

Abstract
This article describes the creation of the first expert manually curated noncoding RNA interaction networks for S. cerevisiae. The RNA–RNA and RNA–protein interaction networks have been carefully extracted from the experimental literature and made available through the IntAct database (www.ebi.ac.uk/intact). We provide an initial network analysis and compare their properties to the much larger protein–protein interaction network. We find that the proteins that bind to ncRNAs in the network contain only a small proportion of classical RNA binding domains. We also see an enrichment of WD40 domains suggesting their direct involvement in ncRNA interactions. We discuss the challenges in collecting noncoding RNA interaction data and the opportunities for worldwide collaboration to fill the unmet need for this data.
Keywords: biocuration, network, interaction, ncRNA, binding domain
INTRODUCTION
Macromolecules within an organism perform many different roles, achieved in a large part through their associations with multiple interacting partners. Many differing types of molecular interaction networks have been studied, including protein–protein interaction (PPI) networks, transcriptional regulatory networks (DNA–protein), signal transduction networks, and metabolic networks. These networks are an essential step in developing a systems-level understanding of an organism. Comparisons of different biological networks can give interesting insights into the nature of cellular evolution, function, and organization. Large-scale RNA sequencing and transcriptomic analyses have revealed that, in contrast with previous hypotheses, most of the genome is transcribed into coding or noncoding RNAs (ncRNAs) (Storz 2002; Clark et al. 2011). Functional studies have shown that ncRNAs are involved in complex regulatory networks, which they modulate by interacting with proteins, nucleic acids, and small molecules.
Any RNA molecule that functions without being translated into a protein is, by definition, a ncRNA, and they have been organized into families with markedly different structural or functional roles (Nawrocki et al. 2015). In addition to the well-characterized ribosomal RNAs and transfer RNAs, several other broad classes have been defined, including microRNAs (miRNA), long noncoding RNAs (lncRNA), and small nuclear and nucleolar RNAs (snRNA and snoRNA) (Amaral et al. 2011; Yoshihama et al. 2013; Kozomara and Griffiths-Jones 2014). These classes are still being continuously added to and enlarged by newly discovered molecules. The whole spectrum of ncRNA function is far from being elucidated. It is likely that they participate in signaling networks, modulating the main cellular processes and regulating gene expression, and also play additional roles via specific RNA–DNA, RNA–RNA, or RNA–protein recognition events (Woolford and Baserga 2013; Yamashita et al. 2016). The sheer quantity of reported data leaves no doubt as to the relevance of ncRNAs in a broad spectrum of signaling pathways, and on the multitude of interactions in which they are involved.
Several high-throughput techniques based on UV cross-linking such as CLASH, CRAC, iCLIP, and PAR-CLIP, have been developed for the in vivo identification of RNA–protein as well as RNA–RNA interactions (Granneman et al. 2009; Hafner et al. 2010; Kudla et al. 2011; Modic et al. 2013). With the help of quantitative mass spectrometry, hundreds of new RNA binding proteins have been recently discovered, gaining novel insights into the nature of RNA–protein recognition specificity and the complexity of the RNA–protein networks (Castello et al. 2012; Mitchell et al. 2013; Beckmann et al. 2015).
The curation of these interactions into the public domain, making them available for query or for network analyses, is therefore of critical importance. However, unlike protein–protein interaction data, no systematic manual curation of the published literature in this area has yet been undertaken. Recently, a few resources have gathered RNA interactions and made them available on the web, such as Clipdb (Yang et al. 2015), NPinter (Hao et al. 2016), RAIN (Junge et al. 2017), and RAID (Yi et al. 2017). An important issue that previously made it difficult for interaction databases to capture and organize ncRNA interactions was the lack of a single central resource providing unambiguous identifiers for each RNA molecule, and linking that identifier to the sequence of that RNA. Over the years, particular ncRNA categories have been collected and classified in specialized databases (miRbase, gtRNAdb, tRNAdb, lncRNAdb, etc.), while other RNAs were still only identified by the gene entry name (Chan and Lowe 2009; Jühling et al. 2009; Amaral et al. 2011; Kozomara and Griffiths-Jones 2014; Li et al. 2014). To consistently identify all ncRNAs, the RNAcentral platform was developed (http://rnacentral.org/), with the aim of incorporating and combining the numerous ncRNA databases to create a unified reference resource that allows the search or comparison of any ncRNA of interest (The RNAcentral Consortium 2015, 2017). RNAcentral is now available and acts as an external reference resource for all ncRNA molecules, as UniProtKB (The UniProt Consortium 2015) does for proteins and ChEBI (Hastings et al. 2016) for small molecules. The linking of identifier to sequence enables the precise mapping of point mutation data, which researchers generate to experimentally demonstrate the effect of these mutants on the interactions of ncRNA with other molecules.
Now that an external reference resource exists for ncRNA molecules, the capture of functional annotations for those molecules has become feasible, and it is possible that information held in disparate databases can be combined. Protein–protein interaction data has been made publicly available in a consistent data format thanks to the combined work of multiple interaction databases and the molecular interaction workgroup of the Human Proteome Organisation Proteomics Standards Initiative (HUPO PSI-MI) (Hermjakob et al. 2004). The community standard data formats enable users to download interaction from multiple resources and readily combine these into a single data set. Members of the IMEx Consortium (Orchard et al. 2012) have gone further than this, managing curation across multiple separately funded resources to give the user a single, consistently annotated data set and make optimum use of public funds by avoiding repetitive capture of the same papers and reducing parallel database and tool development. Members of the IMEx Consortium have already extended their activities into the capture of protein–small molecule data, and the IntAct database also collates transcription factor/transcribed gene interactions.
We present here a study focused on the curation of ncRNA interactions in the IntAct molecular interaction database, and on the analysis of the network subsequently produced. We have selected Saccharomyces cerevisiae—a well-studied model organism—for this work, in order to demonstrate that we can identify those processes and functions we would expect to see in such a network. We have curated over 120 articles and built yeast RNA–protein and RNA–RNA networks. Details concerning the experimental methodology, the interacting molecules’ detection methods, the features of the protein and RNA molecules (tags, point mutations, deletion mutations, etc.), and the organism used to produce or express the molecules, can all be captured by the IntAct data model, and the curation rules and controlled vocabulary (CV) terms have been expanded to encompass this new data type. The use of standard formats and CVs has allowed us to use the existing MIscore algorithm to calculate a confidence score for each interaction, and also has enabled a more detailed characterization of the resulting interaction network (Orchard et al. 2014).
