

Abstract
Aims
Upadacitinib (ABT‐494) is a selective Janus kinase 1 inhibitor being developed for treatment of auto‐immune inflammatory disorders. This work evaluated effects of high‐fat meal, cytochrome P450 (CYP) 3A inhibition, CYP induction, and organic anion transporting polypeptide (OATP) 1B inhibition on upadacitinib pharmacokinetics.
Methods
Two Phase 1 evaluations were conducted, each in 12 healthy subjects. In Study 1, using a randomized, two‐sequence crossover design, a 3 mg dose of upadacitinib (immediate‐release capsules) was administered alone under fasting conditions, after high‐fat meal, or on Day 4 of a 6‐day regimen of 400 mg once‐daily ketoconazole. In Study 2, a 12 mg upadacitinib dose was administered alone, with the first, and with the eighth dose of a 9‐day regimen of rifampin 600 mg once daily. Upadacitinib plasma concentrations were characterized.
Results
Administration of upadacitinib immediate‐release capsules after a high‐fat meal decreased upadacitinib C max by 23% and had no impact on upadacitinib AUC relative to the fasting conditions. Ketoconazole (strong CYP3A inhibitor) increased upadacitinib C max and AUC by 70% and 75%, respectively. Multiple doses of rifampin (broad CYP inducer) decreased upadacitinib C max and AUC by approximately 50% and 60%, respectively. A single dose of rifampin (also an OATP1B inhibitor) had no effect on upadacitinib AUC. Upadacitinib was well tolerated when co‐administered with ketoconazole, rifampin, or after a high‐fat meal.
Conclusions
Strong CYP3A inhibition and broad CYP induction result in a weak and moderate effect, respectively, on upadacitinib exposures. OATP1B inhibition and administration of upadacitinib immediate‐release formulation with food does not impact upadacitinib exposure.
Keywords: high‐fat meal, JAK1 inhibitor, ketoconazole, pharmacokinetics, rifampin, upadacitinib
What is Already Known about this Subject
Upadacitinib (ABT‐494) is a potent and selective Janus kinase 1 inhibitor being developed for treatment of auto‐immune inflammatory diseases.
Upadacitinib exposure is approximately dose‐proportional over the 3–36 mg immediate‐release dose range with no significant accumulation with repeated dosing.
Upadacitinib is expected to be co‐administered in patients with medications that may modulate drug metabolizing enzymes or transporters as well as under different prandial conditions.
What this Study Adds
Strong inhibition of CYP3A results in a limited (~75%) increase in upadacitinib exposures and broad CYP induction results in ~50% reduction in upadacitinib exposures.
Food has no clinically meaningful effect on upadacitinib plasma exposures using the immediate‐release formulation.
OATP1B transporter inhibition does not affect upadacitinib exposure.
Introduction
Upadacitinib (ABT‐494) is a potent and selective JAK1 inhibitor being developed for treatment of systemic auto‐immune inflammatory diseases, including rheumatoid arthritis (RA) and inflammatory bowel disease. In cellular assays, upadacitinib demonstrated higher selectivity for inhibiting JAK1 over JAK2 and JAK3 1. The enhanced JAK1 selectivity of upadacitinib has the potential for an improved therapeutic profile in treating patients with inflammatory diseases compared to non‐selective JAK inhibitors 2. In two recent Phase 2b trials in subjects with RA who were inadequate responders to methotrexate or anti‐TNF therapy, upadacitinib showed improvements in RA signs and symptoms at the evaluated doses of 3–18 mg twice daily or 24 mg once daily, administered as immediate‐release capsules 3, 4. Currently, upadacitinib is being evaluated in six Phase 3 trials in subjects with RA 5, 6, 7, 8, 9 and two Phase 2b trials in subjects with Crohn's disease and ulcerative colitis 10.
In Phase 1 single and multiple ascending dose trials, upadacitinib was generally safe and well tolerated in healthy subjects in single doses up to 48 mg and multiple twice‐daily doses up to 24 mg for 14 days; the maximum tolerated dose for upadacitinib was not identified 11. Upadacitinib pharmacokinetics follows bi‐exponential disposition with a terminal half‐life of 6–16 h and a functional half‐life, calculated from C max to C trough ratio at steady state, of 3–4 h. Upadacitinib exposures were approximately dose‐proportional over the 3–36 mg dose range. Approximately 20% of the upadacitinib dose is eliminated in urine as unchanged upadacitinib. In vitro metabolism studies suggested that upadacitinib is a non‐sensitive substrate for cytochrome P450 (CYP) 3A, with a very minor metabolic contribution from CYP2D6 11.
