

Abstract
The ATP binding cassette (ABC) transporters Pdr11p and its paralog Aus1p are expressed under anaerobic growth conditions at the plasma membrane of the yeast Saccharomyces cerevisiae and are required for sterol uptake. However, the precise mechanism by which these ABC transporters facilitate sterol movement is unknown. In this study, an overexpression and purification procedure was developed with the aim to characterise the Pdr11p transporter. Engineering of Pdr11p variants fused at the C terminus with green fluorescent protein (Pdr11p-GFP) and containing a FLAG tag at the N terminus facilitated expression analysis and one-step purification, respectively. The detergent-solubilised and purified protein displayed a stable ATPase activity with a broad pH optimum near 7.4. Mutagenesis of the conserved lysine to methionine (K788M) in the Walker A motif abolished ATP hydrolysis. Remarkably, and in contrast to Aus1p, ATPase activity of Pdr11p was insensitive to orthovanadate and not specifically stimulated by phosphatidylserine upon reconstitution into liposomes. Our results highlight distinct differences between Pdr11p and Aus1p and create an experimental basis for further biochemical studies of both ABC transporters to elucidate their function.
Introduction
ATP binding cassette (ABC) transporters are members of a superfamily of proteins that mediate ATP-driven unidirectional transport of a variety of molecules across biological membranes. Proteins in this superfamily share a similar domain organization, with two membrane-embedded transmembrane domains (TMDs) and two cytoplasmic nucleotide binding domains (NBDs) with highly conserved amino acid motifs including the Walker A, the Walker B, and the signature C motifs. The four domains are often expressed as separate subunits in prokaryotic ABC proteins. In eukaryotes ABC proteins are organized either as full transporters comprising all four domains or as “half-transporters” with a single TMD and NBD that operate as homo- or heterodimers. Several ABC transporters utilise phospholipids among their substrates (reviewed in [1, 2]). Furthermore, some ABC transporters facilitate transport of sterols [1, 3]. Among these, there are two Saccharomyces cerevisiae ABC transporters, Aus1p and Pdr11p, which belong to the ABCG subfamily and are plasma-membrane-resident full-size transporters. Deletion of both AUS1 and PDR11 genes completely blocks sterol uptake under anaerobic conditions [4, 5] and in yeast mutants deficient in heme biosynthesis [6]. Under these conditions, S. cerevisiae is strictly dependent on the uptake of exogenously supplied sterols to proliferate as these lipids are essential for the cell, and their synthesis requires oxygen. The exact mechanism by which these ABC transporters facilitate sterol movement is unknown. Clarification of this issue is important as previous studies identified an orthologue of Aus1p in the pathogenic fungi Candida glabrata with an essential role in cholesterol uptake [7, 8]. C. glabrata can utilise exogenous cholesterol as a structural analogue and surrogate for ergosterol, thereby diminishing the effect of the ergosterol-specific antifungals.
At least three models, not mutually exclusive, have been proposed to explain how both proteins facilitate sterol uptake. One model is that Aus1p and Pdr11p control the initial insertion of sterol into the plasma membrane upon their passage across the cell wall [4, 6, 9]. In support of this, sterol uptake increases by coexpression of Aus1p and the cell wall protein Dan1p, which (like Aus1p and Pdr11p) is upregulated under anaerobic conditions [10]. Likewise, the presence of bovine serum albumin (BSA) promotes sterol uptake implying that BSA could act as a sterol donor [11]. A second model proposes that both proteins facilitate the removal of sterol from the plasma membrane to a cytosolic acceptor, such as soluble sterol-binding proteins, or closely apposed membranes of the endoplasmic reticulum [5]. Thirdly, Pdr11p and Aus1p have been proposed to flip sterols from the outer to the inner leaflet of the plasma membrane [5]), as spontaneous movement of sterols across the yeast plasma membrane is seemingly too slow for physiological processes to occur [12].
To investigate the function of these transporters, we previously established a purification procedure for Aus1p and reported its activity to be specifically stimulated by phosphatidylserine (PS) and blocked by the classical inhibitors of ABC proteins [13]. Here, we establish a procedure to purify Pdr11p and analysed its ATPase activity in terms of effects of inhibitors and requirements for lipids and sterols. Purified detergent-solubilised and reconstituted Pdr11p showed robust ATPase activity that was remarkably insensitive to orthovanadate and independent of lipid composition. This suggests that despite being part of the same physiological function, these two transporters may contain distinct and hitherto overlooked differences in their functionality.
Materials and methods
Materials
Synthetic dropout medium was from either Sigma-Aldrich (Taufkirchen, Germany) or Bio-Rad (Hercules, California). [γ-32P] ATP (3000 Ci/mmol) was from PerkinElmer (Waltham, Massachusetts). N-dodecyl-β-maltoside (DDM) was from GLYCON Biochemicals (Luckenwalde, Germany). Maltose neopentyl glycol-3 was a gift from Dr. Claus Løland, University of Copenhagen. All other detergents were obtained from Affymetrix (Maumee, Ohio, USA). PageRulerTM prestained or unstained protein ladder from Thermo Scientific (Waltham, Massachusetts) were used for size determination in SDS-PAGE gels. Lipids were purchased from Avanti Polar Lipids (Alabaster, AL). All other chemicals were from Sigma-Aldrich (Taufkirchen, Germany) unless otherwise indicated.
Plasmid construction
The recombinant FLAG-tagged PDR11 expression plasmid was generated by amplification of the PDR11 ORF from genomic DNA by PCR using the primers 5’-GAGAGTCGACATGGATTACAAGGATGACGACGATAAAATCTCTCTTTCCAAATATTTTAATC-3’ and 5’-GAGAGCTAGCTTATACGCTTTGTTCGTTTGG-3’, and subsequent cloning of the amplified product into the Nhe1-Sal1 restriction enzyme sites of the pESC-URA expression plasmid (Agilent Technologies). The resulting expression vector was used as a template for mutagenesis of PDR11 by the QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) employing primers 5’-ATGGGTGAATCTGGTGCTGGTATGACAACTTTGTTGAATGTCTTG and 5’-CAAGACATTCAACAAAGTTGTCATACCAGCACCAGATTCACCCAT according to the manufacturer’s protocol to generate PDR11K788M. For making the FLAG-tagged PDR11-GFP expression plasmid, a PCR fragment containing the GFP tag was amplified from plasmid pYM27 (Euroscarf) using primers that contained homologous sequences for recombination with the Nhe1-digested vector pESC-URA-PDR11; in vivo recombination in yeast yielded plasmid pESC-URA-PDR11-GFP. All PCRs were carried out using Phusion High-Fidelity DNA Polymerase (New England Biolabs) according to the manufacturer’s instructions. All plasmid inserts were fully sequenced, including their junctions, to confirm the expected DNA sequence. The expression plasmid encoding FLAG-tagged Aus1p has been described previously [13].
