ER) signaling from classical to nonclassical pathway via stimulating ER phosphorylation and c-Jun transcription. J. Biol. Chem. 287, 14681–14691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Qi X., Yin N., Ma S., Lepp A., Tang J., Jing W., Johnson B., Dwinell M. B., Chitambar C. R., and Chen G. (2015) p38γ MAPK is a therapeutic target for triple-negative breast cancer by stimulation of cancer stem-like cell expansion. Stem Cells 33, 2738–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meng F., Zhang H., Liu G., Kreike B., Chen W., Sethi S., Miller F. R., and Wu G. (2011) p38γ mitogen-activated protein kinase contributes to oncogenic properties maintenance and resistance to poly (ADP-ribose)-polymerase-1 inhibition in breast cancer. Neoplasia 13, 472–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rosenthal D. T., Iyer H., Escudero S., Bao L., Wu Z., Ventura A. C., Kleer C. G., Arruda E. M., Garikipati K., and Merajver S. D. (2011) p38γ promotes breast cancer motility and metastasis through regulation of RhoC GTPase, cytoskeletal architecture, and a novel leading edge behavior. Cancer Res. 71, 6338–6349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qi X., Xie C., Hou S., Li G., Yin N., Dong L., Lepp A., Chesnik M. A., Mirza S. P., Szabo A., Tsai S., Basir Z., Wu S., and Chen G. (2014) Identification of a ternary protein-complex as a therapeutic target for K-Ras-dependent colon cancer. Oncotarget 5, 4269–4282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shirasawa S., Furuse M., Yokoyama N., and Sasazuki T. (1993) Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science 260, 85–88 [DOI] [PubMed] [Google Scholar]
- 24. Qi X., Tang J., Pramanik R., Schultz R. M., Shirasawa S., Sasazuki T., Han J., and Chen G. (2004) p38 MAPK activation selectively induces cell death in K-ras mutated human colon cancer cells through regulation of vitamin D receptor. J. Biol. Chem. 279, 22138–22144 [DOI] [PubMed] [Google Scholar]
- 25. Mologni L., Dekhil H., Ceccon M., Purgante S., Lan C., Cleris L., Magistroni V., Formelli F., and Gambacorti-Passerini C. B. (2010) Colorectal tumors are effectively eradicated by combined inhibition of β-catenin, KRAS, and the oncogenic transcription factor ITF2. Cancer Res. 70, 7253–7263 [DOI] [PubMed] [Google Scholar]
- 26. Ashktorab H., Daremipouran M., Wilson M., Siddiqi S., Lee E. L., Rakhshani N., Malekzadeh R., Johnson A. C., Hewitt S. M., and Smoot D. T. (2007) Transactivation of the EGFR by AP-1 is induced by helicobacter in gastric cancer. Am. J. Gastroenterol. 102, 2135–2146 [DOI] [PubMed] [Google Scholar]
- 27. Ozanne B. W., Spence H. J., McGarry L. C., and Hennigan R. F. (2007) Transcription factors control invasion: AP-1 the first among equals. Oncogene 26, 1–10 [DOI] [PubMed] [Google Scholar]
- 28. Wang F., Wang S., Wang Z., Duan J., An T., Zhao J., Bai H., Wang J., and Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education) (2012) Phosphorylated EGFR expression may predict outcome of EGFR-TKIs therapy for the advanced NSCLC patients with wild-type EGFR. J. Exp. Clin. Cancer Res. 31, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schlessinger J. (2000) Cell signaling by receptor tyrosine kinases. Cell 103, 211–225 [DOI] [PubMed] [Google Scholar]
- 30. Segovia-Mendoza M., González-González M., Barrera D., Díaz L., and García-Bererra R. (2015) Efficacy and mechanism of action of the tyrosine kinase inhibitors gefitinib, lapatinib and neratinib in the treatment of Her2-posive breast cancer: preclinical and clinial evidence. Am. J. Cancer Res. 5, 2531–2561 [PMC free article] [PubMed] [Google Scholar]
- 31. Hsu J. M., Chen C. T., Chou C. K., Kuo H. P., Li L. Y., Lin C. Y., Lee H. J., Wang Y. N., Liu M., Liao H. W., Shi B., Lai C. C., Bedford M. T., Tsai C. H., and Hung M. C. (2011) Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat. Cell Biol. 13, 174–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Logue J. S., and Morrison D. K. (2012) Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes Dev. 26, 641–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qi X., Tang J., Loesch M., Pohl N., Alkan S., and Chen G. (2006) p38γ MAPK integrates signaling cross-talk between Ras and estrogen receptor to increase breast cancer invasion. Cancer Res. 66, 7540–7547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qi X., Pohl N. M., Loesch M., Hou S., Li R., Qin J. Z., Cuenda A., and Chen G. (2007) p38α antagonizes p38γ activity through c-Jun-dependent ubiquitin-proteasome pathways in regulating Ras transformation and stress response. J. Biol. Chem. 282, 31398–31408 [DOI] [PubMed] [Google Scholar]
- 35. Sabio G., Cerezo-Guisado M. I., Del Reino P., Iñesta-Vaquera F. A., Rousseau S., Arthur J. S. C., Campbell D. G., Centeno F., and Cuenda A. (2010) p38γ regulates interactin of nuclear PSF and RNA with the tumor-suppressor hDlg in response to osmotic shock. J. Cell Sci. 123, 2596–2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mattila E., Pellinen T., Nevo J., Vuoriluoto K., Arjonen A., and Ivaska J. (2005) Negative regulation of EGFR signalling through integrin-α1β1-mediated activation of protein tyrosine phosphatase TCPTP. Nat. Cell Biol. 7, 78–85 [DOI] [PubMed] [Google Scholar]