Ketoconazole, a broad‐spectrum antifungal, is a potent inhibitor of CYP3A that used to be a perpetrator of choice to assess in vivo the impact of inhibition of CYP3A on metabolism of drugs 12, 13. Rifampin, an antibacterial drug, is a potent in vivo inducer of CYP3A and a moderate inducer of other CYP isoforms. Additionally, rifampin is an in vivo inhibitor of the organic anion transporter polypeptide (OATP)1B1 and OAPTP1B3. Rifampin is administered as a single dose in clinical drug interaction studies to evaluate the effect of inhibition of hepatic uptake OATP1B transporters and as multiple doses to evaluate the effect of broad CYP induction on the metabolism of xenobiotics 12, 14.
Patients with systemic inflammatory diseases are likely to use multiple medications for treatment of the inflammatory disease as well as other comorbidities. Therefore, there is a potential for upadacitinib to be used concomitantly with drugs that are modulators of metabolizing enzymes or transporters. Additionally, upadacitinib may be administered under different prandial conditions. Thus, it is important to evaluate the effect of inhibitors or inducers of upadacitinib metabolism as well as the effect of food intake on upadacitinib pharmacokinetics to provide guidance on use in patients. We report the results from two studies that were conducted in healthy subjects to characterize the effects of ketoconazole, rifampin, and high‐fat meal on the pharmacokinetics of upadacitinib.
Methods
Participants and study designs
Two studies were conducted in healthy male and female subjects between the ages of 18 and 55 years, inclusive, with body mass index of 18–29.9 kg m−2, inclusive. Subjects who had a history of diabetes or lymphoproliferative disease, evidence of immunosuppression, evidence of active or latent tuberculosis, or who used tobacco or nicotine‐containing products within 180 days of the first dose of study drug were not allowed to enrol. Use of CYP3A inhibitors or CYP inducers, other than those administered as part of the investigation, was not allowed within 1 month prior to the first dose of study drug and through the end of the study.
The studies were conducted in accordance with Good Clinical Practice guidelines and ethical principles that have their origin in the Declaration of Helsinki. The study protocols were approved by the institutional review boards (RCRC, now known as Salus IRB, Austin, TX, and Vista Medical Center East Institutional Review Board, Waukegan, IL) and subjects provided written informed consent before any study‐related procedures were performed.
The study designs are illustrated in Figure 1. Upadacitinib was administered in both studies as an immediate‐release capsule formulation, the same formulation utilized in upadacitinib Phase 2b trials in subjects with RA.
Figure 1.
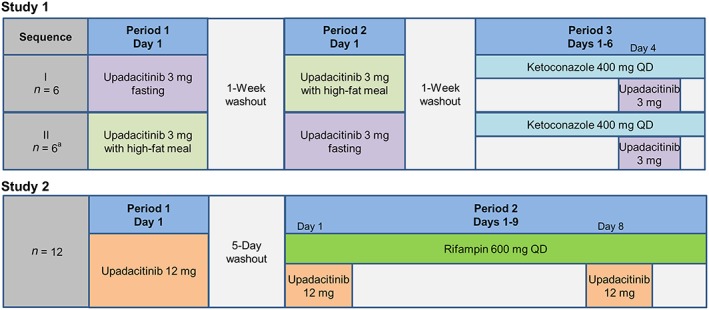
Design of the clinical studies. QD = once daily. a One subject completed dosing in Periods 1 and 2, but did not enter Period 3
Study 1 was an open‐label, randomized, two‐sequence, crossover evaluation in which a single oral dose of 3 mg upadacitinib was administered on three different occasions: in the morning after a 10‐h overnight fast (Day 1 of Period 1 or 2); 30 min after starting a high‐fat breakfast (1000 kilocalories, with approximately 50% of calories from fat 15) (Day 1 of Period 1 or 2), and in the morning on Day 4 of a 6‐day regimen of once‐daily ketoconazole (400 mg; Period 3). Subjects were confined to the study site for approximately 8 days in each period, beginning on Day −1 in each period and ending after the last study procedure on Day 7 of each period.