Yeast strains and growth conditions
Expression and functional complementation were carried out employing the sterol-uptake deficient S. cerevisiae mutant strain W303 hem1Δ::LEU2 pdr11Δ::loxP aus1Δ::loxP-HIS5Sp-loxP, with strain W303 hem1Δ::LEU2 as wild-type [13]. Both strains were derivatives of W303-1α (MATα ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100). In addition, the protease-deficient S. cerevisiae strain BJ1991 (Matα leu2 trp1 ura3-52 pep4-3 prb1-1122 gal2) lacking Pep4p and Prb1p (yeast proteinases A and B) was used in expression studies. Yeast cells were transformed by the lithium acetate method [14] and grown at 30°C in selective synthetic dextrose (SD) or galactose (SG) medium lacking uracil. For the hem1Δaus1Δpdr11Δ strain SD medium was supplemented with 0.3% (w/v) adenine and 20 μg/mL δ-aminolevulinic acid (ALA) while SG media was supplemented with 0.3% (w/v) adenine, 20 μg/mL cholesterol and 0.05% (v/v) Tween 80. For solid media 2% (v/v) agar was added. For complementation tests, cells grown overnight in liquid SD medium were washed twice in sterile water and diluted to 0.2 OD600. Drops (3 μL) of consecutive fivefold dilutions were spotted onto selective SG plates with ALA or cholesterol. The plates were incubated at 30°C for 4 days.
Fluorescence microscopy and flow cytometry
Cells were examined under a Leica DMI4000 B inverted fluorescence microscope using a 63x water objective. GFP was visualised with an excitation filter BP 470/40, beam splitter 500, and BP 525/50 barrier filter. Flow cytometry of cells expressing GFP-tagged Pdr11p was performed as described previously [13].
Membrane preparation and solubilisation
Yeast transformants from a liquid preculture were inoculated in 2 L of SD medium and grown at 30°C with shaking at 140 rev/min until an OD600 of 0.9-1.5 unless otherwise indicated. Cells were harvested, washed (1,000 g, 8 min, room temperature), and induced in 2 L SG medium for 6 h (unless otherwise stated). All subsequent steps were performed at 4°C. Cells were collected (1,000 g, 8 min) and lysed by vortexing 6 x 1 min with acid-washed glass beads (0.5 mm, BioSpec, Bartlesville, OK; 10 g beads per 5 g yeast) in lysis buffer (100 mM NaCl, 20 mM HEPES-NaOH, pH 7.4, 10 ml lysis buffer per 5 g yeast) containing 1 mM dithiothreitol (DTT), protease inhibitors (1 μg/mL aprotinin, 1 μg/mL leupeptin, 1 μg/mL pepstatin, 5 μg/mL antipain and 157 μg/mL benzamidine, 0.25 mM phenylmethylsulfonyl fluoride) and PhosSTOP (Roche Applied Science, Penzberg, Germany). The lysate was cleared by centrifugation (1,000 g, 8 min) and the supernatant centrifuged again at 10,000 or 60,000 g (45 min) for collection of plasma membrane enriched fractions or total membranes in the pellet, respectively. The membrane pellets were homogenised with a douncer in 5 ml glycerol buffer (100 mM NaCl, 20 mM HEPES-NaOH, 20% (w/v) glycerol, pH 7.4) supplemented with protease inhibitors and adjusted to a protein concentration of 1 mg/mL (as measured by Bradford assay). Membranes were solubilised by detergent in the indicated concentrations for 1 h on an end-over-end rotator. Insoluble material was removed by centrifugation (88,000 g, 45 min) and the supernatant used for protein analysis.
Protein purification and analysis
Plasma membrane fractions were isolated and solubilised in 0.6% (w/v) DDM (see above) and incubated with 14 μl anti-FLAG (M2) affinity gel (prewashed twice in glycerol buffer) per 1 ml solubilised membrane followed by end-over-end rotation overnight at 16°C. The gel was washed three times with 25 ml glycerol buffer containing 0.05% (w/v) DDM. Finally the proteins were eluted by use of 400 μg/mL FLAG peptide in glycerol buffer supplemented with 0.05% (w/v) DDM and protease inhibitors without phenylmethylsulfonyl fluoride, frozen in liquid nitrogen, and stored at -80°C. Aliquots of the eluate and cell lysates were analysed by Coomassie blue stained SDS-PAGE and Western blot using mouse monoclonal anti-FLAG M2 and anti-GFP (Roche) antibodies at 1:2000 dilution. Proteins were visualised with alkaline phosphatase-coupled secondary antibody using BCIP/NBT colour development substrate (Promega, Madison) according to manufacturer’s instructions. Concentration of the purified protein was determined by Coomassie Blue staining using bovine serum albumin as a standard via densitometry analysis performed on a Typhoon TRIO (GE Healthcare) equipped with a 633 nm laser (no emission filter) and analysed with ImageQuant TL (GE Healthcare).
Mass spectrometric analysis
The protein preparation containing the recombinant Pdr11p was separated on an 8% SDS gel and stained with Coomassie Blue. Stained gel bands were cut into small pieces (about 1 x 1 mm) and converted to peptides as described by Shevchenko et al. [15]. Peptides were purified using C18 stage-tips according to Rappsilber et al. [16] and measured on a Q-Exactive HF mass spectrometer (Thermo Scientific) coupled to an Ultimate 3000 RSLCnano system (Thermo Scientific) in data-dependent acquisition mode, selecting the top 12 peaks for Higher Energy Collisional Dissociation (HCD) fragmentation. A 66 min gradient (solvent A: 0.1% formic acid; solvent B: HPLC grade acetonitrile in 0.1% formic acid) was applied for the samples using an Acclaim PepMap trap column (2 cm x 75 μm i.d., C18, 3 μm, 100 Å, Thermo Scientific) and an Acclaim PepMap RSLC analytical column (15 cm x 75 μm i.d., C18, 2 μm, 100 Å, Thermo Scientific). A volume of 3 μL sample was injected and the peptides eluted with 66 min gradients of 2 to 35% solvent B at flowrates of 0.3 μL/min. MS1 data acquisition was performed at a resolution of 60,000, using an injection time of 45 ms in the scan range from 375 to 1500 m/z and MS2 at a resolution of 15000 with an injection time of 45 ms. The normalized collision energy was set to 28 eV. The mass window for precursor ion selection was set to 1.2 m/z. The recorded spectra were analysed using the MaxQuant software package (Version 1.5.8.3) [17] by matching the data to the Uniprot yeast database (downloaded on 06.05.2012) with a false discovery rate of 1%.
