

Abstract
We report a genomic selection (GS) study of growth and wood quality traits in an outbred F2 hybrid Eucalyptus population (n=768) using high-density single-nucleotide polymorphism (SNP) genotyping. Going beyond previous reports in forest trees, models were developed for different selection targets, namely, families, individuals within families and individuals across the entire population using a genomic model including dominance. To provide a more breeder-intelligible assessment of the performance of GS we calculated the expected response as the percentage gain over the population average expected genetic value (EGV) for different proportions of genomically selected individuals, using a rigorous cross-validation (CV) scheme that removed relatedness between training and validation sets. Predictive abilities (PAs) were 0.40–0.57 for individual selection and 0.56–0.75 for family selection. PAs under an additive+dominance model improved predictions by 5 to 14% for growth depending on the selection target, but no improvement was seen for wood traits. The good performance of GS with no relatedness in CV suggested that our average SNP density (~25 kb) captured some short-range linkage disequilibrium. Truncation GS successfully selected individuals with an average EGV significantly higher than the population average. Response to GS on a per year basis was ~100% more efficient than by phenotypic selection and more so with higher selection intensities. These results contribute further experimental data supporting the positive prospects of GS in forest trees. Because generation times are long, traits are complex and costs of DNA genotyping are plummeting, genomic prediction has good perspectives of adoption in tree breeding practice.
Introduction
Genomic selection (GS), a method now fully integrated in genetic prediction of breeding values in domestic animals (Van Eenennaam et al., 2014), has been increasingly considered for plant breeding. Its potential applications have been discussed for various crops and forest trees (Desta and Ortiz, 2014; Grattapaglia, 2014) and an exponential growth of published GS studies in several major plant species has taken place in the past 6 years (Lin et al., 2014). However, following the Gartner hype cycle for emerging technologies (Fenn and Raskino, 2008) we are now entering a new phase, where detailed considerations are needed to fully understand the limits and exploit the opportunities of GS (Jonas and de Koning, 2013; Heslot et al., 2015) in order to reach the ‘plateau of productivity’. In the context of long-term tree breeding programs, these include the assessment of the impact of relatedness between training population and selection candidates, the contribution of nonadditive variation to predictive ability and the expected response to GS for different selection targets and selection intensities (Grattapaglia, 2014).
In forest trees, genomic selection was originally approached by simulation studies (Grattapaglia and Resende, 2011; Iwata et al., 2011) and the first experimental reports in Pinus (Resende et al., 2012b) and Eucalyptus (Resende et al., 2012a) demonstrated the encouraging prospects of this new method. Other experimental studies have since then confirmed the potential of GS in conifers and eucalypts (Resende et al., 2012c; Zapata-Valenzuela et al., 2013; Beaulieu et al., 2014a, 2014b; Lima, 2014; El-Dien et al., 2015; Ratcliffe et al., 2015). Recently, genomic prediction models across generations in Pinus pinaster (Bartholomé et al., 2016; Isik et al., 2016) further underpinned the promising perspectives of GS to accelerate breeding of forest trees. Following the developments of GS in animal breeding, early studies in forest trees typically modeled predictions accounting only for the additive component of the genetic variation, whereas the nonadditive contribution only more recently has been contemplated (Munoz et al., 2014; Bouvet et al., 2016; de Almeida Filho et al., 2016; El-Dien et al., 2016). Because genome-wide single-nucleotide polymorphism (SNP) data accurately measure relatedness by capturing the Mendelian sampling term within families, the additive formulation of a genomic relationship matrix (GRM) (G) can be extended to a (G+D) matrix to include dominance effects. Such models were shown to improve the partitioning of genetic variance relative to the pedigree-based approach, and can be used to predict the total genotypic value of an individual in genomic selection (Su et al., 2012; Vitezica et al., 2013; Munoz et al., 2014; Nishio and Satoh, 2014; Wang et al., 2014; Bouvet et al., 2016; de Almeida Filho et al., 2016; El-Dien et al., 2016).
Hybrid eucalypt breeding programs aim at selecting superior trees to be deployed as clones, thus taking advantage of both the additive and nonadditive genetic components. Broadly speaking, these programs usually encompass two main strategies: (1) reciprocal recurrent selection between pure species and (2) multispecies synthetic hybrid populations (SYN) (Assis and de Resende, 2011; Rezende et al., 2014). SYN has gained increasing interest because of its simplicity and higher speed by which generation advancement is made and new elite clones produced, together with the possibility of exploiting a broader range of interspecific variation (Kerr et al., 2004; Assis and de Resende, 2011). SYN is based on relatively small effective size populations composed by 10 to 20 elite parents (hybrids or pure species individuals) that are crossed in incomplete diallel mating designs, aiming to capture desirable features across species into single trees that are ultimately deployed as clones. As discussed earlier (Resende et al., 2012a) it is in the framework of such small, fast-moving specialized Eucalyptus breeding populations that the operational application of GS has an immediate and clear potential of application. However, it is still an open question whether the incorporation of dominance effects in genomic prediction models would effectively improve the predictive ability of phenotypes of single individuals and families for different traits in Eucalyptus.
A second issue relevant to the prospects of GS in tree breeding is the impact of relatedness as a driver of accuracy. Selection candidates closely related to the training population have an advantage in accuracy over distantly related ones (Daetwyler et al., 2013; Lin et al., 2014; Van Eenennaam et al., 2014; Heslot et al., 2015). Increased relatedness reduces the number of independently segregating chromosome segments, therefore increasing the probability that chromosome segments identical by descent sampled in the training population are also found in the selection candidates. If the validation population is more or less related to the training population than the selection candidates, then the prediction accuracy will be over- or underestimated, respectively. Although random assignment of individuals to training or validation sets is prone to inflate the predictive ability because of within-family components, a more realistic approach is to assign whole full-sib or half-sib families to training or validation sets (Saatchi et al., 2011). This expectation was shown experimentally in forest trees where models developed for one population had limited or no ability of predicting phenotypes in an unrelated one (Resende et al., 2012a; Beaulieu et al., 2014a, 2014b). Those studies, however, used a considerably limited marker density, of only a few thousand markers, unlikely to allow capturing any short-range historical linkage disequilibrium (LD), such that prediction models relied essentially on relatedness. It is therefore important to see whether a higher marker density would allow maintaining satisfactory predictive abilities when there is no relatedness between training and validation sets that could be an indicative that short-range LD between SNPs and quantitative trait loci would also be contributing to predictions.