Study 2 was an open‐label, two‐period, sequential design study in which a single oral dose of 12 mg upadacitinib was administered under non‐fasting conditions on three different occasions: in the morning without rifampin (Day 1, Period 1); and in the morning on Days 1 and 8 of a 9‐day regimen of once daily rifampin (600 mg; Period 2). Subjects were confined to the study site for approximately 16 days across Periods 1 and 2, beginning on Day −1 and ending after the last study procedure following dosing in Period 2. Upadacitinib doses on Day 1 of Period 1 and Day 1 of Period 2 were separated by a 5‐day washout period.
During confinement in each study, subjects consumed standardized diets of approximately 2200 kilocalories/day, with 30% of calories from fat and 45% of calories from carbohydrates, for all meals except the high‐fat breakfast in Study 1. Subjects were not allowed to consume alcohol within 48 h, or grapefruit/grapefruit products, Seville oranges, starfruit/starfruit products, or quinine/tonic water within 72 h of study drug administration, or caffeine at any time during confinement.
Pharmacokinetic sampling
In Study 1, serial blood samples for upadacitinib assay were collected by venipuncture before the morning dose (0 h) and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 16, 20, 24, 30, 36, 48 and 72 h after upadacitinib dosing in each period. In Study 2, serial blood samples were collected before the morning dose (0 h) and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 15, 18, 24, 30 and 36 h after upadacitinib dosing in each period.
Plasma concentrations of upadacitinib were determined using a validated liquid chromatography method with mass spectrometric detection at AbbVie (North Chicago, IL) as previously described 11. The lower limit of quantitation for upadacitinib was 0.05 ng ml−1. The mean accuracy of the validated analytical method was 104.5% and the coefficient of variation (%CV; as a measure for precision) was 7.4%.
Pharmacokinetic parameters and statistical analyses
Pharmacokinetic parameters for upadacitinib were estimated by non‐compartmental methods using Phoenix™ WinNonlin® Version 6.3 (Pharsight, A Certara® Company, St. Louis, MO). Upadacitinib pharmacokinetic parameters included the maximum plasma concentration (C max), the time to C max (t max), the area under the plasma concentration–time curve up to the last measureable concentration (AUC t) and from time 0 to infinity (AUC ∞), and the terminal phase elimination half‐life (t 1/2).
Statistical analyses were conducted using SAS®, Version 9.2 (SAS Institute, Inc., Cary, NC). In Study 1, the effect of high‐fat meal on upadacitinib was evaluated by comparing upadacitinib AUC and C max after high‐fat meal vs. under fasting conditions. The effect of ketoconazole on upadacitinib pharmacokinetics was evaluated by comparing upadacitinib AUC and C max on Day 4 of Period 3 to those obtained under fasting conditions in Period 1 or 2. Similarly in Study 2, the effects of single‐ and multiple‐dose rifampin on upadacitinib were evaluated by comparing upadacitinib pharmacokinetic parameters on Days 1 and 8, respectively, of Period 2 relative to those on Day 1 of Period 1.
For each evaluation, an analysis of variance (ANOVA) was performed for the terminal elimination phase rate constant (β), and the natural logarithms of upadacitinib C max and AUC from each respective period. For t max, a nonparametric one‐sample test (Wilcoxon signed‐rank test) was used. For C max and AUC, point estimates and 90% confidence intervals for the “test/reference” ratios of central values were estimated.
Safety and tolerability
Safety and tolerability were assessed throughout each study based on adverse event monitoring, physical examinations, laboratory tests, vital signs measurements, and electrocardiogram assessments.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 16, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 17, 18.
Results
Participants
Twelve subjects were enrolled in each study: ten men and two women in Study 1 and nine men and three women in Study 2. One male subject in Study 1 was prematurely discontinued by the investigator due to non‐clinically significant elevations in creatine phosphokinase after completion of Periods 1 and 2. This subject did not participate in Period 3. All subjects were of white (n = 18) or black (n = 6) race. The mean (± SD) age in Study 1 and Study 2 was 30.1 ± 9.8 and 38.3 ± 10.6 years, respectively, and the mean (± SD) body mass index was 23.6 ± 2.67 and 24.9 ± 2.1 kg m−2, respectively.