Protein reconstitution and vesicle analysis
Reconstitution in preformed liposomes was done as described in [18] using 52 mM octyl glycoside and a protein:lipid molar ratio of 1:50 or 1:100. Preformed liposomes were prepared from synthetic 1-palmitoyl-2-oleoyl (PO) phosphatidylcholine (PC), phosphatidylserine (PS), or phosphatidylglycerol (PG) and contained 0.1 mol% of the fluorescent marker lipid N-rhodamine-dioleoylphosphatidylethanolamine (Rho-PE). For floatation assays, 75 μL proteoliposomes and 75 μL 60% sucrose in lysis buffer were mixed. The sample was overlaid with 150 μL lysis buffer containing 25% sucrose, followed by 150 μL lysis buffer. Sucrose gradients were centrifuged (186,000 g, 1.5 h). Subsequently, three 150-μL fractions were collected (top, 0%; middle, 25%; bottom, 30%), and the pellet was resuspended in 150 μL of lysis buffer. Fluorescence of Rho-PE was detected on SDS/PAGE gels (Typhoon reader, excitation 532 nm, emission at 580 ± 30 nm), and proteins were visualised via silver staining. Lipid composition of the vesicles was confirmed by lipid extraction followed by thin-layer chromatography and lipid-phosphorus determination [19].
ATPase assay
ATPase activity of Pdr11p was measured at 30°C for 30 min as described previously using 1 mM ATP, 5 mM MgCl2, 2 mM DTT, and 2 μCi of [γ-32P] ATP [13]. Release of inorganic phosphate was determined by indirect β-counting (1450 MicroBeta Trilux, Wallac). For determining the pH dependence of ATP hydrolysis, a range of different pH buffers were prepared: 50 mM MES for pH 5.3–6.6, 20 mM HEPES for pH 7–7.4, and 50 mM TRIS-HCl for pH 8–8.8. All were supplemented with 100 mM NaCl. When indicated MgCl2 was replaced by equal concentrations of MnCl2, CoCl2, or NiCl2. Cholesterol and ergosterol were added from an ethanolic stock to a final concentration of 6 μg/mL in presence or absence of 30 μg/mL bovine serum albumin.
Data analysis
Background radiation was subtracted from the data. The ATPase activity versus orthovanadate data (including the 0 mM reference) have been fitted to the function A = min + (max − min)/(1+([V]/IC50)−H) where A is the ATPase activity, [V] is the orthovanadate concentration, IC50 is the half maximal inhibitory concentration, and H is the Hill coefficient. IC50, [V], max, and min values are all approximated with the open source program Gnuplot 4.6. Error bars show the standard deviations (S.D.). All relevant ATPase data are within the paper and its Supporting Information files (S1–S6 Tables).
Results
Overexpression of Pdr11p
For expression analysis and purification of Pdr11p, three different 2-micron—based multi-copy plasmids were constructed to allow for galactose-inducible expression: a N-terminal FLAG-tagged Pdr11p (Pdr11p), a FLAG-tagged Pdr11p fused at the C terminus with green fluorescent protein (Pdr11p-GFP), and a catalytically inactive mutant of FLAG-tagged Pdr11p (Pdr11pK788M) carrying a lysine to methionine substitution in the Walker A region of NBD2 (Table 1) known to block the ATPase activity of ABC proteins [13, 20, 21]. To test the expression and functionality of the tagged proteins, we transformed the constructs into a sterol uptake-deficient mutant hem1Δaus1Δpdr11Δ. Upon galactose induction, immunoblotting of plasma membrane enriched extracts confirmed expression of the proteins (Fig 1A). All three variants of the protein were detected within the expected size range (FLAG-Pdr11p, 162 kDa; Pdr11p-GFP, 189 kDa). Furthermore, we routinely detected an additional band of approximately 60 kDa by the anti-FLAG antibody, indicating some proteolytic breakdown despite the presence of protease inhibitors. In-gel light-activated fluorescence demonstrated equal loading of the four lanes (Fig 1B). In addition, we employed growth complementation assay to verify the functionality of tagged Pdr11p and Pdr11p-GFP (Fig 1C). In line with their previously reported function in sterol transport [4, 5], both versions of the tagged proteins restored the growth defect of the triple mutant hem1Δaus1Δpdr11Δ on sterol-containing medium, while the mutated protein, Pdr11pK788M was unable to sustain viability under these conditions. Collectively, these results confirm successful expression of functional Pdr11p and Pdr11p-GFP.
Table 1. Conserved ABC motifs of Aus1p and Pdr11p.
| Walker A | Signature | Walker B | |
|---|---|---|---|
| Aus1p-NBD1 | GYPTSTLFKT | VSGGE | YLWDNST |
| Aus1p-NBD2 | GESGAGKTT | LNPTQ | LFLDEPT |
| Pdr11p-NBD1 | GNPTSALFKG | VSGGE | YLWDNST |
| Pdr11p-NBD2 | GESGAGKTT | LSPTQ | LFLDEPT |
Amino acids are shown in single letter code. The sequence degeneracy of residues is highlighted in italic.
Fig 1. Overexpression and functionality of tagged Pdr11p variants.