In this study, we report the development of genomic prediction for key traits in Eucalyptus breeding using much higher-density and ‘gold-standard’-quality SNP data than in previous studies in forest tree, generated with a fixed content SNP-chip (EuCHIP60K) (Silva-Junior et al., 2015). To derive more realistic estimates of the value of GS we carried out cross-validation (CV) by removing genetic relatedness between individuals in the training and validation sets. Predictive abilities (PAs) were estimated for different selection targets, namely, families, individuals within family and individuals considering the entire breeding population, using a genomic model that includes dominance (G+D). To provide a more breeder-intelligible assessment of the performance of GS we calculated the expected response to GS (RGS) as the percentage gain over the population average expected genetic value (EGV) for different proportions of individuals selected in the population, based on their genomic expected genotypic value (GEGV). In addition, the response to genomic selection was compared with the response to phenotypic selection (PS) considering the application of GS at early age for different proportions of individuals selected.
Materials and methods
Plant material and SNP data
Genomic prediction models were developed for a multispecies synthetic breeding population composed by 856 trees distributed across 37 full-sib families derived from an incomplete diallel mating design among 10 unrelated elite Eucalyptus grandis × Eucalyptus urophylla F1 hybrids (Ne=10) (Supplementary Table S1) such that the target trees of our study were equivalent to outbred F2 individuals. Phenotypes for diameter at breast height (DBH), height growth (HEI), basic wood density (BWD) and screened pulp yield (SPY) and genotypes at 24 806 polymorphic SNPs were collected for 768 of the 856 trees as described in a previous genome-wide association study of this same population (Resende et al., 2017). SNP genotypes were obtained using the Illumina Infinium EuCHIP60K (Silva-Junior et al., 2015) that includes 47 069 SNPs located inside or at <10 kb distance of 30 444 of the 36 376 (84%) annotated gene models. SNP genotypes were called from intensity files obtained through GENESEEK (Lincoln, NE, USA) using GenomeStudio 2011.1 (Illumina Inc., San Diego, CA, USA) following standard genotyping and quality control procedures with no manual editing of clusters (Silva-Junior et al., 2015). The 24 806 SNPs had call frequency >90%, sample call rates >95%, minimum allele frequency >0.01 and provided an average genome-wide density of one SNP every 24.3 kb. BWD was measured by the water displacement method using a 3–5-cm-thick wood disk sampled at breast height and SPY was estimated by batch kraft digestion of 150 g of wood chips at 15–18% effective alkali. For this study, however, DBH and HEI were combined to estimate tree volume (VOL) in cubic meters calculated by Equation (1) (Schumacher and Hall, 1933) where f is the taper factor (assumed to be 0.45), and π is the ratio between the circumference and diameter of a circle.
VOL was in turn used to estimate the mean annual increment (MAI), the actual trait used in selection decisions, by extrapolating the volume of individual trees in one hectare divided by the age at measurement.
Phenotypic model
Phenotypic evaluation between and within families, components of phenotypic variation and individual selection accuracies, were calculated using the mixed-effects procedure with model no. 35 of the free software Selegen-REML/BLUP (Resende, 2016) described as follows:
where y is the observed phenotypic values; β is the controlled fixed effects corresponding to the field trial; b is the random effect of experimental blocks; a is the random additive effect of individual; s is the random effect of specific combining ability associated with full-sib families; ε is the residual; X is the incidence matrix for fixed effects; W, Z and T are the incidence matrices for random effects. The EGV was calculated based on Equation (2) where EGV = â + ŝ. The covariance structures are:
with A corresponding to the matrix of additive genetic relationships (also known as identity-by-descent matrix) and I is an identity matrix. Trait heritabilities were obtained by calculating 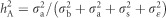 for each one of three traits under consideration MAI, BWD and SPY. Total genotypic heritabilities were obtained by calculating
for each one of three traits under consideration MAI, BWD and SPY. Total genotypic heritabilities were obtained by calculating 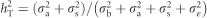 .
.
Genomic model
Genomic evaluations were carried out using a genomic best linear unbiased prediction (GBLUP) approach, using the Sommer R package (Covarrubias-Pazaran, 2016) using the full (G+D) model in Equation (3)
and the reduced (G) model that is equivalent to Equation (3) without the dominance term. The input data were the adjusted phenotypes obtained with Equation (2), corrected for fixed effects of experiment and random effects of blocks, by means of the expression 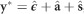 , where
, where 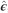 ,
,  and
and  are estimated values of residual, individual and full-sib families. Following, g is the genomic random effect with covariance structure given by
are estimated values of residual, individual and full-sib families. Following, g is the genomic random effect with covariance structure given by 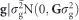 ; d is the dominant random effect with covariance structure given by
; d is the dominant random effect with covariance structure given by 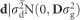 ; μ is the intercept (fixed effect of overall mean); ε is the residual of the genomic model, with covariance structure given by
; μ is the intercept (fixed effect of overall mean); ε is the residual of the genomic model, with covariance structure given by 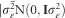 ; X and Z are the incidence matrices for fixed and random effects of the genomic model, respectively. The G matrix stands for the full genomic relationships matrix associated to the random effect accounting for relatedness and family structure (also known as identity-by-state matrix), and D is the genomic matrix of the dominant effects (Vitezica et al., 2013), represented respectively by Equations (4) and (5):
; X and Z are the incidence matrices for fixed and random effects of the genomic model, respectively. The G matrix stands for the full genomic relationships matrix associated to the random effect accounting for relatedness and family structure (also known as identity-by-state matrix), and D is the genomic matrix of the dominant effects (Vitezica et al., 2013), represented respectively by Equations (4) and (5):
where W is the SNP marker incidence matrix assuming 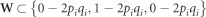 , pi is the allele frequency of the ith SNP marker present in the W matrix columns (Legarra et al., 2008);
, pi is the allele frequency of the ith SNP marker present in the W matrix columns (Legarra et al., 2008); 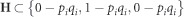 , with qi=1−pi. Genomic (or molecular) heritabilities were obtained for MAI, BWD and SPY for the two alternative models, the reduced additive-only model (G) and the full additive+dominance (G+D) model by calculating
, with qi=1−pi. Genomic (or molecular) heritabilities were obtained for MAI, BWD and SPY for the two alternative models, the reduced additive-only model (G) and the full additive+dominance (G+D) model by calculating 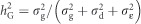 and
and 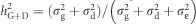 .
.
Cross-validations
To validate the GS models three jackknife CV schemes were used involving different levels of relatedness between training and validation populations based on the existing half-sib and full-sib family relationships in the population (Supplementary Figure S1). In the first one (CV+Relatedness) each family was used as a validation population, whereas the remaining 36 families were used jointly as a training population. Thus, in this scheme, 37 validation sets were tested and only half-sib relationships between training and validation were present. In the second one (CV−Relatedness) all half-sib relationships between training and validation populations were removed such that individuals used for validation were totally unrelated to individuals in training. Finally, a third scheme (CV_Random) was used as a control where individuals were randomly assigned to training and validation populations such that variable levels of half and full-sib relationships were present in the different fold validation sets (Supplementary Figure S1). Training populations had between 740 and 758 individuals for CV+Relatedness, and between 400 and 603 individuals for CV−Relatedness, whereas validation populations had between 10 and 28 individuals for both schemes. For adequate comparisons, a 37-fold validation was used in the CV_Random scheme such that the number of individuals in training and validation populations were the same as in CV+Relatedness.