Effect of high‐fat meal on upadacitinib pharmacokinetics
The mean plasma concentration vs. time profiles for upadacitinib when administered under fasting conditions and after high‐fat breakfast are presented in Figure 2. Upadacitinib pharmacokinetic parameters are listed in Table 1. Upadacitinib was rapidly absorbed, with a median t max of approximately 1 and 3 h under fasting conditions and after high‐fat meal, respectively. Administration of upadacitinib immediate‐release capsules after high‐fat meal resulted in a 23% decrease in upadacitinib C max relative to fasting conditions, but had no effect on AUC ∞ (point estimate of ~1 and 90% confidence interval within the no effect bioequivalence bounds of 0.8–1.25).
Figure 2.
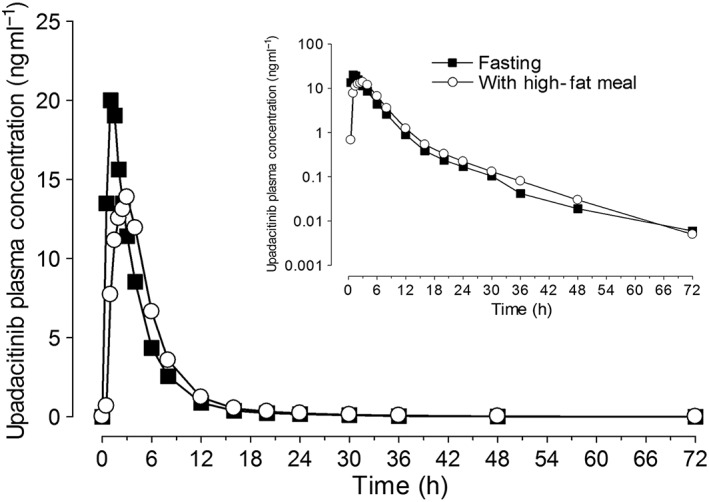
Upadacitinib mean plasma concentrations vs. time profiles following administration of upadacitinib under fasting conditions and with high‐fat meal
Table 1.
Pharmacokinetic parameters of upadacitinib when administered under fasting conditions, after high‐fat breakfast, and with ketoconazole in healthy volunteers
| Parameter | Upadacitinib 3 mg fasting | Upadacitinib 3 mg after high‐fat breakfast | Ratio of central values a after high‐fat breakfast vs. fasting (90% CI) | Upadacitinib 3 mg fasting + ketoconazole 400 mg | Ratio of central values a with vs. without ketoconazole (90% CI) |
|---|---|---|---|---|---|
| C max (ng ml −1 ) | 21.4 (4.2) | 16.4 (3.6)* | 0.77 (0.66–0.89) | 36.3 (6.34)* | 1.70 (1.55–1.89) |
| AUC ∞ (ng•h ml −1 ) | 87.7 (13) | 86.9 (13) | 0.99 (0.93–1.06) | 156 (31.8)* | 1.75 (1.62–1.88) |
| t max (h) | 1.0 (0.5–1.5) | 3.0 (1.0–4.0)* | – | 1.0 (0.5–1.0) | – |
| t 1/2 (h) | 8.5 (3.8) | 7.6 (4.1) | – | 7.4 (3.0) | – |
Estimates are expressed as mean (SD) for all parameters except t max and t 1/2. t max is presented as median (range), t 1/2 is presented as harmonic mean (pseudo‐SD).
Point estimate is the antilogarithm of the difference (test minus reference) of the least squares means for logarithms.
Statistically significantly different from upadacitinib 3 mg fasting; P < 0.05
P‐values were 0.49, and 0.40 for comparisons of upadacitinib β (as a test for change in t 1/2) and 0.002, and 0.06 for comparisons of t max after high‐fat breakfast and after ketoconazole, respectively, compared to when upadacitinib was administered alone.
Effect of ketoconazole on upadacitinib pharmacokinetics
Mean plasma concentration vs. time profiles for upadacitinib administered with and without ketoconazole are presented in Figure 3. Mean upadacitinib C max and AUC ∞ values increased by 70% and 75%, respectively when administered on Day 4 of a 6‐day regimen of ketoconazole 400 mg once daily (Table 1). There was no statistically significant change in upadacitinib β (as a test for change in t 1/2) or t max when upadacitinib was administered with or without ketoconazole.
Figure 3.