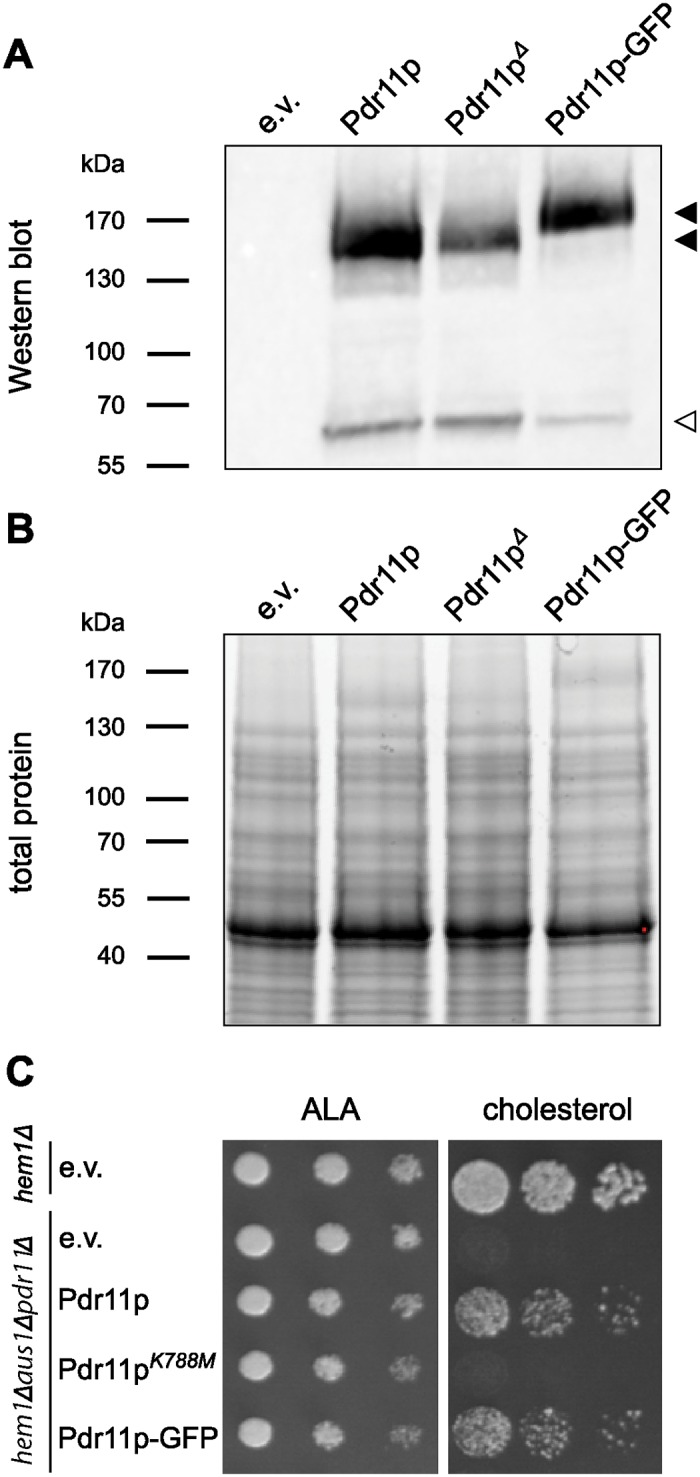
S. cerevisiae cells were transformed with empty vector (e.v.) or pESC-URA carrying PDR11, PDR11-GFP, or PDR11K788M. Sizes of the molecular mass standards are given to the left when relevant. A: Western blot illustrating the induced expression of FLAG-tagged variants of Pdr11p (filled arrowheads) and breakdown product (open arrowhead). Cells carrying the empty vector served as control. The blot was stained with anti-FLAG antibody. B: In-gel fluorescence detection using Criterion TGX stain-free gel. C: Serial dilutions of transformed heme-deficient hem1Δ and sterol uptake-deficient triple mutant hem1Δaus1Δpdr11Δ strains spotted onto standard synthetic galactose plates supplemented with δ-aminolevulinic acid (ALA) or cholesterol.
To determine the optimal induction time, we followed the level of Pdr11p-GFP expression in hem1Δaus1Δpdr11Δ cells by flow cytometry and fluorescence microscopy (Fig 2). After 6 h of induction 36 ± 9% of the cells expressed the gene and the GFP-specific fluorescence was found at the plasma membrane and unidentified internal structures (Fig 2A, S7 Table). Increasing the induction time to 17 h resulted in a similar population (37 ± 5%) of expressing cells but caused an increased intracellular accumulation of the protein, indicating protein mistargeting and degradation. Analysis of cells grown from single colonies to different densities in repressing (SD) medium followed by culturing in inducing (SG) medium revealed a large variability in the number of expressing cells, i.e. induction of pre-cultures grown to OD600 <1 and >2.5 for 6 h resulted in 10–20% and 45% Pdr11p-GFP expressing cells, respectively (Fig 2B). A similar strong dependency in the number of expressing cells was also observed for the protease-deficient strain BJ1991 (Fig 2B, S8 Table). These results show a correlation between cell density/growth state in the repressing media and the level of Pdr11p-GFP expression in the inducing media. Thus, for all subsequent experiments, cells were therefore pre-cultured to OD600 >2.5 followed by induction for 6 h.
Fig 2. Expression analysis utilising GFP-tagged Pdr11p.

A: Effect of induction time on Pdr11p-GFP expression in hem1Δaus1Δpdr11Δ. Panels I-IV, Representative flow cytometry based histograms of cells transformed with plasmids carrying PDR11-GFP (green) or empty vector (black) after 6 and 17 h in SD medium (non-induced) or SG medium (induced). Each data set consists of minimum 20,000 cells. Panels V-VIII, Representative fluorescence microscopy images of cells expressing Pdr11p-GFP after 6 and 17 h induction. The intensities of the fluorescent signals cannot be compared between the images. Bars equal 10 μm. B: Percentage of cells expressing Pdr11p-GFP after galactose induction as a function of optical density at 600 nm (OD600) and cell concentration of the glucose pre-culture just prior to media change. All cultures in each repetition grew from a single colony. Lines are included solely to guide the eye. hem1Δaus1Δpdr11Δ strain, expression after 6 h induction in 3 cultures from the same selected high-expressing colony. BJ1991 strain, expression after 8 (open triangles) and 10 h (open diamonds) from the same selected colony.
Solubilisation and purification of Pdr11p
For solubilisation, we prepared total membranes from Pdr11-GFP expressing cells (Fig 3A) and exposed them to a panel of nonionic and zwitterionic detergents. Among the tested detergents, we found DDM to be very effective in solubilising the protein even at a concentration of 0.6% (Fig 3B and 3C, lane P2 and S3) and consequently chose this mild non-ionic detergent for purification of Pdr11p from plasma membrane-enriched fractions by anti-FLAG affinity chromatography. Upon elution with a buffer containing FLAG peptide, the protein was highly enriched and readily detectable by Western blot analysis using anti-FLAG antibody and by Coomassie Blue staining (Fig 3C). Densitometric analysis of the Coomassie Blue-stained gels revealed at least a 95% degree of protein purity. We estimated the amount of purified protein to be up to 0.5 mg from 1 L culture, based on SDS-PAGE analysis along with standards of known amounts of bovine serum albumin. The preparation contains a second band migrating at 60 kDa. Mass-spectrometric analysis of the excised band at 160 kDa identified 47 peptides of Pdr11p (S9 Table).
Fig 3. Solubilisation and purification of Pdr11p from hem1Δaus1Δpdr11Δ cells.