The results of this pilot study demonstrate the value of the expert curation of ncRNA networks, allowing the subsequent analysis both of overall network properties and the detailed interactions between individual molecules. As stated above, Saccharomyces cerevisiae was chosen as an appropriate model organism for this initial work because it contains a limited number of genes that encode ncRNAs. While we are not claiming this network is complete, we have achieved good coverage of the existing literature and keeping the network up to date with new findings will be a relatively easy task. The real value of this work is to highlight the importance of starting to tackle the collection of such data in more complex organisms, such as human. The required data resources and data exchange formats exist; the analysis tools originally developed for PPIs have been shown to be applicable to other molecule types, but funding and expert input from the ncRNA community will be required to tackle this important data collection effort.
RESULTS
General network properties
Protein–protein interaction (PPI) networks have long been studied in detail in various organisms, either as complete proteomes or as specific modules (proteins that belong to a certain pathway or complex). RNA–protein interaction (RPI) networks have been described in detail for very few organisms, with the exception of human and yeast (Nishtala et al. 2016), and little information is available regarding network analysis of RNA–RNA interactions. One of the reasons noncoding RNA–RNA interaction (RRI) networks are less well studied could be the limited availability of experimentally validated large-scale RNA–RNA interaction data.
Here we have taken a comparative approach to studying various network properties of PPI, RPI, and RRI networks to identify any similarities or differences between them.
The yeast PPI network modeled in this study comprises 77,620 unique interactions among 6091 proteins. The RPI network consists of 596 interactions between 105 ncRNAs and 153 proteins, and the RRI network comprises 195 interactions among 102 ncRNAs.
We computed the degree distribution to measure the probability of nodes to interact with k, other nodes. As seen in Figure 1, the degree distribution of the PPI network follows a power law P(k) ∼ k−γ, wherein a large number of nodes interact with a few partners, and a small number of nodes, called hubs, interact with a large number of partners. We observe a similar trend in RPI and RRI networks. Hub proteins are critical for the functioning of a network and their removal can result in failure of the system (Jeong et al. 2001). On average there are 12.74 interactions per node in the PPI network and 651 hubs identified that interact with more than 50 proteins; the major hubs include heat shock proteins Ssb1 (UniProt: P11484), Ssa1 (UniProt: P10591), and Ssa2 (UniProt: P10592) with 3493, 2751, and 2444 interactors, respectively. In comparison, the RPI and RRI networks are sparsely connected with an average 2.13 and 1.90 interactions per node, respectively. The Gar1 protein, a subunit of the H/ACA ribonucleoprotein complex (UniProt: P28007), and the small nucleolar RNA U3a (IntAct: EBI-10821792, RNAcentral: URS0000444F9B) form the major protein and RNA hubs in the RPI network with 32 and 51 edges, respectively. On the other hand, the 18S and 25S ribosomal RNAs (rRNAs) dominate the RRI network (Fig. 2). Comparison of yeast networks with human networks show similar trends of degree distribution (Supplemental Fig. S1). The nodes in the three networks are closely linked with a mean shortest path length of 2.58, 3.27, and 2.35 between any two nodes in PPI, RPI, and RRI networks, respectively.
FIGURE 1.

Degree distribution of a random interaction network (nodes: 6091, edges 77620), protein–protein interaction network (PPI), RNA–protein interaction network (RPI), and RNA–RNA interaction network (RRI). Degree distribution of all three biological networks exhibits a power law. The degree exponent γ is shown in red text.
FIGURE 2.

The yeast noncoding RNA–RNA interaction network (RRI) shows ribosomal RNA subunits as the major hubs interacting with tRNAs and snoRNAs. The spliceosomal module comprising spliceosomal RNAs is circled in red. The ribosomal RNAs are highlighted in pink and tRNAs are highlighted in yellow. The snoRNA nodes SNR8 and SNR81, with high betweenness but low centrality, are shown in orange.
Other network properties, such as clustering coefficient and neighborhood connectivity, show similar trends between the PPI, RPI, and RRI networks (Table 1). The betweenness and closeness centrality of PPI, RPI, and RRI networks were also found to show similar distributions (not shown).
TABLE 1.
Summary of network properties
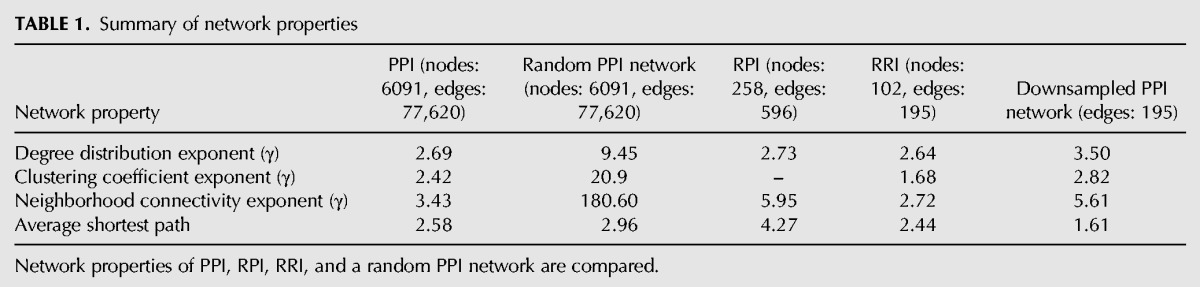
Node centrality measures such as betweenness and closeness centrality evaluate the crucial role of a node as a mediator of interactions within the network. A node is central to the network if the shortest paths of a large number of nodes pass through it. Hubs cover a large number of paths that connect nodes within a network and therefore tend to show high betweenness and closeness centralities. Interestingly, both the PPI and RRI networks contain nodes that have low connectivity (degree) but relatively higher betweenness centralities. In the RRI network these nodes are represented by snoRNAs SNR8 (RNAcentral: URS00001F7017) and SNR81 (RNAcentral: URS00003377F1). SnoRNAs guide chemical modifications of ribosomal, transfer, and other small nuclear RNAs. In the RRI network (Fig. 2), snoRNAs SNR8 and SNR81 interact with nodes from both the ribosomal and spliceosomal RNA subnetworks. The H/ACA box SNR81 guides pseudouridylation of the 25S rRNA subunit and U2 (LSR1) snRNA (Schattner et al. 2004; Wu et al. 2011). Two distinct hairpin sites of SNR81 modify 25S rRNA and U2; the 3′ pseudouridylation pocket guides the pseudouridylation of 25S rRNA at position 1051 (Schattner et al. 2004), whereas the 5′ pseudouridylation pocket guides pseudouridylation of U2 snRNA at positions 42 (Ma et al. 2005) and 93 (Wu et al. 2011). SnoRNA SNR8 modifies the large ribosomal subunit and the actin pre-mRNA. The two snoRNAs form a structural link between the ribosomal subnetwork and the spliceosome module of the RRI network. The five snRNAs U1, U2, U4, U5, and U6 comprise the spliceosome module.