- 37. Xu Y., Tan L. J., Grachtchouk V., Voorhees J. J., and Fisher G. J. (2005) Receptor-type protein-tyrosine phosphatase-κ regulates epidermal growth factor receptor function. J. Biol. Chem. 280, 42694–42700 [DOI] [PubMed] [Google Scholar]
- 38. Moran N. (2011) p38 kinase inhibitor approved for idiopathic pulmonary fibrosis. Nat. Biotechnol. 29, 301. [DOI] [PubMed] [Google Scholar]
- 39. King T. E. Jr., Bradford W. Z., Castro-Bernardini S., Fagan E. A., Glaspole I., Glassberg M. K., Gorina E., Hopkins P. M., Kardatzke D., Lancaster L., Lederer D. J., Nathan S. D., Pereira C. A., Sahn S. A., Sussman R., et al. (2014) A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 370, 2083–2092 [DOI] [PubMed] [Google Scholar]
- 40. Ozes O., Blatt L. M., and Seiwert S. D. (August 5, 2008) Use of pirfenidone in therapeutic regimens. United States Patent 7,407,973 B2 [Google Scholar]
- 41. Noble P. W., Albera C., Bradford W. Z., Costabel U., Glassberg M. K., Kardatzke D., King T. E. Jr., Lancaster L., Sahn S. A., Szwarcberg J., Valeyre D., du Bois R. M., et al. (2011) Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 377, 1760–1769 [DOI] [PubMed] [Google Scholar]
- 42. Frank D., Jumonville A., Loconte N. K., Schelman W. R., Mulkerin D., Lubner S., Richter K., Winterle N., Wims M. B., Dietrich L., Winkler J. M., Volk M., Kim K., and Holen K. D. (2012) A phase II study of capecitabine and colorectal adenocarcinoma: a Wisconsin oncology network study. J. Gastrointest. Oncol. 3, 90–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pao W., Wang T. Y., Riely G. J., Miller V. A., Pan Q., Ladanyi M., Zakowski M. F., Heelan R. T., Kris M. G., and Varmus H. E. (2005) KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chong C. R., and Jänne P. A. (2013) The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 19, 1389–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yamaguchi H., Chang S. S., Hsu J. L., and Hung M. C. (2014) Signaling cross-talk in the resistance to Her family receptor targeted therapy. Oncogene 33, 1073–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gangarosa L. M., Sizemore N., Graves-Deal R., Oldham S. M., Der C. J., and Coffey R. J. (1997) Raf-independent epidermal growth factor receptor autocrine loop is necessary for ras transformation of rat intestinal epethelial cells. J. Biol. Chem. 272, 18926–18931 [DOI] [PubMed] [Google Scholar]
- 47. Grana T. M., Sartor C. I., and Cox A. D. (2003) Epidermal growth factor receptor autocrine signaling in RIE-1 cells transformed by the Ras oncogene enhances radiation resistance. Cancer Res. 63, 7807–7814 [PubMed] [Google Scholar]
- 48. Matallanas D., Romano D., Al-Mulla F., O'Neill E., Al-Ali W., Crespo P., Doyle B., Nixon C., Sansom O., Drosten M., Barbacid M., and Kolch W. (2011) Mutant K-Ras activation of the proapoptotic MST2 pathway is antagonized by wild-type K-Ras. Mol. Cell 44, 893–906 [DOI] [PubMed] [Google Scholar]
- 49. Zhu Z., Aref A. R., Cohoon T. J., Barbie T. U., Imamura Y., Yang S., Moody S. E., Shen R. R., Schinzel A. C., Thai T. C., Reibel J. B., Tamayo P., Godfrey J. T., Qian Z. R., Page A. N., et al. (2014) Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 4, 452–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Arteaga C. L. (2002) Epidermal growth factor receptor dependence in human tumors: more than just expression? Oncologist 7, 31–49 [DOI] [PubMed] [Google Scholar]
- 51. Dolloff N. G., Mayes P. A., Hart L. S., Dicker D. T., Humphreys R., and El-Deiry W. S. (2011) Off-target lapatinib activity sensitizes colon cancer cells through trail death receptor up-regulation. Sci. Transl. Med. 3, 86ra50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. van Houdt W. J., Hoogwater F. J. H., de Bruijn M. T., Emmink B. L., Nijkamp M. W., Raats D. A., van der Groep P., van Diest P., Borel Rinkes I. H., and Kranenburg O. (2010) Oncogenic KRAS decsensitizes colorectal tumor cells to epidermal growth factor receptor inhibition and activation. Neoplasia 12, 443–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hou S., Lepp A., and Chen G. (2010) p38γ MAP kinase UCSD-Nat. Mol. Pages 10.1038/mp.a001720.01 [DOI] [Google Scholar]
- 54. Zhi H. Y., Hou S. W., Li R. S., Basir Z., Xiang Q., Szabo A., and Chen G. (2011) PTPH1 cooperates with vitamin D receptor to stimulate breast cancer growth through their mutual stabilization. Oncogene 30, 1706–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yin N., Qi X., Tsai S., Lu Y., Basir Z., Oshima K., Thomas J. P., Myers C. R., Stoner G., and Chen G. (2016) p38γ MAPK is required for inflammation-associated colon tumorigenesis. Oncogene 35, 1039–1048 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.