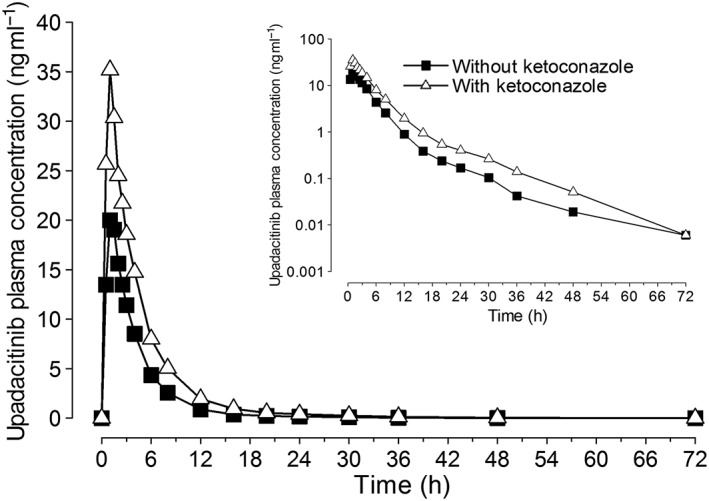
Upadacitinib mean plasma concentrations vs. time profiles following administration of upadacitinib (3 mg) alone or on day 4 of a 6‐day regimen of ketoconazole (400 mg)
Effect of single and multiple doses of rifampin on upadacitinib pharmacokinetics
Mean plasma concentrations vs. time profiles for upadacitinib administered alone and with a single dose or after multiple doses of rifampin are presented in Figure 4. Administration of a single dose of rifampin had minimal effects on upadacitinib C max (14% increase) and no effect on upadacitinib AUC ∞ (point estimate of ~1 and 90% confidence interval within the no effect equivalence bounds of 0.8–1.25). Administration of multiple doses of rifampin decreased upadacitinib C max and AUC ∞ values by approximately 50% and 60%, respectively (Table 2). There was no significant change in upadacitinib t max or β with administration of single or multiple doses of rifampin.
Figure 4.
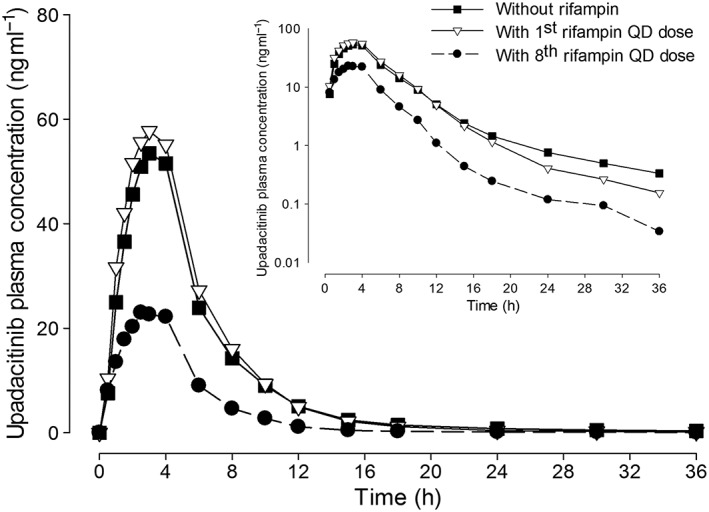
Upadacitinib mean plasma concentrations vs. time profiles following administration of upadacitinib (12 mg) alone and with single and multiple 600 mg once‐daily doses of rifampin
Table 2.
Upadacitinib pharmacokinetic parameters in the presence of a single dose or multiple doses of rifampin in healthy volunteers
| Parameter | Upadacitinib 12 mg alone | Upadacitinib 12 mg + rifampin 600 mg single dose | Ratio of central values a with vs. without a single dose of rifampin (90% CI) | Upadacitinib 12 mg + rifampin 600 mg QD | Ratio of central values a with vs. without QD doses of rifampin (90% CI) |
|---|---|---|---|---|---|
| C max (ng ml −1 ) | 62.0 (10.9) | 71.3 (15.5) | 1.14 (1.02–1.28) | 31.7 (11.5)* | 0.49 (0.44–0.55) |
| AUC ∞ (ng•h ml −1 ) | 334 (76.1) | 357 (80.9) | 1.07 (1.01–1.14) | 131 (27.9)* | 0.39 (0.37–0.42) |
| t max (h) | 2.8 (1.0–4.0) | 2.8 (0.5–4.0) | – | 3.0 (1.0–4.0) | – |
| t 1/2 (h) | 6.5 (3.0) | 5.9 (3.1) | – | 4.9 (2.7) | – |
Estimates are expressed as mean (SD) for all parameters except t max and t 1/2. t max is presented as median (range), t 1/2 is presented as harmonic mean (pseudo‐SD). A total of 12 subjects were evaluated using a sequential study design.