“High betweenness but low connectivity” nodes were identified in a previous study of the yeast proteome (Joy et al. 2005). It was shown that nodes in the PPI network with “high betweenness but low connectivity” tend to be important connectors that link various modules (or clusters) within the network and are essential proteins of recent evolutionary origin (Joy et al. 2005). It has been proposed that “high betweenness but low connectivity” nodes in PPI networks evolve by the addition of nodes with edges and random rewiring of these edges, as a result of gene duplication and point mutations (Joy et al. 2005). Although node addition (gene duplications) and random rewiring (mutations) may explain the presence of “high betweenness low connectivity” nodes in PPI networks, the same evolutionary model cannot be extended to RPI and RRI networks. Protein-coding genes and noncoding genes evolve by different mechanisms and under different evolutionary constraints; while duplication and divergence are suggested as the major mechanism for expanding the protein-coding gene repertoire (Guan et al. 2007; Dujon 2010), mechanisms such as retroposition (Weber 2006; Schmitz et al. 2008), intragenic duplications (Shao et al. 2009), and de novo emergence (Meunier et al. 2013) are suggested to drive the expansion of short ncRNA genes. Moreover, mutation rates of protein-coding genes and noncoding RNAs, which potentially can rewire interactions in the network, are different, thereby affecting edge dynamics of the networks.
Network features
The IMEx curation process captures the full contextual details of experiments generating molecular interactions data, and this resource has previously been successfully used for computational analyses of PPI interactomes (Porras et al. 2015). Most of the controlled vocabulary (CV) terms that describe PPIs were also applicable to RPI and RRI interactions; however, new CV terms were introduced, when required, to enable the curation of specific interaction detection methods or participant detection methods. As shown in Figure 3B, more than 50% of the curated RNA–protein interactions were detected by affinity chromatography, anti-tag or anti-bait coimmunoprecipitation, and tandem affinity purification; however, new approaches developed to specifically capture protein-binding RNAs, such as CRAC and PAR-CLIP (Granneman et al. 2009; Hafner et al. 2010), were included to describe 19% and 1% of the RPI interactions, respectively. The yeast RNA–RNA interactions were demonstrated by cosedimentation (19%), EMSA (5%), pull down (5%), and cross-linking (5%), but the majority (55%) of these interactions were shown with RNA–RNA specific techniques, such as CLASH (Kudla et al. 2011). RNA–RNA interactions are often derived from sequence complementarity that can be predicted with bioinformatics analyses and confirmed by genetic interference. This approach was used to demonstrate most of the yeast snoRNA–rRNA interactions, using methodology that leads to a chemical modification of the ribosomal RNA. To formalize the representation of this approach, the term “chemical RNA modification plus base pair prediction” (Fig. 3A) was introduced into the CV. Information concerning the techniques used to identify the molecules participating in each interaction are annotated into the PSI-MI data exchange format as “participant identification methods.” Figure 3C shows that most of the RNAs were identified by sequence analysis (17%), Northern blot (29%), or primer extension assay (42%).
FIGURE 3.

Distribution of experimental methods used to demonstrate the yeast RRI and RPI interactions (A,B) or the identity of the RNA molecules (C). In D the percentage of curated interactions for each MIscore interval is reported for RPI (orange bar) and RRI (blue bar).
Moreover, to assess the validity of each interaction pair, a confidence score is calculated in IntAct (MIscore) that is reliant on the annotation of experimental, predicted, or inferred data, from which each interacting binary pair is supported (Villaveces et al. 2015). MIscore, as implemented in IntAct, provides a normalized score between 0 and 1 calculated from the weighted sum of the three different subscores: the number of publications, the experimental detection methods, and the interaction types found for the interaction. The higher the score, the more reliable the interaction is seen as being. According to this scoring system, more than 20% of the RNA–protein interactions have a score greater than 0.5, while most of the RNA–RNA interactions scores are in the range of 0.2–0.5, and only 8% of the interactions have a score higher than 0.5 (Fig. 3D). These low values are presumably due to the limited characterization of the RNA interactomes.
Using the Cytoscape software (Shannon et al. 2003), interactions can be filtered according to confidence values to limit the visualization only to the most reliable interactions or to those validated with a specific method. Figure 4 shows a subgraph of the RPI network, including only high-confidence interactions with a score higher than 0.5.
FIGURE 4.

RPI subgraph showing interactions with a MIscore value equal or superior to 0.5. Proteins are colored in yellow and ncRNAs in blue. Edge line width is proportional to the score value.
Gene Ontology analysis of ncRNA interactome
One important use of interaction data is to enable producers of large-scale Omics data to perform network analysis, looking for proteins that cluster in the interactome of a particular organism, or a given cell type, and that are all up- or down-regulated. Clusters of interacting proteins are often involved in the same biological process or are present in the same macromolecular complex. Gene Ontology is commonly used to identify commonalities of function and process. This relies on the proteins in the network having first been annotated to the appropriate GO terms, an ongoing process that has been the work of many curation teams for almost 20 years (The Gene Ontology Consortium 2015). The GO was designed for the annotation of gene products, and the inclusion of RNA within the scope of the GO project was always intended and predates our current understanding of the size and complexity of such an undertaking. The annotation of ncRNAs into interaction networks, and the ability to include their functional annotations into analyses will improve the analytical power of our current algorithms (Huntley et al. 2016); and the existence of RNAcentral to provide reference molecular identifiers for GO term curation of ncRNAs now makes this possible. We have evaluated the occurrence of GO terms of both proteins and ncRNAs of the yeast RPI interactome. Unfortunately, most analytical tools do not perform yeast RNA GO terms enrichment analyses as yet, so for ncRNAs we have used GO SLIM terms associated with the molecules based on curation undertaken by SGD (Cherry et al. 2012), and have analyzed the frequencies of the most represented terms. Protein GO terms enrichment analysis was carried out using BiNGO (Maere et al. 2005). As expected, nucleic acid/RNA binding molecular function was largely overrepresented in the RPI in respect to the yeast proteome, as well as enzymatic activities involved in RNA metabolism (see Fig. 5), while RNA processing, ribosome biogenesis, and maturation of RNAs are the most common biological processes terms.