For each CV scheme, PAs for the (G+D) model were estimated by the correlation between the GEGV and the corresponding phenotypic values (y*) corrected for environmental and experimental effects estimated by Equation (2). GEGVs were obtained by Equation (5)
where W is the SNP marker matrix (with order i markers by j individuals of the validation population), such as described for Equation (4), and 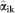 is the additive effect of ith marker of kth validation group and
is the additive effect of ith marker of kth validation group and 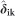 is the dominance effect of ith marker of kth validation group. PAs were estimated for three different selection targets: (1) for overall individual selection among all 768 trees considered jointly, a single PA by the correlation between the vectors of GEGV and y* for all 768 individuals; (2) for family selection a single PA was estimated by the correlation between the family average GEGV and family average y*, such that the PA was a correlation with 37 entries; (3) for individual selection within family a PA for each one of the 37 families by estimating the correlation of the vectors of GEGV and y* for the individuals of each family separately. For comparison, PAs were also estimated for the reduced, additive-only model (G) by estimating the genomic expected breeding value whose expression is equivalent to Equation (5) without the dominance term.
is the dominance effect of ith marker of kth validation group. PAs were estimated for three different selection targets: (1) for overall individual selection among all 768 trees considered jointly, a single PA by the correlation between the vectors of GEGV and y* for all 768 individuals; (2) for family selection a single PA was estimated by the correlation between the family average GEGV and family average y*, such that the PA was a correlation with 37 entries; (3) for individual selection within family a PA for each one of the 37 families by estimating the correlation of the vectors of GEGV and y* for the individuals of each family separately. For comparison, PAs were also estimated for the reduced, additive-only model (G) by estimating the genomic expected breeding value whose expression is equivalent to Equation (5) without the dominance term.
Expected performance of genomic selection
The expected performance of GS compared with standard PS was evaluated only for the full (G+D) model by calculating the RGS as a percentage of the population average EGV as follows:
where 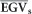 is the average of the selected population proportion (or individual EGV when a single individual is selected) and
is the average of the selected population proportion (or individual EGV when a single individual is selected) and 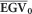 is the population average. For example, suppose a truncation selection of the top 40 individual trees of the population for a trait based on the GEGVs. We took the corresponding EGV for these individuals and calculated their
is the population average. For example, suppose a truncation selection of the top 40 individual trees of the population for a trait based on the GEGVs. We took the corresponding EGV for these individuals and calculated their 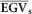 and from that, the RGS as a percentage gain over the average population EGV according to Equation (6). Because the result of
and from that, the RGS as a percentage gain over the average population EGV according to Equation (6). Because the result of 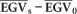 corresponds to the selection differential and the heritability is already embedded in the calculation of the EGVs, RGS corresponds to the response to selection as described in Falconer (1989). Furthermore, because GS can be practiced in Eucalyptus at seedling stage, 1 year or less, whereas PS requires obtaining phenotypes for trees at least at age 3 years, a conservative RGS(%)/year was obtained by dividing the RGS(%) by three such that the response to selection/year of GS and PS could be compared. RGS was then estimated for increasingly higher proportions of individuals selected.
corresponds to the selection differential and the heritability is already embedded in the calculation of the EGVs, RGS corresponds to the response to selection as described in Falconer (1989). Furthermore, because GS can be practiced in Eucalyptus at seedling stage, 1 year or less, whereas PS requires obtaining phenotypes for trees at least at age 3 years, a conservative RGS(%)/year was obtained by dividing the RGS(%) by three such that the response to selection/year of GS and PS could be compared. RGS was then estimated for increasingly higher proportions of individuals selected.
Results
Genomic predictions
Trait heritabilities under the additive (h2A) and additive+dominance (h2T) models were essentially the same for BWD and SPY but showed a considerable difference for MAI increasing from 0.33 to 0.51, and the same was seen when genomic heritabilities were compared. For MAI a considerable increase of 84% was seen in the estimate of genomic heritability when the dominance component was included, going from h2G=0.26 to h2G+D=0.48 (Table 1). The genomic heritability (h2G+D) explained 94.1, 95.7 and 87.5% of the total heritability (h2T) estimated from phenotypic data (Table 1). Genomic PAs for BWD were slightly higher than for MAI and SPY, irrespective of the selection target and CV scheme, consistent with the higher heritability of BWD. For comparison, we report the PAs for the additive-only model (G), and the additive+dominance model (G+A) (Table 1). Only for MAI the PAs for the additive+dominance model (G+A) were somewhat higher than for the additive-only model (G), but this increase was relatively modest between 5 and 14%. Henceforth, we will only consider the PA estimates for the (G+A) model that is the more relevant one for tropical Eucalyptus amenable to clonal propagation. For all three traits, PAs were higher as more relatedness was present between training and validation sets (Table 1 and Figure 1). As expected, higher PAs were seen in the CV_Random scheme (full-sib and half-sib relationship present) when compared with CV by minimizing or removing relatedness (Table 1 and Figure 1). PAs decreased between 9 and 25% when CV was carried out removing all relatedness between training and validation (CV−Relatedness) when compared with CV_Random for individual selection, 17 to 28% for family selection and 14 to 32% for within-family individual selection. Reductions were always the highest for MAI (23, 30 and 25%) for the three selection targets, consistent with the lower heritability of this trait (Table 1). Nevertheless, PAs when no relatedness was present in CV were still quite satisfactory at 0.40, 0.57 and 0.48 for individual selection and 0.56, 0.75 and 0.65 for family selection for MAI, BWD and SPY, respectively. When the selection target was the individual within family, for over 50% of the full-sib families PAs were greater than 0.41, 0.56 and 0.45 for the three traits, MAI, BWD and SPY, respectively, reaching values ⩾0.82 for BWD and SPY in some families.
Table 1. Accuracy of phenotypic selection and predictive abilities (PAs) of genomic selection for different cross-validation (CV) schemes and selection targets for MAI, BWD and SPY in the Eucalyptus breeding population.