Point estimate is the antilogarithm of the difference (with rifampin minus without rifampin) of the least squares means for logarithms.
Statistically significantly different from upadacitinib 12 mg alone; P < 0.05
P‐values were 0.67 and 0.15 for comparisons of upadacitinib β (as a test for change in t 1/2) and 0.90, and 0.92 for comparisons of t max with single and QD doses of rifampin, respectively, compared to when upadacitinib was administered alone.
Safety and tolerability
No serious adverse events or adverse events that led to discontinuation from a study were reported. The majority of adverse events were mild in severity. In Study 1, adverse events reported by more than one subject in a regimen were headache (two subjects) following administration of upadacitinib under fasting conditions and nausea, dizziness, and headache (two subjects each) following administration of upadacitinib with ketoconazole. In Study 2, the only adverse event reported by more than one subject in any regimen was chromaturia, which was reported by all subjects after receiving a single dose of rifampin and upadacitinib. Rifampin is known to cause orange discoloration of body fluids (including urine) and therefore the reports of chromaturia during administration of rifampin were not unexpected.
One male subject was prematurely discontinued from Study 1 by the investigator due to elevations in creatine phosphokinase (approximately twice the upper limit of normal) after completing Periods 1 and 2. The increases in creatine phosphokinase were not clinically significant, were not reported as adverse events, and were not considered to be related to study drug by the investigator.
There were no clinically meaningful changes in any other laboratory test values, vital signs values, or electrocardiogram parameters in either study.
Discussion
Upadacitinib showed weak (< twofold increase in AUC) interaction with the strong CYP3A inhibitor ketoconazole and moderate (~50% decrease in AUC) interaction with the broad CYP inducer rifampin following multiple doses of ketoconazole and rifampin, respectively. A single dose of rifampin inhibiting OATP1B had no effect on upadacitinib AUC. High‐fat meal decreased upadacitinib C max by only 23% and had no effect on upadacitinib AUC from immediate‐release capsules.
The doses of upadacitinib administered in these evaluations were 3 mg in Study 1 and 12 mg in Study 2. In two Phase 2b studies in subjects with RA, upadacitinib doses of 3–18 mg twice‐daily showed significant improvement in therapeutic response assessed as the American College of Rheumatology 20% improvement criteria compared to placebo, with the maximum efficacy reached at doses of 6 or 12 mg twice‐daily 3, 4. Therefore, upadacitinib doses evaluated in Study 1 and Study 2 are clinically relevant. A upadacitinib dose of 3 mg was selected for the ketoconazole and food effect assessments to ensure that a sufficient safety margin for upadacitinib exposures was maintained in the case of a significant increase in exposures when upadacitinib was co‐adminstered with ketoconazole. The dose of 12 mg was selected for the assessment of the effect of rifampin to ensure that upadacitinib concentrations were still quantifiable with induction.
At the time of conducting Study 1, ketoconazole was the gold standard strong CYP3A inhibitor used to determine the impact of CYP3A inhibition on the clearance of CYP3A substrates in vivo 12, 13. To characterize the maximal effect of CYP3A inhibition, ketoconazole is typically administered as either a 400 mg once‐daily regimen or as a 200 mg twice‐daily regimen for several days depending on the disposition characteristics of the victim drugs 19. The regimen of 200 mg twice‐daily is recommended when the substrate has a longer half‐life compared to that of ketoconazole (3–5 h) 19. Given the short half‐life of upadacitinib, ketoconazole was administered as 400 mg once‐daily. Therefore, it is expected that the maximal effect of CYP3A inhibition on upadacitinib clearance has been adequately characterized in Study 1. It is worth noting that ketoconazole is no longer recommended to be used in clinical studies as a CYP3A inhibitor due to the risk of hepatotoxicity and adrenal insufficiency, and alternatives (e.g. itraconazole) have been suggested 20, 21.
The limited (~75%) increase in upadacitinib exposures (C max and AUC) in the presence of ketoconazole indicates that CYP3A‐mediated metabolism is not a major route of elimination for upadacitinib. There was no observed effect of ketoconazole on upadacitinib terminal half‐life, suggesting primarily inhibition of upadacitinib first‐pass metabolism by ketoconazole and/or the moderate nature of the effect of ketoconazole on upadacitinib clearance which was less pronounced on terminal half‐life, a relatively variable measure, than on AUC.