FIGURE 5.

Gene Ontology enrichment analysis of yeast RPI proteins. Statistically significant overrepresented Gene Ontology terms in the yeast ncRNA–protein interactome were calculated with the BinGO tool.
Notably, a significant enrichment of metabolic enzymes was observed in yeast mRNA binding proteins (Matia-Gonzalez et al. 2015), including glycolytic enzymes not directly involved with RNA metabolism, which may affect RNA stability. Most of the ncRNAs in RPI function as guides for methylation and pseudouridylation of rRNA during its maturation process, or have triplet codon adaptor activity, so that rRNA methylation and pseudouridylation are among the most represented biological processes terms (see Fig. 6). These terms are also the most represented in the RRI interactome (not shown), probably due to the lack in Saccharomyces of a relevant set of regulatory ncRNAs.
FIGURE 6.

Yeast RPI ncRNAs Gene Ontology terms. Slim ncRNAs Gene Ontologies were downloaded from SGD (Cherry et al. 2012) and frequencies were calculated as the occurrence of each term in RPI with respect to the number of considered ncRNAs.
RNA binding proteins analysis
RNA–protein interactions are often mediated by a small number of protein domains (RBD) that specifically recognize RNA structures and/or sequences (Lunde et al. 2007; Cook et al. 2015). We analyzed the proteins associated with ncRNAs in the yeast RPI to retrieve the occurrence of RBDs. We included in the list both “direct interactions” and “association” edges, which means that the proteins have been found in complexes with ncRNAs, but for some of them direct binding to ncRNA has not been demonstrated. We queried both Pfam and SMART databases with the list (Letunic et al. 2015; Finn et al. 2016). As shown in Figure 7, some canonical RBDs, such as RRM, DEXDC, and S1, were enriched in comparison with the yeast proteome; however, most of the proteins do not contain any of them, but contain specific regions selectively found in single proteins (such as Nop10 domain in Nop10p, UniProtKB: Q6Q547; or Utp8 domain in Utp8p, UniProtKB: P53276; or Mpp10 domain in Mpp10p, UniProtKB: P47083; and so forth), or are composed of disordered regions only (such as SF3B1, UniProtKB: P49955 or UTP9, UniProtKB: P38882, or Nsa1, UniProtKB: P53136), suggesting the evolution of multiple RNA binding strategies. It has been recently reported that yeast mRNA binding proteins often lack typical RBDs (Beckmann et al. 2015; Matia-Gonzalez et al. 2015; Brannan et al. 2016; Castello et al. 2016a). We compared the occurrence of the most common RNA binding domains in yeast ncRNA binding proteins (BP) versus a list of yeast mRNA BP (Mitchell et al. 2013), and we observed that ncRNAs are recognized by unconventional modules similarly to or even more often than mRNAs (Fig. 7C).
FIGURE 7.

Putative RNA binding domains in yeast ncRNA binding proteins (BP). Frequencies were calculated as the percentage of proteins that contain a certain domain in the yeast proteome/6000 (occurrence in the yeast proteome A,D), proteins that contain a domain in the ncRNA interactome/173 protein nodes (occurrence in yeast RPI B,D), or proteins that contain a domain/120 mRNA BP from Mitchell et al. (2013) (C). (D) Frequencies of the most represented protein domains in the yeast ncRNA binding proteins. (Note that in A, KH, DSRM, PUMILIO, C3H1, and S1 are barely visible, due to their frequencies being 0.1% or less, while in B, C3H1 and Pumilio are absent, and in C, C2H2 is absent.)
Interestingly, the two most enriched domains in RPI are SM and WD40. SM domains (http://www.ebi.ac.uk/interpro/entry/IPR010920) are known to be associated with RNA binding proteins; however, WD40 repeat domains (http://www.ebi.ac.uk/interpro/entry/IPR017986) are mainly described as protein–protein interaction modules. This domain has also been reported to be enriched in mRNA binding proteins (Castello et al. 2012), which, together with our observation and with the characteristics of the domain, suggest that it may also in some cases mediate ncRNA–protein interactions.
DISCUSSION
Expert manual curation of the literature and high-quality annotation of described interactions has become a challenge for interaction databases, made more difficult by the increase of experimental results generated by high-throughput techniques. In the last decade, standardized formats and quality controls have been developed to help data sharing in proteomics and protein networks, in order to allow the user to score the reliability of each interaction (Orchard and Hermjakob 2015). In contrast, no specific standards have yet been developed for the capture of RNA interactions. This has meant that databases have collected RNA interactions using subjective rules for reliability and scores that do not allow a direct comparison of the data, while sometimes the source of the experiment and the conditions it was carried out with are not made clear to the reader. As recalled in the Introduction, the main issue previously preventing the biocuration community from collecting ncRNA data was the problems in assigning an unambiguous identifier to the molecules, recently resolved by the creation of RNAcentral. As a result, however, RNA molecules have been extensively annotated in only a few model organisms, and in some cases it can be hard to associate the transcript referred to in the article with a specific identifier, with the result that, for example, different mRNAs transcribed from the same gene cannot always be distinguished. This is also true for some precursors of ncRNAs. Another issue has been the classification of different experimental approaches as to their suitability, or otherwise, for building physical (as opposed to predicted or genetic) networks. Members of the PSI-MI community have defined a controlled vocabulary capable of describing experimental approaches, transcript metadata, point mutations, and expression levels that are extensively used to produce PPI networks and are crucial to the assembly of consistent and reproducible models. This framework is now being extended to encompass RNA–RNA and RNA–protein interactions, allowing the curation of the yeast ncRNAs interactome according to the curation principles developed by the IMEx Consortium. The IntAct data model has proven able to hold these additional data via a web-based editing tool to which one of the authors (S.P.) was given an account and training. IntAct curators additionally provided quality control checks on the resulting annotations. The interaction data collected in the present study are publicly available and represent a bona fide gold standard that can be utilized in prediction analyses.