A: Flowchart of the procedure. Supernatant S1 was centrifuged at 10,000 or 60,000 g (45 min) for collection of plasma membrane enriched fractions or total yeast membranes, respectively. B: Solubilisation of Pdr11p from total yeast membranes. Equal amounts of membranes (P2) were solubilised at the given DDM concentrations. After ultracentrifugation, the amounts of solubilised and non-solubilised Pdr11p were estimated in aliquots of supernatants (S3) and pellets (P3), respectively, via immunoblotting with anti-FLAG antibodies. Pdr11p, filled arrowheads; breakdown product, open arrowhead. C, original membrane fraction not been subjected to solubilisation (positive control). C: Purification of Pdr11p from plasma membrane enriched fractions. Representative Western blot analysis with anti-FLAG antibodies and Coomassie stained SDS-PAGE (SDS) of selected purification fractions. The loaded purification fractions are normalised with respect to volume. Sizes of the molecular mass standards are indicated on the sides.
Functional characterisation of purified Pdr11p
Next, we analysed the ATPase activity of purified Pdr11p in detergent-containing buffer. To prevent possible interference from detergent, lipid, and reagents, we used an assay based on radiolabelled ATP. We estimated an ATPase activity of about 60 ± 24 nmol of ATP/min/mg of protein for the purified protein with Mg2+ as the catalysing metal ion. Pdr11p purified from the BJ1991 strain displayed a slightly reduced activity compared to protein purified from hem1Δaus1Δpdr11Δ cells (88 ± 19%, 2 independent measurements on the same purification).
The purified transporter was active in a broad pH range (Fig 4A) with an optimum around 7.4 similar to other purified ABC transporters [22–24]. Depletion of Mg2+ by addition of EDTA abolished the activity demonstrating that ATP hydrolysis is dependent on the divalent cation. ATPase activity was unaffected in the presence of ouabain (an inhibitor of the Na+/K+-ATPase) or azide (an inhibitor of the F-ATPase) but was strongly inhibited by the classical inhibitors of ABC proteins beryllium fluoride and aluminium fluoride (Fig 4B). Based on this, we conclude that the observed ATPase activity is Pdr11p-dependent. Surprisingly, Pdr11p activity was barely sensitive to orthovanadate, with a calculated IC50 of 4 ± 2 mM (Fig 4C), which is about three orders of magnitude higher than other ABC transporters [23, 25–27]. The purified ATPase-inactive mutant Pdr11pK788M contained comparable levels of contaminating proteins. A parallel analysis showed only very low background ATPase activity (7 ± 2% relative to wild type). We conclude that the observed ATPase activity is Pdr11p-dependent.
Fig 4. ATPase activity of solubilised Pdr11p.
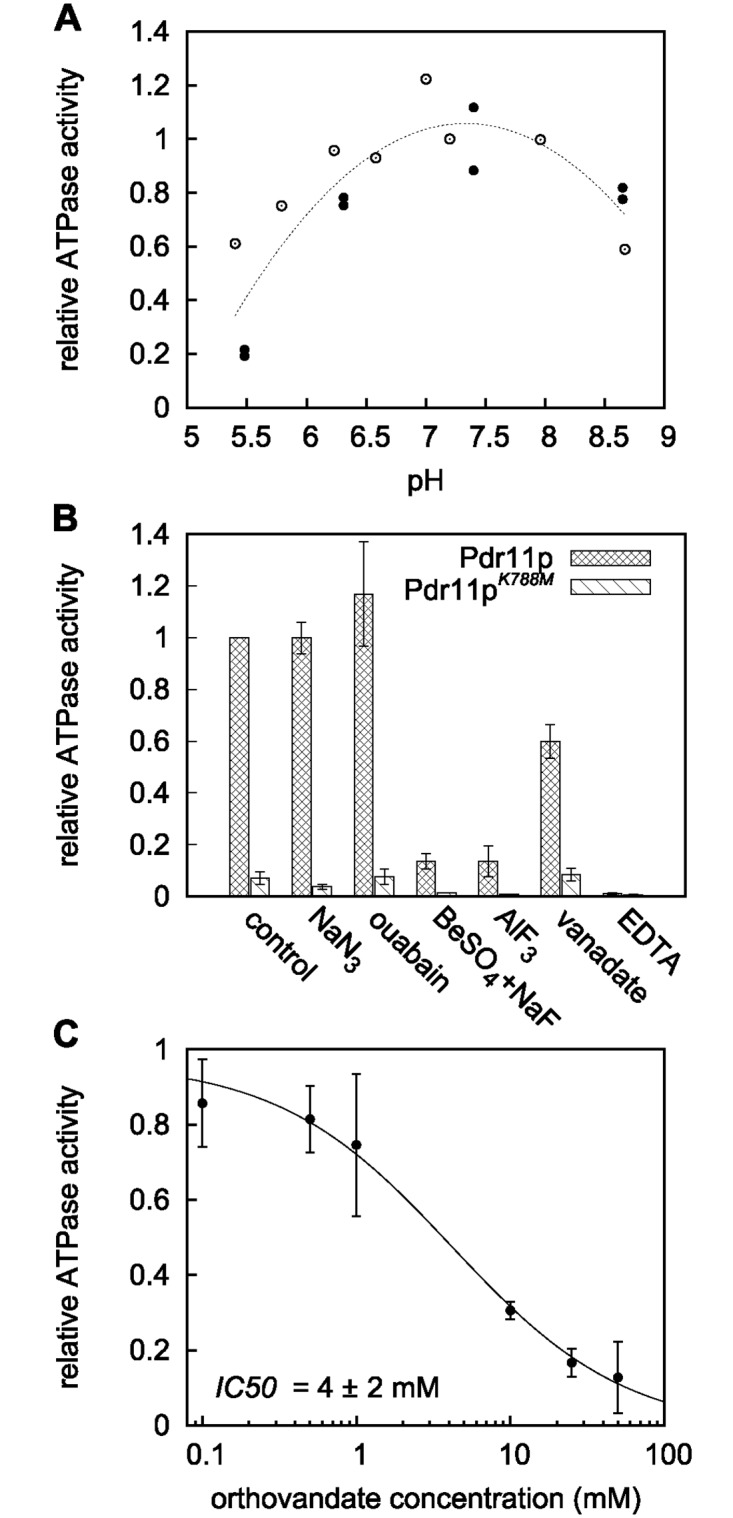
ATPase activity of the purified detergent-solubilised transporter was assayed as described under “Materials and Methods” using [γ-32P] ATP. A: ATPase activity as a function of pH. Open and filled circles are data from two independent experiments. Values are normalised with respect to the values at pH 7.2 (open circles) or pH 7.4 (closed circles). The dashed line is included to guide the eye. B: Effect of various inhibitors: NaN3, 5 mM; ouabain, 5 mM; BeSO4, 1 mM; NaF, 5 mM; AlF3, 1 mM; orthovanadate, 1 mM; EDTA, 1 mM. C: ATPase activity as a function of orthovanadate concentration. Fitting of data to a dose-response/activity curve (see Material and methods) gives IC50 = 4 ± 2 mM, and a Hill coefficient = 0.8 ± 0.2. Results in B and C are the mean ± S.D. from at least two independent experiments relative to the value obtained for the purified detergent-solubilised protein in the absence of inhibitors (control).