| Attribute | MAI | BWD | SPY | |
|---|---|---|---|---|
| Phenotypic | ||||

|
35.28±16.87 | 471.64±33.38 | 50.76±2.33 | |
| h2A | 0.33 | 0.69 | 0.46 | |
| h2T | 0.51 | 0.70 | 0.48 | |
| Accuracy of individual selection | 0.70 | 0.80 | 0.74 | |
| Accuracy of family selection | 0.78 | 0.85 | 0.83 | |
| Genomic | ||||
| h2G | 0.26 | 0.67 | 0.37 | |
| h2G+D | 0.48 | 0.67 | 0.42 | |
| Explained h2T | 94.1% | 95.7% | 87.5% | |
| CV_Random: cross-validation including full- and half-sib individuals | ||||
| PA individual selection | 0.49/0.52 | 0.65/0.64 | 0.53/0.51 | |
| PA between-family selection | 0.78/0.80 | 0.84/0.83 | 0.79/0.79 | |
| PA within-family individual selection | Min | 0.16/0.35 | 0.36/0.36 | 0.23/0.20 |
| Mean | 0.48/0.52 | 0.64/0.63 | 0.53/0.53 | |
| Median | 0.52/0.55 | 0.63/0.63 | 0.54/0.53 | |
| Max | 0.72/0.79 | 0.84/0.84 | 0.88/0.88 | |
| CV+Relatedness: cross-validation including only half-sib individuals | ||||
| PA individual selection | 0.40/0.50 | 0.64/0.64 | 0.50/0.51 | |
| PA between-family selection | 0.76/0.79 | 0.74/0.75 | 0.68/0.66 | |
| PA within-family individual selection | Min | 0.04/−0.03 | 0.31/0.31 | 0.14/0.00 |
| Mean | 0.41/0.47 | 0.60/0.61 | 0.47/0.46 | |
| Median | 0.41/0.48 | 0.63/0.64 | 0.51/0.48 | |
| Max | 0.80/0.78 | 0.84/0.85 | 0.84/0.84 | |
| CV−Relatedness: cross-validation with no relatedness | ||||
| PA individual selection | 0.37/0.40 | 0.56/0.57 | 0.48/0.48 | |
| PA between-family selection | 0.52/0.56 | 0.63/0.75 | 0.65/0.65 | |
| PA within-family individual selection | Min | −0.21/−0.15 | 0.20/0.20 | 0.09/0.11 |
| Mean | 0.34/0.39 | 0.54/0.52 | 0.46/0.46 | |
| Median | 0.34/0.41 | 0.57/0.56 | 0.45/0.45 | |
| Max | 0.73/0.74 | 0.82/0.82 | 0.87/0.85 | |
Abbreviations: BWD, basic wood density (kg m−3); h2, trait heritability; h2G, genomic heritability; MAI, mean annual increment (m3 ha−1 per year); Max, maximum; Min, minimum; SPY, screened pulp yield (%);  , phenotypic mean and their s.d.
, phenotypic mean and their s.d.
PAs are reported for the additive-only model (G)/additive+dominance model (G+D).
Figure 1.

Box plots of the predictive abilities for within-family individual selection obtained with the three different CV schemes for the additive-only (G) and additive+dominance (G+D) models for the three traits: MAI(m3 ha−1 per year), BWD (kg m−3) and SPY (%). Thick lines within the box plots indicate the median PA of the distribution for the 37 families.
Expected response to genomic selection
The expected performance of GS compared with standard PS was evaluated by calculating the response to genomic selection (RGS%). The performance of GS was compared using only for the most rigorous CV scheme, that is, without relatedness (CV−Relatedness). Scatter diagrams between the GEGV and the corresponding phenotypic value (y*) (corrected for environmental and experimental effects) were plotted for the three traits and for individual and family selection. GEGV is centered to a zero average, and the phenotypic value in actual units of measurement (Figure 2). In addition, a heatmap was applied to each dot (individual or family) to indicate the response (RGS%) that would result from selecting the individual or family. Dots colored toward blue indicate individuals or families that would result in higher gain in EGV. As expected, better performance of GS would result from family than individual selection and for BWD and SPY than MAI. Truncation genomic selection of the top 40 and 20 individuals based on their GEGVs (under CV−Relatedness) were performed to evaluate the response (Figure 3). Such numbers of selected individuals in this population of 768 trees correspond to ∼5 and 2.5% selection intensities, respectively, that are the usual proportions of trees selected to be taken to the next step of clonal trial in a Eucalyptus breeding program. Boxplots show how would these individuals selected based on their GEGV correspond to the ranking in terms of phenotype-based total expected EGV. All selected individuals by GS are ranked at the top of the distribution of EGV with the exception of a few outliers indicated by the dots outside the boxplot range. The boxplots also show that for all traits in both selection intensities ~50% of the individuals selected by GEGV would rank among the top 100 individuals selected by EGV, exception made for SPY where the median was at the 116th individual ranked by EGV. To check whether GS would in fact perform better than random selection we built a null distribution with the same selection intensities (40 and 20 individuals) using a resampling permutation test with 10 000 iterations. The null hypothesis (Ho) considers that GS would select individuals at random, whereas the alternative (Ha) would be that GS correctly selects individuals with higher average EGV. The dashed line indicates the probability of rejecting Ho at α=1% and the solid line the average EGV of the selected individuals. Results clearly show that GS would successfully select individuals with an average EGV much higher than the population average (Figure 4). Finally, because GS would be carried out at seedling stage (less than age 1 year) whereas PS, in the fastest scenario, at age 3 years, a conservative response per year (RGS(%)/year) was calculated to compare GS with PS for variable proportions of individuals selected by GS. When compared with PS, GS provides much larger response per year for all three traits. Taking the results for MAI for example, if the top 10% individuals were selected by GS the graph shows that the response to selection over the population average EGV would be 40% but only 20% by PS, that is, a relative increase of 100% gain per year. Equivalent gain in response would be seen for BWD and SPY (Figure 5). The relative gains would evidently become smaller as a larger proportion of individuals were selected that is not what is done in breeding. Conversely, if PS would be practiced at ages later than 3 years, for example age 6 years, the relative gain of GS over PS illustrated by the spread between the GS curve and the PS dotted curve would be even larger.
Figure 2.

Scatter plots of the performance of genomic selection for the (G+D) model with no relatedness between training and validation sets. The x and y axes are respectively the adjusted phenotypic values (y*) and genomic breeding value (GEGV). Top charts: individual selection over the entire population; bottom charts: between-family selection. Heatmaps indicate the response to genomic selection as a percentage gain over the population average expected breeding value (EGV) obtained when selecting the individual or family. MAI (m3 ha−1 per year), BWD (kg m−3) and SPY (%).
Figure 3.

Boxplots of the GEGV of the top 40 and 20 genomically selected individuals scaled to the total EGV ranking of all 768 individuals in the population. Dashed line is the median EGV of the population. The EGV rankings of the median (thick line inside boxplots) and extreme individuals selected are indicated; dots outside the boxplot range are outliers. MAI (m3 ha−1 per year), BWD (kg m−3) and SPY (%).
Figure 4.

Average of the EGV of 40 and 20 top selected individuals based on their GEGV (solid vertical lines) compared with the null distributions of the same numbers of randomly selected individuals. Dashed lines indicate probabilities of rejecting Ho at 1%. MAI (m3 ha−1 per year), BWD (kg m−3) and SPY (%).
Figure 5.

Response to GS expressed as a percentage gain of the average population EGV per year (solid line) compared with PS (dashed line) calculated for different proportions of individuals selected by GS.