We have chosen Saccharomyces cerevisiae as a model system, which does not express many ncRNAs with transcription regulation functions compared to higher eukaryotes, so the ncRNA network we built consists mainly of interactions involved in the RNA maturation process or in the assembly of functional complexes such as the ribosome, spliceosome, and telomerase, etc. The RRI interactome in yeast is relatively small and most of the ncRNA interactions concern the ribosomal RNA processing. In comparison, mammalian cells express a very large number of ncRNAs that interact with both mRNAs and ncRNAs. While PPI networks have been extensively studied in the last decade, little is known about RPI and RRI network characteristics, and this pilot study is one of the first to allow for a direct comparison of the three networks. Interestingly, they show similar parameters, with a few hubs dominating the networks (Table 1). We also analyzed ontology frequencies for ncRNA involved in RRI and RPI (see Fig. 6). The ontological representation of networks often facilitates functional analyses; however, comprehensive annotation of Gene Ontology terms for ncRNAs is still largely incomplete, and common tools to determine statistically overrepresented GO categories have not yet been implemented for all ncRNAs (Huntley et al. 2016).
The analysis of ncRNA–protein interactions in yeast reveals that the limited number of canonical RNA binding domains classified in 2007 (Lunde et al. 2007) mediates only a small fraction of them, while in some processes the protein–ncRNA-sequence recognition is helped by the binding of a second RNA carrying a complementary region. For example, some snoRNAs help Cbf5 in recognizing specific sites of the ribosomal RNA and modifying them (Reichow et al. 2007). Notably, the occurrence of the WD40 binding domain in yeast ncRNA binding proteins is clearly overrepresented in respect to the yeast proteome (see Fig. 7), while no other protein-binding domain is. The WD40 repeats usually act as a scaffold for assemblies of complexes, which can explain their presence in some of the associations. However, the strong enrichment of WD40 domains suggests that they may also have a more direct role in ncRNA binding, which is also in agreement with its versatility in binding very different classes of ligands. Recently, the crystal structure of gemin5 WD40 domain binding an RNA oligomer was reported (Xu et al. 2016).
Macromolecular structures provide a growing source of ncRNA interaction data. To date, the PDB contains 1808 entries that represent complexes between an RNA and a protein. Of those, 137 have S. cerevisiae listed as one of its source organisms. Most of these structures are large macromolecular complexes: More than 40 structures are variants of the 80S and 60S ribosome, 10 structures concern the spliceosome complex, five the preinitiation 40S and 48S complexes, and a few others the exosome complex, while only a small number involve tRNA–protein interactions. With the advances in cryoEM techniques, we hope that an increasing number of RNA–protein interactions will be defined at atomic resolution.
This study presents the first attempt at creating a noncoding RNA network for yeast in IntAct. Other resources have started to collect RNA interactions (Li et al. 2014; Hao et al. 2016; Junge et al. 2017; Yi et al. 2017). However, new interactions are continuously published for yeast and mammalian mRNAs (Baltz et al. 2012; Baejen et al. 2014; Farazi et al. 2014; Gerstberger et al. 2014; Castello et al. 2016b) and for less characterized classes of ncRNAs such as SUT, CUT, and XUT (Tuck and Tollervey 2013). We believe that it is extremely timely to begin to collate RNA interaction data from the literature and high-throughput experimental screens. This will be greatly aided through collaboration between the world's noncoding RNA interaction databases and the adoption of standards by them. The widespread availability of protein–protein interaction data is critical for scientific research and the same must happen for RNA interaction data to fill the gap in scientific data provision.
MATERIALS AND METHODS
Data collection and curation: yeast noncoding RNAs
The yeast Saccharomyces cerevisiae cell represents an excellent model system to perform analyses on ncRNA interaction networks, since it contains a limited number of ncRNAs, most of which are well-characterized in literature and annotated in databases. As reported in Table 2, the majority of the yeast ncRNAs belong to the small nucleolar/nuclear classes of RNAs, which regulate ribosomal and messenger RNA metabolism. Other well-known classes include the ribosomal and transfer RNAs, the telomerase RNA component, the SRP complex RNA 7S, and the catalytic introns of groups 1 and 2. No microRNAs have been reported in S. cerevisiae to date, and few lncRNA have been described (Martens et al. 2004; Bumgarner et al. 2009; van Werven et al. 2012). Numerous cryptic unstable transcripts (CUT), Xrn1-sensitive unstable transcripts (XUT), and other noncoding transcripts are synthesized in yeast and immediately targeted for degradation (Neil et al. 2009; van Dijk et al. 2011). They are still poorly characterized and we did not consider them in our study (Tisseur et al. 2011). To select the RNAcentral identifier for each ncRNA, the database was searched with the SGD identifier or, for tRNAs, with the GTRNAdb identifier. For a few tRNA precursors, the RNA sequence search was used.
TABLE 2.
Summary of yeast noncoding RNAs and their interactions
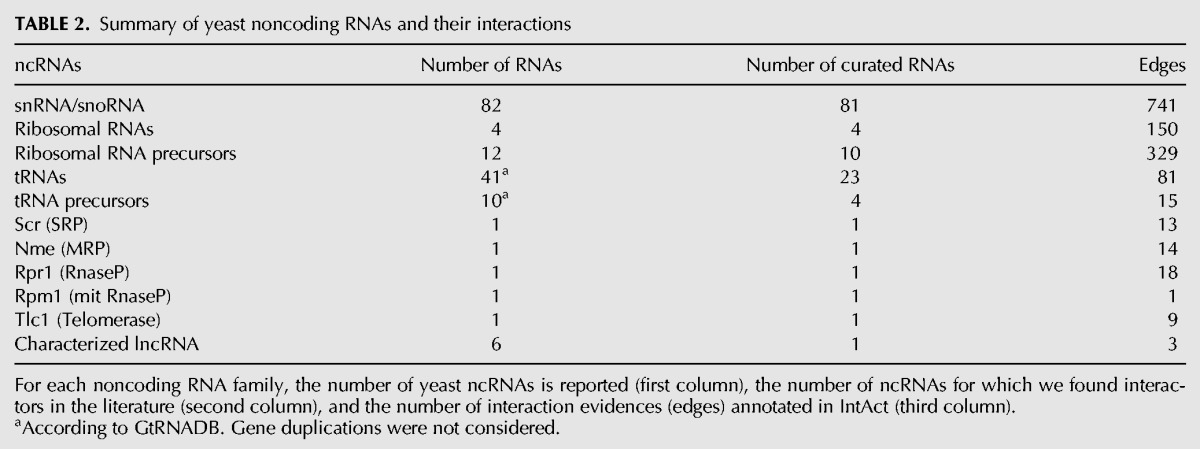
Both transfer and ribosomal RNAs are transcribed as precursor molecules that undergo a series of post-transcriptional events required for the biogenesis of the functional molecules, and these precursors are sometimes not annotated as independent entries in RNA databases. During the curation process, we considered precursor and mature RNAs as different interactors whenever it was clearly specified which molecular form was involved in an interaction (Table 2).