To ensure that the lack of vanadate inhibition was not due to slow binding, we preincubated the purified Pdr11p with 1 mM orthovanadate in presence of 1 mM ADP and analysed the ATPase activity upon addition of 1 mM ATP. The overall activity reduced to half (as expected in presence of equal amounts of ADP and ATP) while the low sensitivity towards vanadate inhibition remained (Table 2). Replacement of Mg2+ with other divalent cations can alter the activity and sensitivity to vanadate [28, 29]. We therefore repeated the ATPase assay in presence of MnCl2, NiCl2, and CoCl2. DTT was omitted from the experiments with the metal salts as it caused precipitation. Except Ni2+, all tested metal ions supported Pdr11p ATPase activity but with a maximal inhibition of little more than 50% for 1 mM vanadate neither Co2+ nor Mn2+ effectively altered the sensitivity to vanadate (Table 2).
Table 2. Effect of various compounds on ATPase activity of purified Pdr11pa.
| Compound | Relative ATPase activity (%) | Inhibition by vanadateb (%) |
|---|---|---|
| Control | 100 | 27 ± 15 |
| ADP | 55 ± 30c | 40 ± 4 |
| Mn2+ | 150 ± 28d | 29 ± 16 |
| Co2+ | 41 ± 19d | 54 ± 8 |
| Ni2+ | 10 ± 19d | –e |
| Ergosterol | 94 ± 5 | ND |
| Cholesterol | 105 ± 12 | ND |
| Ergosterol/albumin | 96 ± 1 | ND |
| Cholesterol/albumin | 104 ± 13 | ND |
| Albumin | 107 ± 5 | ND |
a ATP hydrolysis of the purified detergent-solubilised transporter was assayed in the absence and presence of 1 mM orthovanadate. Indicated divalent cations replaced Mg2+. Data are based on at least duplicate determinations on one purification. ND, not determined.
b Inhibition was expressed relative to assays containing the same divalent ion.
c Relative to control without 1 mM ADP.
d The divalent cation replaced Mg2+ in the assay buffer.
e Activity too low to measure vanadate inhibition.
It is notable that addition of PhosSTOP to the ATPase assay with Mn2+ as a catalyst caused an increase in ATPase activity (150 ± 28%) that was not observed without PhosSTOP (82 ± 7%). The reasons are unclear at present.
Many ABC transporters have been shown to possess intrinsic ATPase activity that is stimulated in the presence of transported substrates [1, 24, 30]. As sterol uptake is mediated by Pdr11p [4] and facilitated by albumin [11], we tested the effect of ergosterol, cholesterol, and albumin on ATPase activity. Neither ergosterol nor cholesterol in absence and presence of albumin stimulated the ATPase activity of purified Pdr11p (Table 2).
Analysis of vesicle-reconstituted Pdr11p
To corroborate the effects of inhibitors and test the effect of the lipid environment, we reconstituted Pdr11p in unilamellar vesicles of different lipid compositions. In all cases we observed co-migration of the majority of Pdr11p and liposomes to the top of a sucrose gradient (Fig 5A), which confirms successful reconstitution. ATPase activity of liposome-reconstituted Pdr11p was assayed based on the release of inorganic phosphate from ATP as described for the purified protein. Similarly to the purified protein, lipid-reconstituted Pdr11p demonstrated robust ATPase activity that was efficiently inhibited by beryllium fluoride but not by orthovanadate (Fig 5B). ATPase activity was independent of lipid composition of the liposomes; Pdr11p reconstituted into PC, PC/PS, and PC/PG liposomes exhibited similar ATPase activity. Under the same conditions, reconstitution of Aus1 in PC/PS liposomes resulted in a stimulation of the ATPase activity, confirming previous results [13]. In addition, we observed a slight stimulation the ATPase activity of Aus1p upon reconstitution into PC/PG liposomes.
Fig 5. ATPase activity of liposome-reconstituted Pdr11p and Aus1p.