Administration of multiple doses of rifampin resulted in moderate decreases in upadacitinib exposures. Enzyme induction by rifampin reaches the full effect within approximately 1 week of dosing 22. Upadacitinib was coadministered with rifampin on Day 8 of a 9‐day regimen; thus, the 50–60% reduction in upadacitinib exposures observed in Study 2 represents the maximal effect of rifampin‐mediated CYP3A induction on upadacitinib. This indicates that administration of upadacitinib with a strong CYP3A inducer would be expected to reduce upadacitinib plasma concentrations by approximately half.
OATP1B1 and OATP1B3 influx transporters are expressed on the sinusoidal membrane of hepatocytes and play an important role in the hepatic uptake of their substrate drugs. In vivo inhibitors of OATP1B1 or OATP1B3 include atorvastatin, cyclosporine, gemfibrozil, and rifampin 23. Evaluation of upadacitinib exposures after the first dose of rifampin was used in Study 2 to evaluate the effect of OATP1B inhibition on upadacitinib exposures. There was no clinically meaningful effect of a single dose of rifampin on upadacitinib exposures, indicating lack of interaction between upadacitinib and inhibitors of OATP1B transporters.
Compared to the fasting conditions, high‐fat meal delayed upadacitinib t max by approximately 2 h and as a result decreased C max by only 23%, with no effect on AUC. Upadacitinib was administered in this study as an immediate‐release capsule formulation, which was used later in the Phase 2b studies of upadacitinib in RA and Crohn's disease 3, 4, 10. Based on results from Study 1, upadacitinib was administered without regard to food in these Phase 2 trials.
In conclusion, food has no clinically relevant impact on upadacitinib pharmacokinetics from immediate‐release capsules. Strong CYP3A inhibition results in a limited (~75%) increase in upadacitinib AUC while the broad CYP inducer rifampin reduces upadacitinib exposures by approximately half. No interaction is expected between upadacitinib and OATP1B inhibitors.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare that the studies were funded by AbbVie. M.‐E.F.M., S.J., A.A., P.J., and A.A.O. are employees of AbbVie and may hold AbbVie stocks or options.
The studies were funded by AbbVie. AbbVie contributed to the study design, research, and interpretation of data, and reviewing and approving the publication. Medical writing support was provided by Allison Kitten from AbbVie.
Contributors
M.‐E.F.M., S.J., A.A., P.J. and A.A.O. designed the study, collected, analyzed, and interpreted the data, and drafted, revised, and approved the manuscript.
Mohamed, M.‐E. F. , Jungerwirth, S. , Asatryan, A. , Jiang, P. , and Othman, A. A. (2017) Assessment of effect of CYP3A inhibition, CYP induction, OATP1B inhibition, and high‐fat meal on pharmacokinetics of the JAK1 inhibitor upadacitinib. Br J Clin Pharmacol, 83: 2242–2248. doi: 10.1111/bcp.13329.
References
- 1. Voss J, Graff C, Schwartz A, Hyland D, Argiriadi M, Camp H, et al. Pharmacodynamics of a novel JAK1 selective inhibitor in rat arthritis and anemia models and in healthy human subjects. Arthritis Rheum 2013; 65: 1.23045170 [Google Scholar]
- 2. Norman P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert Opin Investig Drugs 2014; 23: 1067–1077. [DOI] [PubMed] [Google Scholar]
- 3. Genovese MC, Smolen JS, Weinblatt ME, Burmester GR, Meerwein S, Camp HS, et al. Efficacy and safety of ABT‐494, a selective JAK‐1 inhibitor, in a phase IIb study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol 2016; 68: 2857–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kremer JM, Emery P, Camp HS, Friedman A, Wang L, Othman AA, et al. A phase IIb study of ABT‐494, a selective JAK‐1 inhibitor, in patients with rheumatoid arthritis and an inadequate response to anti‐tumor necrosis factor therapy. Arthritis Rheumatol 2016; 68: 2867–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. AbbVie . A study comparing ABT‐494 to placebo and to adalimumab in subjects with rheumatoid arthritis who are on a stable dose of methotrexate and who have an inadequate response to methotrexate (SELECT‐COMPARE) [ClinicalTrials.gov identifier NCT02629159]. In: US National Institutes of Health, ClinicalTrials.gov.