Noncoding RNA interactions were manually curated from the literature, following the curation standards established by the IMEx Consortium. First we consulted the Rfam, SGD, the LncRNAdb, and the yeast snoRNA databases (Piekna-Przybylska et al. 2007; Cherry et al. 2012; Nawrocki et al. 2015; Quek et al. 2015) in addition to the published literature, to draw up a complete list of S. cerevisiae ncRNAs. We then retrieved relevant articles by searching PubMed for abstracts containing at least one ncRNA name and “yeast” or “S. cerevisiae” terms. We also searched PubMed with other keywords, such as “CLIP,” “CLIP-seq,” “CLASH,” “rna rna interaction” “rna protein interaction,” etc. We collected several hundreds of papers that were manually filtered to a final total of approximately 120 actually containing interactions that were manually curated. We annotated interactions for each type of yeast ncRNAs, although there were a few exceptions (such as regulatory RNAs Pwr1, Icr1, and Irt 1) for which we have not found any physical interactions to date. RNA–RNA and RNA–protein interactions were manually curated using the IntAct editor, according to the IMEx standard. All data are available for download from the IntAct home page in both PSI-MI XML and MITAB tabular format, including all relevant information about the constructs used, the detection methods, and the host organism.
To download the RNA interactomes, the IntAct database can be queried with (ptypeA:RNA AND ptypeB:RNA) or [(ptypeA:RNA AND ptypeB:protein) or (ptypeA:protein AND ptypeB:RNA)] (to limit the search to yeast, add Saccharomyces cerevisiae in the advanced search), or it can be queried with the list of ncRNAs (Supplemental Table S1), using EBI identifiers or URS identifiers followed by _559292.
Network analysis
Protein–protein, ncRNA–protein, and noncoding RNA–RNA interaction data for Saccharomyces cerevisiae (taxonomy ID: 559292) were downloaded from IntAct database (November 2016). Only a single edge was kept for examples of duplicate edges and self-interaction (loops) edges were removed before computing network properties. The complete yeast proteome PPI network was inferred from 77,620 unique interactions among 6091 proteins, the RPI network was inferred from 596 interactions between 105 ncRNAs and 153 proteins, and the RRI network from 195 interactions among 102 ncRNAs (tRNA–rRNA interactions were added to the RNA–RNA interactome). To compare network properties between the RRI network and the PPI network, the PPI network was downsampled such that the downsampled network comprised the same number of edges as the RRI network, and mean values from 100 downsampled networks were considered. To compare network properties of biological networks with a random network, a random network was generated using the Erdös–Renyi model with the same number of nodes and edges as in the PPI network (nodes: 6091, edges: 77620). Network analyses were carried out using the igraph package (Csardi and Nepusz 2006) in R.
Degree distribution
The degree of a node n is the number of edges linked to it. Number of nodes ordered by their increasing degree gives the degree distribution of a network.
Clustering coefficient
The clustering coefficient of a node is n defined as C(n) = 2e/[k(k−1)], where e is the number of edges between neighbors of node n, and k is the number of neighbors of n. The value of the clustering coefficient lies between values 0 and 1 and is highest if all the neighbors of the node directly interact with each other (i.e., edges form triangles) and 0 when none of the neighbors are connected with each other.
Betweenness centrality
Betweenness centrality of a node n is defined as B(n) = Σa≠n≠b [σab(n)/σab], where σab is the shortest number of paths between nodes a, b; and σab(n) is the shortest number of paths between nodes a, b through node n. Betweenness centrality is divided by normalizing factor (N−1)(N−2)/2, where N is the total number of nodes in the connected network.
Closeness centrality
Closeness centrality of a node n is defined as the reciprocal of the average shortest path length, K(n) = 1/average [L(n,a)], where L(n,a) is the shortest path between two nodes n and a. The closeness centrality measure was computed on subgraphs with the highest number of interconnected nodes.
Neighborhood connectivity
Neighborhood connectivity of a node n is defined as the average connectivity (degree) of all its neighbor nodes.
Gene ontology enrichment analysis
The RPI network was downloaded from IntAct database (November 2016) and the BiNGO tool was used to search ontology files. For proteins, the hypergeometric test with 0.01 significance level was used to evaluate the statistical significance of enrichment of each term in comparison to the yeast proteome. No data are available for the whole-yeast transcriptome to date, so that for ncRNAs we calculated the occurrence of GO terms in the RPI network with respect to the set of yeast ncRNAs.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Anton Petrov and Benedikt Beckmann for critical reading of the manuscript. This work was funded by IntAct–BBSRC MIDAS grant BB/L024179/1. A.P. is funded by EMBL International PhD Programme. S.P. was partially funded by Unical grant DR1568. S.P. would like to thank Margaret Duesbury, Pablo Porras, and all the IntAct team for invaluable help during the curation process.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.060996.117.
Freely available online through the RNA Open Access option.