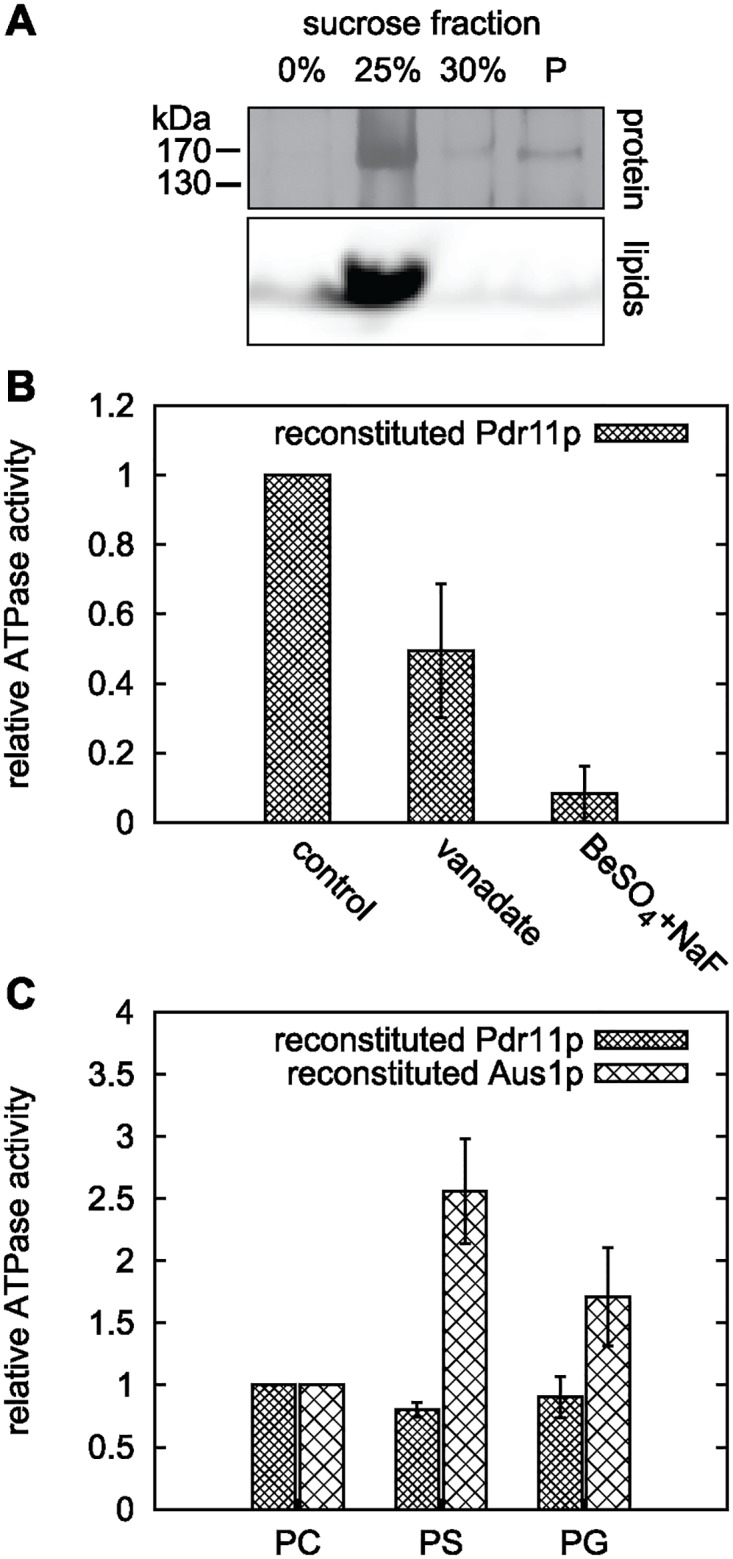
Purified Pdr11p and Aus1p were reconstituted into different liposomes (containing Rho-PE as fluorescent lipid marker) and assayed for ATPase activity using [γ-32P] ATP. A: SDS PAGE analysis of a flotation assay of Pdr11p proteoliposomes in a sucrose gradient. Detection of lipids and protein in the same low density fraction validated successful reconstitution. Proteins are visualised by silver staining and lipids by fluorescence from Rho-PE. B: Relative ATPase activity of Pdr11p reconstituted in PS liposomes in presence of the indicated inhibitors: orthovanadate, 1 mM; BeSO4, 1 mM; NaF, 5 mM. Data is based on at least two reconstitutions from one purification batch. C: Lipid effect on ATPase activity of reconstituted Pdr11p and Aus1p. All activities are corrected for protein amount in the proteoliposomes. Data is based on two reconstitutions from one purification batch of each protein. PC, PC only; PS, PC/PS (1:1); PG, PC/PG (7:3).
Discussion
The study of membrane transporters at the molecular level is hampered by the complexity of native membranes in which they are embedded. Thus, an important step towards a detailed functional understanding of membrane proteins is their purification and reconstitution into defined, tuneable systems such as liposomes and nanodiscs. One important prerequisite for such analysis is access to adequate amounts of functional, purified protein. In this study, we describe a successful purification protocol and the biochemical characterisation of the yeast ABC transporter Pdr11p.
Expression of a FLAG tagged version of Pdr11p from a multicopy plasmid under the control of the strong inducible promoter, GAL10, allowed for effective purification to near homogeneity in yields high enough for subsequent biochemical studies. Further modification of Pdr11p—by addition of GFP fused at its C terminus—retained the functionality of the ABC transporter and facilitated the analysis of its expression and solubilisation. For expression, we tested two different host strains: the triple mutant strain W303 hem1Δaus1Δpdr11Δ and the protease deficient strain lacking yeast proteinases A (Pep4p) and B (Prb1p). In both strains, the number of expressing cells and intensity of the expression signal varied substantially for Pdr11p upon galactose induction. Several studies report similar results of heterogeneous expression within the cell culture for other proteins expressed from multicopy plasmids [11, 31–33]. A likely reason is fluctuating plasmid copy numbers within the cell population [34]. Interestingly, we observed a strong dependence of Pdr11p expression from the growth state of the cells in the repressing pre-culture medium; the highest expression was observed upon galactose induction of late logarithmic phase cells. In line with this, Mead et al. [34] observed less plasmid-free cells in cultures passing through the stationary phase. Microscopic analysis of the cells revealed that a short induction time was critical to prevent increased intracellular accumulation and degradation of Pdr11p-GFP.
Purification and reconstitution of Pdr11p into liposomes permitted us to study the biochemical properties of the transporter. Several biochemical features were similar to Aus1p [13]. First, purified Pdr11p exhibited a stable ATPase activity of about 60 ± 24 nmol of ATP/min/mg of protein even before reconstitution. For comparison, Aus1p has a specific activity of 56 nmol/min/mg under these conditions [13]. These activities correspond to ∼ 10 ATP molecules/min/protein and are within the range of values reported for a number of other purified eukaryotic ABC transporters, such as Ste6p, ABCR, transporter associated with antigen processing, and ABCA1 [35–38]. Second, mutagenesis of the conserved lysine to methionine (K788M) in the Walker A motif abolished ATP hydrolysis. Finally, neither ergosterol nor cholesterol stimulated the ATPase activity. In sharp contrast to Aus1p, whose activity is inhibited by low concentrations of orthovanadate and specifically stimulated by PS [13], Pdr11p ATPase activity was was barely sensitive to orthovanadate and not specifically stimulated by phosphatidylserine upon reconstitution into liposomes. These results highlight distinct differences between Pdr11p and Aus1p and create an experimental basis for further studies on both ABC transporters to elucidate their regulation by the lipid environment.
Similar to other fungal ABCG-family transporters Pdr11p and Aus1p have uniquely conserved asymmetric NBDs [39, 40]. The N-terminal NBD motif (NBD1) of both transporters contains a well-conserved ABC signature sequence, VSGGE, but possesses degenerated Walker A and Walker B motifs (Table 1). In contrast, the Walker A and Walker B motifs of the C-terminal NBD2 are conserved but the ABC signature motif is degenerated. The similarity of the ABC transporter motifs of Pdr11p and Aus1p indicates that an explanation for the different responses to orthovanadate should be found in other parts of the sequence (potentially around the Q-loop or H-loop) or by differences in the environment of the ATP binding sites. In line with this, a lack of orthovanadate inhibition has also been reported for other asymmetric ABC transporters such as ABCA1 [29] and ABCG5/G8 [41] and for symmetric ABC transporters such as ABCG1 [24], ruling out that this feature is connected to the non-consensus amino acids in one of the two binding sites. In some cases insensitive ABC transporters have recovered orthovanadate sensitivity by replacing Mg2+ with other divalent metal ions. This has been demonstrated for ABCA1 using Mn2+[29] and ABCC6 using Ni2+[28]. ABCG2 show orthovanadate-dependent adenine nucleotide trapping in the presence of Co2+ but not Mg2+[21]. None of the above ions restored orthovanadate sensitivity of Pdr11p. It remains to be established why some ABC transporters are only inhibited by beryllium and aluminium fluoride and not by orthovanadate.