Discussion
In this study, we provide original results regarding the expected response of genomically selecting individual trees and families in terms of gain over the population average total expected genetic value in hybrid Eucalyptus. Because tree breeding generally advances based on the application of truncation selection of specific proportions of individuals or families, more than learning the overall predictive ability, it is relevant to estimate the actual response to selection and consequently the gain that GS would provide over PS. Furthermore, our results advance beyond most previous reports in forest trees in general (see below), and Eucalyptus in particular, by estimating genomic predictive ability including dominance in the genomic model and removing relatedness in CV.
Pedigree-based heritabilities estimated for growth and wood quality traits were moderate to high with BWD showing higher values than MAI and SPY. Heritabilities for MAI were generally similar to estimates reported earlier for other E. grandis × E. urophylla hybrid populations (Bouvet and Vigneron, 1995; Bouvet et al., 2009; Lima, 2014) with a higher value when dominance effects were included in the model. Genomic heritabilities either under an additive-only (h2G) or an additive+dominance (h2G+D) model, captured large proportions (>87%) of the pedigree-based estimates and showed a considerable difference only for MAI (Table 1). This result is consistent with reports based on pedigree data indicating that dominance had substantial contribution to genetic variance for growth in E. grandis × E. urophylla hybrids but was unimportant for BWD (Bison et al., 2006, 2009), and in line with the documented heterosis for growth exploited in clonally propagated hybrid eucalypts (Rezende et al., 2014). The slightly larger heritabilities obtained from pedigrees could be because of the inability of the pedigree model to establish the true genetic relationship among the individuals, and its ineffectiveness in disentangling the nonadditive genetic component from the additive one, particularly so in some studies that were carried out on half-sib families (Beaulieu et al., 2014a; Munoz et al., 2014; El-Dien et al., 2015).
Random allocation of individuals in CV has been the common way to estimate PAs in most GS studies to date with two noteworthy exceptions (Beaulieu et al., 2014a; El-Dien et al., 2016). For example, the initial report of GS for Eucalyptus (Resende et al., 2012a) disregarded the degree of kinship in CV and provided estimates of PA that are equivalent to our estimates obtained by CV_Random (Table 1). Although legitimate, this approach tends to overestimate the positive perspective of GS. Predictive abilities when removing relatedness between training and validation sets typically showed a significant reduction (Resende et al., 2012a; Beaulieu et al., 2014a, 2014b; El-Dien et al., 2016), consistent with theoretical studies (Habier et al., 2007; Daetwyler et al., 2013) on the role of relationship as one of the main drivers of genomic selection. In our data set, however, relatively modest reductions between 9 and 30% were seen when no relationship existed between training and validation, depending on trait and selection target (Table 1). Doing the inverse calculation, when performing CV by randomly sampling individuals irrespective of genetic relatedness, PAs 10 to 47% greater than those seen when removing relatedness were obtained, with the largest difference observed for MAI. Nevertheless, from the standpoint of the prospects of GS, PAs obtained when relatedness was removed (CV−Relatedness) were still largely satisfactory (Table 1). We had previously estimated the average LD for this breeding population (r2=0.06) and reported that the LD decayed below the usable LD threshold (r2=0.2) at ∼20–30 kb in the majority of the chromosomes (Resende et al., 2017). Such an estimated extent of LD is consistent with the SNP density in this experiment, on average one SNP every 25 kb (605 Mb/24 806 SNPs) (Supplementary Figure S2), suggesting that in this experiment the SNP density used was able to capture usable short-range LD to drive good predictions despite the lack of kinship. These results are important because they suggest that GS under such circumstances should perform satisfactorily across unrelated families, although most likely limited to parents that have at least a common provenance origin to the ones sampled in this study such that historical LD is shared. Moreover, in theory, if short-range LD is in fact having a key contribution to predictions, PAs estimated in this generation would better persist across subsequent ones (Habier et al., 2013), and this will need to be assessed experimentally across generations.
We used GBLUP as the method to model predictions. We chose not to use alternative methods because it has recurrently been shown that they perform similarly for complex traits with a large number of quantitative trait loci involved (Lorenz et al., 2011; Daetwyler et al., 2013). In fact, growth and wood quality traits in eucalypts, pines and spruces have adequately fit the assumptions of the infinitesimal model in previous reports, such that GBLUP provides a sound compromise between computation time and prediction efficiency (Resende et al., 2012c; Lima, 2014; El-Dien et al., 2015; Ratcliffe et al., 2015; Isik et al., 2016). GS provided considerably higher PA to select families when compared with selecting individuals consistent with the higher accuracies for family versus individual selection either over the entire population or within family when using phenotypic data alone (Table 1 and Figures 1 and 2). To the best of our knowledge, genomic prediction of average family GEGV has not been yet considered by previous studies in forest trees. These first estimates of PA for GS of families in Eucalyptus should be an interesting alternative to breeders when the selection unit for commercial deployment is an elite family and not an individual. Genomically selected families of high GEGV could be amplified by large-scale mating between the parents involved, and additionally scaled up by clonal propagation. Although such a family forestry deployment scheme is common in pines, this has not been implemented yet for tropical eucalypts, mostly because individual clones usually provide higher gains (Rezende et al., 2014). However, with the recent increased occurrence of a number of physiological- and pathogen-related problems of previously adapted clones in Brazil following extended droughts and climatic fluctuations (Gonçalves et al., 2013), the use of family forestry would provide a considerably higher buffering capacity while still providing high productivities. GS of elite families with high predictive ability would therefore find great fit in breeding toward such a deployment strategy in eucalypts.
Predictive abilities under an additive+dominance model improved predictions by 5 to 14% for growth (MAI) when compared with an additive-only model, but no improvement was seen for wood quality traits (Table 1 and Figure 1). Interestingly, although a 54% increase in heritability was seen when dominance was included (h2A=0.33 going to h2T=0.51), only a relatively small increase in predictive ability was realized. Our experimental results agree with a recent study (Bouvet et al., 2016) showing that despite the documented importance of dominance for height growth in F1 hybrid eucalypts (Vigneron and Bouvet, 2000; Bison et al., 2006; Volker et al., 2008; Rezende et al., 2014) adding dominance or epistatic effects did not improve genomic prediction. In other words, in our population that is actually equivalent to an outbred F2 as the parents were themselves hybrids, the inclusion of nonadditive genetic effects did not improve the prediction to the extent that would be expected based on the increase in heritability. Similar conclusions were found in experimental studies in loblolly pine for height growth (Munoz et al., 2014; de Almeida Filho et al., 2016), and in line with these experimental data, simulations have shown that the inclusion of nonadditive effects in the model only improves prediction ability when they are prominent (Denis and Bouvet, 2013; de Almeida Filho et al., 2016). Our results comparing the (G) with the (G+D) models and the available literature prompted us not to spend time testing any more complex model including higher-order interactions (epistasis). In summary, the additive infinitesimal model in the context of genomic prediction of growth and wood quality traits in Eucalyptus, and likely in forest trees in general, seems to be a useful abstraction that closely fits the trait architecture, except for traits involving a few large-effect nonadditive loci (de los Campos et al., 2015).