REFERENCES
- Amaral PP, Clark MB, Gascoigne DK, Dinger ME, Mattick JS. 2011. lncRNAdb: a reference database for long noncoding RNAs. Nucleic Acids Res 39: D146–D151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baejen C, Torkler P, Gressel S, Essig K, Söding J, Cramer P. 2014. Transcriptome maps of mRNP biogenesis factors define pre-mRNA recognition. Mol Cell 55: 745–757. [DOI] [PubMed] [Google Scholar]
- Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al. 2012. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 46: 674–690. [DOI] [PubMed] [Google Scholar]
- Beckmann BM, Horos R, Fischer B, Castello A, Eichelbaum K, Alleaume AM, Schwarzl T, Curk T, Foehr S, Huber W, et al. 2015. The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat Commun 6: 10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannan KW, Jin W, Huelga SC, Banks CA, Gilmore JM, Florens L, Washburn MP, Van Nostrand EL, Pratt GA, Schwinn MK, et al. 2016. SONAR discovers RNA-binding proteins from analysis of large-scale protein-protein interactomes. Mol Cell 64: 282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumgarner SL, Dowell RD, Grisafi P, Gifford DK, Fink GR. 2009. Toggle involving cis-interfering noncoding RNAs controls variegated gene expression in yeast. Proc Natl Acad Sci 106: 18321–18326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al. 2012. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149: 1393–1406. [DOI] [PubMed] [Google Scholar]
- Castello A, Fischer B, Frese CK, Horos R, Alleaume AM, Foehr S, Curk T, Krijgsveld J, Hentze MW. 2016a. Comprehensive identification of RNA-binding domains in human cells. Mol Cell 63: 696–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A, Horos R, Strein C, Fischer B, Eichelbaum K, Steinmetz LM, Krijgsveld J, Hentze MW. 2016b. Comprehensive identification of RNA-binding proteins by RNA interactome capture. Methods Mol Biol 1358: 131–139. [DOI] [PubMed] [Google Scholar]
- Chan PP, Lowe TM. 2009. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res 37: D93–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, Christie KR, Costanzo MC, Dwight SS, Engel SR, et al. 2012. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res 40: D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MB, Amaral PP, Schlesinger FJ, Dinger ME, Taft RJ, Rinn JL, Ponting CP, Stadler PF, Morris KV, Morillon A, et al. 2011. The reality of pervasive transcription. PLoS Biol 9: e1000625; discussion e1001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook KB, Hughes TR, Morris QD. 2015. High-throughput characterization of protein-RNA interactions. Brief Funct Genomics 14: 74–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csardi G, Nepusz T. 2006. The igraph software package for complex network research. InterJournal, Complex Systems 1695 http://igraph.org.
- Dujon B. 2010. Yeast evolutionary genomics. Nat Rev Genet 11: 512–524. [DOI] [PubMed] [Google Scholar]
- Farazi TA, Leonhardt CS, Mukherjee N, Mihailovic A, Li S, Max KE, Meyer C, Yamaji M, Cekan P, Jacobs NC, et al. 2014. Identification of the RNA recognition element of the RBPMS family of RNA-binding proteins and their transcriptome-wide mRNA targets. RNA 20: 1090–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, et al. 2016. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44: D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium. 2015. Gene Ontology Consortium: going forward. Nucleic Acids Res 43: 1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstberger S, Hafner M, Tuschl T. 2014. A census of human RNA-binding proteins. Nat Rev Genet 15: 829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granneman S, Kudla G, Petfalski E, Tollervey D. 2009. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proc Natl Acad Sci 106: 9613–9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Dunham MJ, Troyanskaya OG. 2007. Functional analysis of gene duplications in Saccharomyces cerevisiae. Genetics 175: 933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Wu W, Li H, Yuan J, Luo J, Zhao Y, Chen R. 2016. NPInter v3.0: an upgraded database of noncoding RNA-associated interactions. Database 2016: baw057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings J, Owen G, Dekker A, Ennis M, Kale N, Muthukrishnan V, Turner S, Swainston N, Mendes P, Steinbeck C. 2016. ChEBI in 2016: improved services and an expanding collection of metabolites. Nucleic Acids Res 44: D1214–D1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermjakob H, Montecchi-Palazzi L, Bader G, Wojcik J, Salwinski L, Ceol A, Moore S, Orchard S, Sarkans U, von Mering C, et al. 2004. The HUPO PSI's molecular interaction format—a community standard for the representation of protein interaction data. Nat Biotechnol 22: 177–183. [DOI] [PubMed] [Google Scholar]
- Huntley RP, Sitnikov D, Orlic-Milacic M, Balakrishnan R, D'Eustachio P, Gillespie ME, Howe D, Kalea AZ, Maegdefessel L, Osumi-Sutherland D, et al. 2016. Guidelines for the functional annotation of microRNAs using the Gene Ontology. RNA 22: 667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Mason SP, Barabási AL, Oltvai ZN. 2001. Lethality and centrality in protein networks. Nature 411: 41–42. [DOI] [PubMed] [Google Scholar]
- Joy MP, Brock A, Ingber DE, Huang S. 2005. High-betweenness proteins in the yeast protein interaction network. J Biomed Biotechnol 2005: 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jühling F, Mörl M, Hartmann RK, Sprinzl M, Stadler PF, Pütz J. 2009. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res 37: D 159–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge A, Refsgaard JC, Garde C, Pan X, Santos A, Alkan F, Anthon C, von Mering C, Workman CT, Jensen LJ, et al. 2017. RAIN: RNA–protein Association and Interaction Networks. Database (Oxford) 2017: baw167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. 2014. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42: D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla G, Granneman S, Hahn D, Beggs JD, Tollervey D. 2011. Cross-linking, ligation, and sequencing of hybrids reveals RNA–RNA interactions in yeast. Proc Natl Acad Sci 108: 10010–10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Doerks T, Bork P. 2015. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res 43: D257–D260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JH, Liu S, Zhou H, Qu LH, Yang JH. 2014. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-seq data. Nucleic Acids Res 42: D92–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde BM, Moore C, Varani G. 2007. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol 8: 479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Yang C, Alexandrov A, Grayhack EJ, Behm-Ansmant I, Yu YT. 2005. Pseudouridylation of yeast U2 snRNA is catalyzed by either an RNA-guided or RNA-independent mechanism. EMBO J 24: 2403–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, Heymans K, Kuiper M. 2005. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21: 3448–3449. [DOI] [PubMed] [Google Scholar]
