
Fig. 3.
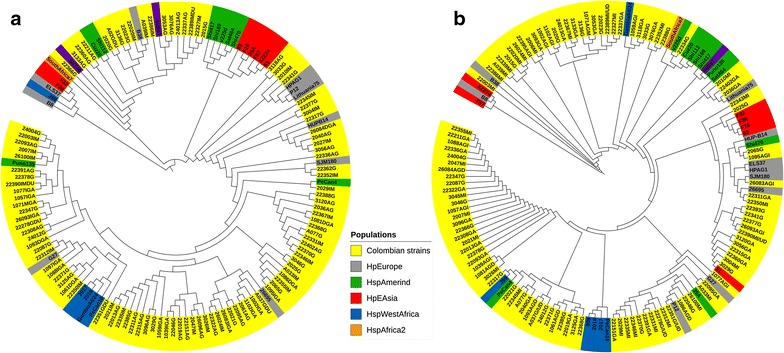
Phylogenetic analysis of virulence factors. a VacA, the phylogeny was inferred from 138 sequences using maximum likelihood based on the GTR+G model. Only the first positions were included, and all gaps were removed. b HorB, the phylogeny was inferred from 140 sequences using the NJ algorithm, and the distance was computed using the Kimura 2-parameter method with a gamma distribution of 1; all gaps were removed. A total of 1000 bootstrap repetitions were used for all the reconstructions as statistical support