The scatter plots (Figure 2) are illustrative in showing the dispersion of the GEGV × adjusted phenotypic values (y*) and what would be the expected response in terms of EGV (heatmaps of RGS%) when individuals or families would be selected based on genomic data. In predicting the GEGVs, the phenotypes of individuals are regressed on genetic markers for training. Although the ideal phenotype would be the true total genetic value, this is unknown. Instead, adjusted phenotypes are used to estimate marker effects. Plots therefore compare a genomic expected total genetic value that involves additive+ dominance effects with the phenotypic value (y*) that involves a ‘package’ including additive, dominance, epistasis and uncontrollable environmental variation. Consistent with the magnitudes of the estimates of PA (Table 1), these dot plots and the heatmap inside each dot show that when applying GS one would miss individuals or families that would in principle contribute a higher response and include some poor individuals or families. However, from the standpoint of the actual practice of GS, the relevant issue is to evaluate the average response when applying truncation genomic selection in terms of the average EGV of the selected proportion. The distribution of the GEGV-selected individuals compared very favorably both with their EGV ranking (Figure 3) and with the null distributions of equally sized randomly selected proportions of individuals (Figure 4). Clearly, the GEGV-selected individuals, either the simulated top 20 or 40 individuals, would display a significantly higher average EGV for all three traits, and thus provide a strongly positive response to selection.
Besides the fact that GS would largely identify individuals or families well ranked in terms of EGV, it is when response to selection per year is calculated that the advantage of GS becomes more evident. We calculated a conservative comparative estimate of RGS%/year between GS and PS assuming that PS would be practiced at age 3. In tropical Eucalyptus age–age correlations for growth are typically high between age 3 and rotation age at 6 years, such that age 3 has been consistently shown to be an adequate although minimum biological age for selection (Osorio et al., 2003; Pinto et al., 2014). For wood quality traits such as BWD and SPY however, selection usually has to wait until age 6 for robust phenotyping of adult wood (Rezende et al., 2014). Still, even adopting early PS as a benchmark, responses to genomic selection were considerably higher and more so as a higher selection intensity was applied (Figure 5). The nominally higher percent gains in MAI reflect the significantly larger phenotypic variance for growth when compared with BWD and SPY. It is important to point out, however, that from the economic standpoint a gain of 5% in BWD or 1% in pulp yield (SPY) may have a greater overall impact on the total efficiency of a pulp production operation than a gain of 20% in MAI (Borralho et al., 1993). In the only study that presented a similar approach of evaluating the response to truncation genomic selection on a per year basis in tree breeding, significant gains from selecting the top 5% individuals based on their genomic expected breeding value were also predicted (Beaulieu et al., 2014a, 2014b).
Based on our results we proposed a comparative flowchart of a typical Eucalyptus multispecies recurrent breeding cycle that releases new clones in 18 years compared with two alternative GS schemes (Figure 6). In a more conservative one, where progeny trial is precluded and only the preliminary and the expanded clonal trial are deployed, elite clones would be released in 14 years. A more aggressive one where GS selected seedlings would be immediately deployed in an expanded clonal trial would allow selecting elite clones in 9 years. The recurrent selection breeding cycle would in turn be reduced from 9 to 5 years, depending on the efficiency of flower induction in young plants, usually attained between ages 1 and 2 using growth regulators (Hasan and Reid, 1995). However, not only time would be saved by GS, but also costs involved in the clonal testing phase could be optimized by increasing selection intensity in the clonal trials. GS provides an important and frequently overlooked additional advantage of selecting individuals for all traits of interest simultaneously thus exploiting the entire spectrum of genetic variation available. This further emphasizes the key importance of investing heavily in phenotyping the training population for all traits of interest at the most adequate age in the target environment (Grattapaglia, 2014).
Figure 6.

Comparative timeline of two alternative genomic selection strategies aiming to reduce the breeding cycle length and release of new elite clones of Eucalyptus compared with the conventional breeding cycle. The release of new clones can be reduced to 14 years by precluding the progeny trial or down to 9 years when the initial clonal trial is also eliminated. The recurrent breeding cycle is reduced from 9 to 5 years by precluding the progeny trial.
When breeding for individuals to be deployed as clones, a critical issue that has emerged is how good is the correspondence between the performance of an individual tree in a progeny trial, that is, based on a single observation, and the performance of the same tree as a clone. Unless this correspondence is high, the breeding program efficiency can be severely affected. Experimental studies with E. grandis × E. urophylla hybrids have shown strikingly contrasting results. Whereas in one study the correspondence for wood volume was >80% (Fonseca et al., 2010), in another the coincidence across different selection intensities was only 27% (Reis et al., 2011). This low correspondence was attributed in large part to a significant genotype × year interaction. Although this issue is also present in conventional breeding, it becomes particularly challenging when GS is to be implemented. Assuring that the target environment where phenotypes are collected for model training will be the same one for the forthcoming selection candidates may be much more critical for GS than conventional PS (Heslot et al., 2015). In GS if a particular year of data are a distorted sample of the target environment due to climate fluctuations, it may affect genetic gain over a much longer period of time unless model updating is carried out. This risk could be mitigated by clonally propagating the entire training population. This would open the possibility of replicating the training population in different environments to build environment-specific prediction models. In addition, historical data of prior and existing clonal trials could be very valuable as complementary training sets providing very robust phenotype data. Field data typically comprising hundreds of clones across multiple years and locations together with Geographic Information System (GIS) data (Marcatti et al., 2017) could be integrated in model training and validation to optimize recommendations of Eucalyptus genotype deployment across environments.
In conclusion, this work provides novel data on the expected response to selection in the context of a breeding program that aims at clonal deployment of elite genotypes. More specifically, performance of GS was evaluated with a genomic model including dominance aiming at different selection targets and adopting a more rigorous CV scheme with no relatedness between training and validation sets. These experimental results further highlight the fact that GS has great prospects for accelerating tree breeding, although we are aware that other important issues still remain to be addressed for the practical implementation of genomic prediction, most notably the performance of GS across subsequent generations (Grattapaglia, 2014). The groundbreaking advance of GS in dairy cattle is frequently used as an example of the economically successful use of this technology. Although gains from GS may still be elusive for annual crops (Bassi et al., 2016) it does not seem so for forest trees where GS was considered to be potentially even more successful than in dairy cattle (Jonas and de Koning, 2013). The convergence of genomics and quantitative genetics is bound to transform how we breed trees. Because generation times are long, traits are complex, tree breeders are fully familiar with BLUP methods and costs of DNA genotyping are plummeting, genomic prediction is likely to be adopted in standard tree breeding practice. Strategic and logistics aspects for the adoption of GS are now the challenges to fully integrate this new breeding technology into routine tree improvement.