- 6. AbbVie . A study comparing ABT‐494 to placebo in subjects with rheumatoid arthritis on a stable dose of conventional synthetic disease‐modifying antirheumatic drugs (csDMARDs) who have an inadequate response to csDMARDs alone (SELECT‐NEXT) [ClinicalTrials.gov identifier NCT02675426]. In: US National Institutes of Health, ClinicalTrials.gov.
- 7. AbbVie . A study comparing ABT‐494 monotherapy to methotrexate (MTX) monotherapy in subjects with rheumatoid arthritis (RA) who have an inadequate response to MTX (SELECT‐MONOTHERAPY) [ClinicalTrials.gov identifier NCT02706951]. In: US National Institutes of Health, ClinicalTrials.gov.
- 8. AbbVie . A study to Compare ABT‐494 to placebo in subjects with rheumatoid arthritis on stable dose of conventional synthetic disease‐modifying antirheumatic drugs (csDMARDs) who have an inadequate response or intolerance to biologic DMARDs (SELECT‐BEYOND) [ClinicalTrials.gov identifier NCT02706847]. In: US National Institutes of Health, ClinicalTrials.gov.
- 9. AbbVie . A study in healthy adults and adult subjects with rheumatoid arthritis to evaluate the safety, tolerability and pharmacokinetics after multiple doses of ABT‐494 [ClinicalTrials.gov identifier NCT01741493]. In: US National Institutes of Health, ClinicalTrials.gov.
- 10. AbbVie . A multicenter, randomized, double‐blind, placebo‐controlled study of ABT‐494 for the induction of symptomatic and endoscopic remission in subjects with moderately to severely active Crohn's disease who have inadequately responded to or are intolerant to anti‐TNF therapy (Celest study) [ClinicalTrials.gov identifier NCT02365649]. In: US National Institutes of Health, ClinicalTrials.gov.
- 11. Mohamed MF, Camp HS, Jiang P, Padley RJ, Asatryan A, Othman AA. Pharmacokinetics, safety and tolerability of ABT‐494, a novel selective JAK 1 inhibitor, in healthy volunteers and subjects with rheumatoid arthritis. Clin Pharmacokinet 2016; 55: 1547–1558. [DOI] [PubMed] [Google Scholar]
- 12. Guidance for Industry: Drug interaction studies – Study design, data analysis, implications for dosing, and labeling recommendations. Draft Guidance. In: US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, February 2012.
- 13. Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V, Gan L, Grimm S, et al., Pharmaceutical Research and Manufacturers of America Drug Metabolism/Clinical Pharmacology Technical Working Group, FDA Center for Drug Evaluation and Research . The conduct of in vitro and in vivo drug–drug interaction studies: a Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab Dispos 2003; 31: 815–832. [DOI] [PubMed] [Google Scholar]
- 14. Ho RH, Kim RB. Transporters and drug therapy: implications for drug disposition and disease. Clin Pharmacol Ther 2005; 78: 260–277. [DOI] [PubMed] [Google Scholar]
- 15. Guidance for Industry: Food‐effect bioavailability and fed bioequivalence studies. In: United States Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, December 2002.
- 16. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, et al. The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 2015; 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao P, Ragueneau‐Majlessi I, Zhang L, Strong JM, Reynolds KS, Levy RH, et al. Quantitative evaluation of pharmacokinetic inhibition of CYP3A substrates by ketoconazole: a simulation study. J Clin Pharmacol 2009; 49: 351–359. [DOI] [PubMed] [Google Scholar]
- 20. Ke AB, Zamek‐Gliszczynski MJ, Higgins JW, Hall SD. Itraconazole and clarithromycin as ketoconazole alternatives for clinical CYP3A inhibition studies. Clin Pharmacol Ther 2014; 95: 473–476. [DOI] [PubMed] [Google Scholar]
- 21. Liu L, Bello A, Dresser MJ, Heald D, Komjathy SF, O'Mara E, et al. Best practices for the use of itraconazole as a replacement for ketoconazole in drug‐drug interaction studies. J Clin Pharmacol 2016; 56: 143–151. [DOI] [PubMed] [Google Scholar]
- 22. Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivisto KT. Pharmacokinetic interactions with rifampicin: clinical relevance. Clin Pharmacokinet 2003; 42: 819–850. [DOI] [PubMed] [Google Scholar]
- 23. Kalliokoski A, Niemi M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol 2009; 158: 693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]