- Martens JA, Laprade L, Winston F. 2004. Intergenic transcription is required to repress the Saccharomyces cerevisiae SER3 gene. Nature 429: 571–574. [DOI] [PubMed] [Google Scholar]
- Matia-Gonzalez AM, Laing EE, Gerber AP. 2015. Conserved mRNA-binding proteomes in eukaryotic organisms. Nat Struct Mol Biol 22: 1027–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J, Lemoine F, Soumillon M, Liechti A, Weier M, Guschanski K, Hu H, Khaitovich P, Kaessmann H. 2013. Birth and expression evolution of mammalian microRNA genes. Genome Res 23: 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SF, Jain S, She M, Parker R. 2013. Global analysis of yeast mRNPs. Nat Struct Mol Biology 20: 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modic M, Ule J, Sibley CR. 2013. CLIPing the brain: studies of protein-RNA interactions important for neurodegenerative disorders. Mol Cell Neurosci 56: 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki EP, Burge SW, Bateman A, Daub J, Eberhardt RY, Eddy SR, Floden EW, Gardner PP, Jones TA, Tate J, et al. 2015. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res 43: D130–D137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil H, Malabat C, d'Aubenton-Carafa Y, Xu Z, Steinmetz LM, Jacquier A. 2009. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature 457: 1038–1042. [DOI] [PubMed] [Google Scholar]
- Nishtala S, Neelamraju Y, Janga SC. 2016. Dissecting the expression relationships between RNA-binding proteins and their cognate targets in eukaryotic post-transcriptional regulatory networks. Sci Rep 6: 25711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S, Hermjakob H. 2015. Shared resources, shared costs—leveraging biocuration resources. Database (Oxford) 2014: bav009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S, Kerrien S, Abbani S, Aranda B, Bhate J, Bidwell S, Bridge A, Briganti L, Brinkman FS, Cesareni G, et al. 2012. Protein interaction data curation: the International Molecular Exchange (IMEx) consortium. Nat Methods 9: 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, Campbell NH, Chavali G, Chen C, del-Toro N, et al. 2014. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res 42: D358–D363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piekna-Przybylska D, Decatur WA, Fournier MJ. 2007. New bioinformatic tools for analysis of nucleotide modifications in eukaryotic rRNA. RNA 13: 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porras P, Duesbury M, Fabregat A, Ueffing M, Orchard S, Gloeckner CJ, Hermjakob H. 2015. A visual review of the interactome of LRRK2: using deep-curated molecular interaction data to represent biology. Proteomics 15: 1390–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quek XC, Thomson DW, Maag JL, Bartonicek N, Signal B, Clark MB, Gloss BS, Dinger ME. 2015. lncRNAdb v2.0: expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res 43: D168–D173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichow SL, Hamma T, Ferré-D'Amaré AR, Varani G. 2007. The structure and function of small nucleolar ribonucleoproteins. Nucleic Acids Res 35: 1452–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The RNAcentral Consortium. 2015. RNAcentral: an international database of ncRNA sequences. Nucleic Acids Res 43: D123–D129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The RNAcentral Consortium. 2017. RNAcentral: a comprehensive database of non-coding RNA sequences. Nucleic Acids Res 4: D128–D134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattner P, Decatur WA, Davis CA, Ares M Jr, Fournier MJ, Lowe TM. 2004. Genome-wide searching for pseudouridylation guide snoRNAs: analysis of the Saccharomyces cerevisiae genome. Nucleic Acids Res 32: 4281–4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz J, Zemann A, Churakov G, Kuhl H, Grutzner F, Reinhardt R, Brosius J. 2008. Retroposed SNOfall—a mammalian-wide comparison of platypus snoRNAs. Genome Res 18: 1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao P, Yang JH, Zhou H, Guan DG, Qu LH. 2009. Genome-wide analysis of chicken snoRNAs provides unique implications for the evolution of vertebrate snoRNAs. BMC Genomics 10: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G. 2002. An expanding universe of noncoding RNAs. Science 296: 1260–1263. [DOI] [PubMed] [Google Scholar]
- Tisseur M, Kwapisz M, Morillon A. 2011. Pervasive transcription—lessons from yeast. Biochimie 93: 1889–1896. [DOI] [PubMed] [Google Scholar]
- Tuck AC, Tollervey D. 2013. A transcriptome-wide atlas of RNP composition reveals diverse classes of mRNAs and lncRNAs. Cell 154: 996–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium. 2015. UniProt: a hub for protein information. Nucleic Acids Res 43: D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk EL, Chen CL, d'Aubenton-Carafa Y, Gourvennec S, Kwapisz M, Roche V, Bertrand C, Silvain M, Legoix-Ne P, Loeillet S, et al. 2011. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature 475: 114–117. [DOI] [PubMed] [Google Scholar]
- van Werven FJ, Neuert G, Hendrick N, Lardenois A, Buratowski S, van Oudenaarden A, Primig M, Amon A. 2012. Transcription of two long noncoding RNAs mediates mating-type control of gametogenesis in budding yeast. Cell 150: 1170–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villaveces JM, Jimenez RC, Porras P, Del-Toro N, Duesbury M, Dumousseau M, Orchard S, Choi H, Ping P, Zong NC, et al. 2015. Merging and scoring molecular interactions utilising existing community standards: tools, use-cases and a case study. Database (Oxford) 2015: bau131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MJ. 2006. Mammalian small nucleolar RNAs are mobile genetic elements. PLoS Genet 2: e205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolford JL Jr, Baserga SJ. 2013. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 195: 643–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Xiao M, Yang C, Yu YT. 2011. U2 snRNA is inducibly pseudouridylated at novel sites by Pus7p and snR81 RNP. EMBO J 30: 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Ishikawa H, Izumikawa K, Li L, He H, Nobe Y, Yamauchi Y, Shahjee HM, Wu XH, Yu YT, et al. 2016. Structural insights into Gemin5-guided selection of pre-snRNAs for snRNP assembly. Genes Dev 30: 2376–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Shichino Y, Yamamoto M. 2016. The long non-coding RNA world in yeasts. Biochim Biophys Acta 1859: 147–154. [DOI] [PubMed] [Google Scholar]
- Yang YC, Di C, Hu B, Zhou M, Liu Y, Song N, Li Y, Umetsu J, Lu ZJ. 2015. CLIPdb: a CLIP-seq database for protein-RNA interactions. BMC Genomics 16: 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Y, Zhao Y, Li C, Zhang L, Huang H, Li Y, Liu L, Hou P, Cui T, Tan P, et al. 2017. RAID v2.0: an updated resource of RNA-associated interactions across organisms. Nucleic Acids Res 4: 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihama M, Nakao A, Kenmochi N. 2013. snOPY: a small nucleolar RNA orthological gene database. BMC Res Notes 6: 426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.