Data archiving
Genotype and phenotype data have been submitted to the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.ms580.
Acknowledgments
This work was supported by PRONEX/FAPDF/CNPq Grant 2009/00106-8 ‘NEXTREE’ and EMBRAPA Grant 03.11.01.007.00.00 to DG. RTR and OBS-J had CNPq and EMBRAPA doctoral fellowships respectively; DG, MDVR and FFS hold CNPq research fellowships. We thank the CENIBRA SA field and lab technical crew for collecting the phenotypic data.
Footnotes
Supplementary Information accompanies this paper on Heredity website (http://www.nature.com/hdy)
The authors declare no conflict of interest.
Supplementary Material
References
- Assis TF, de Resende MDV. (2011). Genetic improvement of forest tree species. Crop Breed Appl Biotechnol 11: 44–49. [Google Scholar]
- Bartholomé J, Van Heerwaarden J, Isik F, Boury C, Vidal M, Plomion C et al. (2016). Performance of genomic prediction within and across generations in maritime pine. BMC Genomics 17: 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassi FM, Bentley AR, Charmet G, Ortiz R, Crossa J. (2016). Breeding schemes for the implementation of genomic selection in wheat (Triticum spp.). Plant Sci 242: 23–36. [DOI] [PubMed] [Google Scholar]
- Beaulieu J, Doerksen T, Clement S, Mackay J, Bousquet J. (2014. a). Accuracy of genomic selection models in a large population of open-pollinated families in white spruce. Heredity 113: 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu J, Doerksen TK, MacKay J, Rainville A, Bousquet J. (2014. b). Genomic selection accuracies within and between environments and small breeding groups in white spruce. BMC Genomics 15: 1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bison O, Ramalho MAP, Rezende G, Aguiar AM, de Resende MDV. (2006). Comparison between open pollinated progenies and hybrids performance in Eucalyptus grandis and Eucalyptus urophylla. Silvae Genet 55: 192–196. [Google Scholar]
- Borralho NMG, Cotterill PP, Kanowski PJ. (1993). Breeding objectives for pulp production of Eucalyptus globulus under different industrial cost structures. Can J For Res 23: 648–656. [Google Scholar]
- Bouvet JM, Makouanzi G, Cros D, Vigneron P. (2016). Modeling additive and non-additive effects in a hybrid population using genome-wide genotyping: prediction accuracy implications. Heredity 116: 146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvet JM, Saya A, Vigneron P. (2009). Trends in additive, dominance and environmental effects with age for growth traits in Eucalyptus hybrid populations. Euphytica 165: 35–54. [Google Scholar]
- Bouvet JM, Vigneron P. (1995). Age trends in variances and heritabilities in Eucalyptus factorial mating designs. Silvae Genet 44: 206–216. [Google Scholar]
- Covarrubias-Pazaran G. (2016). Genome-assisted prediction of quantitative traits using the R package Sommer. PLoS One 11: e0156744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daetwyler HD, Calus MPL, Pong-Wong R, de los Campos G, Hickey JM. (2013). Genomic prediction in animals and plants: simulation of data, validation, reporting, and benchmarking. Genetics 193: 347–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida Filho JE, Guimarães JFR, e Silva FF, de Resende MDV, Muñoz P, Kirst M et al. (2016). The contribution of dominance to phenotype prediction in a pine breeding and simulated population. Heredity 117: 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de los Campos G, Sorensen D, Gianola D. (2015). Genomic heritability: what is it? PLoS Genet 11: e1005048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis M, Bouvet JM. (2013). Efficiency of genomic selection with models including dominance effect in the context of Eucalyptus breeding. Tree Genet Genomes 9: 37–51. [Google Scholar]
- Desta ZA, Ortiz R. (2014). Genomic selection: genome-wide prediction in plant improvement. Trends Plant Sci 19: 592–601. [DOI] [PubMed] [Google Scholar]
- El-Dien OG, Ratcliffe B, Klapste J, Chen C, Porth I, El-Kassaby YA. (2015). Prediction accuracies for growth and wood attributes of interior spruce in space using genotyping-by-sequencing. BMC Genomics 16: 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Dien OG, Ratcliffe B, Klápště J, Porth I, Chen C, El-Kassaby YA. (2016). Implementation of the realized genomic relationship matrix to open-pollinated white spruce family testing for disentangling additive from nonadditive genetic effects. G3 (Bethesda) 6: 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer DS. (1989) Introduction to Quantitative Genetics. Third edn, Longman Scientific and Technical: Essex, England. [Google Scholar]
- Fenn J, Raskino M. (2008) Mastering the Hype Cycle: How to Choose the Right Innovation at the Right Time. Harvard Business Press: Boston, Massachusetts. [Google Scholar]
- Fonseca RRG, Goncalves FMA, Rosse LN, Ramalho MAP, Bruzi AT, Reis CAF. (2010). Realized heritability in the selection of Eucalyptus spp. trees through progeny test. Crop Breed Appl Biotechnol 10: 160–165. [Google Scholar]
- Gonçalves JLdM, Alvares CA, Higa AR, Silva LD, Alfenas AC, Stahl J et al. (2013). Integrating genetic and silvicultural strategies to minimize abiotic and biotic constraints in Brazilian eucalypt plantations. For Ecol Manage 301: 6–27. [Google Scholar]
- Grattapaglia D. (2014) Breeding forest trees by genomic selection: current progress and the way forward Chap 26, In: Tuberosa R, Graner A, Frison E (eds). Advances in Genomics of Plant Genetic Resources. Springer: New York, pp 652–682. [Google Scholar]
- Grattapaglia D, Resende MDV. (2011). Genomic selection in forest tree breeding. Tree Genet Genomes 7: 241–255. [Google Scholar]
- Habier D, Fernando RL, Dekkers JCM. (2007). The impact of genetic relationship information on genome-assisted breeding values. Genetics 177: 2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habier D, Fernando RL, Garrick DJ. (2013). Genomic BLUP decoded: a look into the black box of genomic prediction. Genetics 194: 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan O, Reid JB. (1995). Reduction of generation time in eucalyptus-globulus. Plant Growth Regul 17: 53–60. [Google Scholar]
- Heslot N, Jannink JL, Sorrells ME. (2015). Perspectives for genomic selection applications and research in plants. Crop Sci 55: 1–12. [Google Scholar]
- Isik F, Bartholome J, Farjat A, Chancerel E, Raffin A, Sanchez L et al. (2016). Genomic selection in maritime pine. Plant Sci 242: 108–119. [DOI] [PubMed] [Google Scholar]
- Iwata H, Hayashi T, Tsumura Y. (2011). Prospects for genomic selection in conifer breeding: a simulation study of Cryptomeria japonica. Tree Genet Genomes 7: 747–758-758. [Google Scholar]
- Jonas E, de Koning DJ. (2013). Does genomic selection have a future in plant breeding? Trends Biotechnol 31: 497–504. [DOI] [PubMed] [Google Scholar]
- Kerr RJ, Dieters MJ, Tier B. (2004). Simulation of the comparative gains from four different hybrid tree breeding strategies. Can J Forest Res 34: 209–220. [Google Scholar]
- Legarra A, Robert-Granie C, Manfredi E, Elsen JM. (2008). Performance of genomic selection in mice. Genetics 180: 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima BM. (2014) Bridging genomics and quantitative genetics of Eucalyptus: genome-wide prediction and genetic parameter estimation for growth and wood properties using high-density SNP data. Available in English at: http://www.teses.usp.br/teses/disponiveis/11/11137/tde-25062014-085814/pt-br.php Dr thesis University of São Paulo: Piracicaba, Brazil. [Google Scholar]
- Lin Z, Hayes BJ, Daetwyler HD. (2014). Genomic selection in crops, trees and forages: a review. Crop Pasture Sci 65: 1177–1191. [Google Scholar]
- Lorenz AJ, Chao SM, Asoro FG, Heffner EL, Hayashi T, Iwata H et al. (2011). Genomic selection in plant breeding: knowledge and prospects. Adv Agron 110: 77–123. [Google Scholar]
- Marcatti GE, Resende RT, Resende MDV, Ribeiro CAAS, dos Santos AR, da Cruz JP et al. (2017). GIS-based approach applied to optimizing recommendations of Eucalyptus genotypes. For Ecol Manage 392: 144–153. [Google Scholar]
- Munoz PR, Resende MFR, Gezan SA, Resende MDV, de los Campos G, Kirst M et al. (2014). Unraveling additive from nonadditive effects using genomic relationship matrices. Genetics 198: 1759–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio M, Satoh M. (2014). Including dominance effects in the genomic BLUP method for genomic evaluation. PLoS One 9: e85792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio L, White T, Huber D. (2003). Age-age and trait-trait correlations for Eucalyptus grandis Hill ex Maiden and their implications for optimal selection age and design of clonal trials. Theor Appl Genet 106: 735–743. [DOI] [PubMed] [Google Scholar]
- Pinto DS, Resende RT, Mesquita AGG, Rosado AM, Cruz CD. (2014). Early selection in tests for growth traits of Eucalyptus urophylla clonestest. Sci For 42: 251–257. [Google Scholar]
- Ratcliffe B, El-Dien OG, Klapste J, Porth I, Chen C, Jaquish B et al. (2015). A comparison of genomic selection models across time in interior spruce (Picea engelmannii x glauca) using unordered SNP imputation methods. Heredity 115: 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis CAF, Goncalves FMA, Rosse LN, Costa R, Ramalho MAP. (2011). Correspondence between performance of Eucalyptus spp trees selected from family and clonal tests. Genet Mol Res 10: 1172–1179. [DOI] [PubMed] [Google Scholar]
- Resende MDV. (2016). Software Selegen-REML/BLUP: a useful tool for plant breeding. Crop Breed Appl Biotechnol 16: 330–339. [Google Scholar]
- Resende MDV, Resende MFR, Sansaloni CP, Petroli CD, Missiaggia AA, Aguiar AM et al. (2012. a). Genomic selection for growth and wood quality in Eucalyptus: capturing the missing heritability and accelerating breeding for complex traits in forest trees. New Phytol 194: 116–128. [DOI] [PubMed] [Google Scholar]
- Resende MFR, Munoz P, Acosta JJ, Peter GF, Davis JM, Grattapaglia D et al. (2012. b). Accelerating the domestication of trees using genomic selection: accuracy of prediction models across ages and environments. New Phytol 193: 617–624. [DOI] [PubMed] [Google Scholar]
- Resende MFR, Munoz P, Resende MDV, Garrick DJ, Fernando RL, Davis JM et al. (2012. c). Accuracy of genomic selection methods in a standard data set of loblolly pine (Pinus taeda L.). Genetics 190: 1503–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resende RT, Resende MDV, Silva FF, Azevedo CF, Takahashi EK, Silva-Junior OB et al. (2017). Regional heritability mapping and genome-wide association identify loci for complex growth, wood and disease resistance traits in Eucalyptus. New Phytol 213: 1287–1300. [DOI] [PubMed] [Google Scholar]
- Rezende GDSP, Resende MDV, Assis TF. (2014) Eucalyptus breeding for clonal forestry. In: Fenning T (ed). Challenges and Opportunities for the World's Forests in the 21st Century. Springer Netherlands: Dordrecht, pp 393–424. [Google Scholar]
- Saatchi M, McClure MC, McKay SD, Rolf MM, Kim J, Decker JE et al. (2011). Accuracies of genomic breeding values in American Angus beef cattle using K-means clustering for cross-validation. Genet Sel Evol 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher FX, Hall FS. (1933). Logarithmic expression of timber-tree volume. J Agric Res 47: 719–734. [Google Scholar]
- Silva-Junior OB, Faria DA, Grattapaglia D. (2015). A flexible multi-species genome-wide 60K SNP chip developed from pooled resequencing 240 Eucalyptus tree genomes across 12 species. New Phytol 206: 1527–1540. [DOI] [PubMed] [Google Scholar]
- Su G, Christensen OF, Ostersen T, Henryon M, Lund MS. (2012). Estimating additive and non-additive genetic variances and predicting genetic merits using genome-wide dense single nucleotide polymorphism markers. PLoS One 7: e45293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eenennaam AL, Weigel KA, Young AE, Cleveland MA, Dekkers JCM. (2014). Applied Animal Genomics: Results from the Field. Annu Rev Anim Biosci 2: 105–139. [DOI] [PubMed] [Google Scholar]
- Vigneron P, Bouvet J. (2000) Eucalypt hybrid breeding in Congo. In: Nikles DG (ed). Hybrid Breeding and Genetics of Forest Trees. Proceedings of QFRI/CRC-SPF Symposium, 9-14th April 2000 Noosa, Queensland, Australia. Department of Primary Industries: Brisbane, pp 14–26. [Google Scholar]
- Vitezica ZG, Varona L, Legarra A. (2013). On the additive and dominant variance and covariance of individuals within the genomic selection scope. Genetics 195: 1223–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volker PW, Potts BM, Borralho NMG. (2008). Genetic parameters of intra- and inter-specific hybrids of Eucalyptus globulus and E-nitens. Tree Genet Genomes 4: 445–460. [Google Scholar]
- Wang C, Prakapenka D, Wang S, Pulugurta S, Runesha HB, Da Y. (2014). GVCBLUP: a computer package for genomic prediction and variance component estimation of additive and dominance effects. BMC Bioinformatics 15: 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapata-Valenzuela J, Whetten RW, Neale D, McKeand S, Isik F. (2013). Genomic estimated breeding values using genomic relationship matrices in a cloned population of loblolly pine. G3 (Bethesda) 3: 909–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.