In summary, we developed a relatively simple affinity-tag purification and reconstitution procedure yielding sufficient amounts of pure and active Pdr11p for future biochemical and biophysical studies that will potentially lead to elucidation of the mechanism(s) by which this transporter contributes to sterol uptake.
Supporting information
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
We are grateful to Anne-Mette Bjerg Petersen for expert technical assistance, Jonathan Erling Nagl for help with detergent screening; Søren Roi Midtgaard for technical instructions, Knud Jørgen Jensen for giving access to his laboratory, and Tonni G. Andersen for critical comments to the manuscript.
Data Availability
All ATPase data are within the paper and in the Supporting Information. Flow cytometry files are available under Figshare: DOI 10.6084/m9.figshare.5259457 and DOI 10.6084/m9.figshare.5259454.
Funding Statement
This work was supported by the Research Centre ‘bioSYNergy’ at University of Copenhagen funded by the UCPH Excellence Program for Interdisciplinary Research and the Villium Fonden (grant 022868 to TGP). AB was supported by FNR grant FNR/P09/01. TK was funded by the Helmholtz Virtual Institute of Complex Molecular Systems in Environmental Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Neumann J, Rose-Sperling D, Hellmich UA. Diverse relations between ABC transporters and lipids: An overview. Biochimica et Biophysica Acta (BBA)—Biomembranes. 2017;1859(4):605–618. 10.1016/j.bbamem.2016.09.023 [DOI] [PubMed] [Google Scholar]
- 2. Aye ILMH, Singh AT, Keelan JA. Transport of lipids by ABC proteins: Interactions and implications for cellular toxicity, viability and function. Chemico-Biological Interactions. 2009;180(3):327–339. 10.1016/j.cbi.2009.04.012 [DOI] [PubMed] [Google Scholar]
- 3. Phillips Michael C. Molecular Mechanisms of Cellular Cholesterol Efflux. Journal of Biological Chemistry. 2014;289(35):24020–24029. 10.1074/jbc.R114.583658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wilcox L, Balderes D, Wharton B, Tinkelenberg A, Rao G, Sturley S. Transcriptional profiling identifies two members of the ATP-binding cassette transporter superfamily required for sterol uptake in yeast. Journal of Biological Chemistry. 2002;277(36):32466–32472. 10.1074/jbc.M204707200 [DOI] [PubMed] [Google Scholar]
- 5. Li Y, Prinz W. ATP-binding cassette (ABC) transporters mediate nonvesicular, raft-modulated sterol movement from the plasma membrane to the endoplasmic reticulum. Journal of Biological Chemistry. 2004;279(43):45226–45234. 10.1074/jbc.M407600200 [DOI] [PubMed] [Google Scholar]
- 6. Reiner S, Micolod D, Zellnig G, Schneiter R. A Genomewide Screen Reveals a Role of Mitochondria in Anaerobic Uptake of Sterols in Yeast. Molecular Biology of the Cell. 2006;17(1):90–103. 10.1091/mbc.E05-06-0515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakayama H, Tanabe K, Bard M, Hodgson W, Wu S, Takemori D, et al. The Candida glabrata putative sterol transporter gene CgAUS1 protects cells against azoles in the presence of serum. Journal of Antimicrobial Chemotherapy. 2007;60(6):1264–1272. 10.1093/jac/dkm321 [DOI] [PubMed] [Google Scholar]
- 8. Nagi M, Tanabe K, Ueno K, Nakayama H, Aoyama T, Chibana H, et al. The Candida glabrata sterol scavenging mechanism, mediated by the ATP-binding cassette transporter Aus1p, is regulated by iron limitation. Molecular Microbiology. 2013;88(2):371–381. 10.1111/mmi.12189 [DOI] [PubMed] [Google Scholar]
- 9. Kohut P, Wüstner D, Hronska L, Kuchler K, Hapala I, Valachovic M. The role of ABC proteins Aus1p and Pdr11p in the uptake of external sterols in yeast: Dehydroergosterol fluorescence study. Biochemical and Biophysical Research Communications. 2011;404(1):233–238. 10.1016/j.bbrc.2010.11.099 [DOI] [PubMed] [Google Scholar]
- 10. Alimardani P, Regnacq M, Moreau-Vauzelle C, Ferreira T, Rossignol T, Blondin B, et al. SUT1-promoted sterol uptake involves the ABC transporter Aus1 and the mannoprotein Dan1 whose synergistic action is sufficient for this process RID B-9398-2008. Biochemical Journal. 2004;381:195–202. 10.1042/BJ20040297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marek M, Silvestro D, Fredslund MD, Andersen TG, Pomorski TG. Serum albumin promotes ATP-binding cassette transporter-dependent sterol uptake in yeast. FEMS Yeast Research. 2014;14(8):1223–1233. 10.1111/1567-1364.12219 [DOI] [PubMed] [Google Scholar]
- 12. Georgiev A, Sullivan DP, Kersting MC, Dittman JS, Beh CT, Menon AK. Osh proteins regulate membrane sterol organization but are not required for sterol movement between the ER and PM. Traffic. 2011;12(10):1341–1355. 10.1111/j.1600-0854.2011.01234.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marek M, Milles S, Schreiber G, Daleke DL, Dittmar G, Herrmann A, et al. The Yeast Plasma Membrane ATP Binding Cassette (ABC) Transporter Aus1 PURIFICATION, CHARACTERIZATION, AND THE EFFECT OF LIPIDS ON ITS ACTIVITY. Journal of Biological Chemistry. 2011;286(24):21835–21843. 10.1074/jbc.M111.244525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. Journal of Bacteriology. 1983;153(1):163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shevchenko A, Tomas H, Havlis J, Olsen J V, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nature Protocols. 2006;1:2856–2860. 10.1038/nprot.2006.468 [DOI] [PubMed] [Google Scholar]
- 16. Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature Protocols. 2007;2(8):1896–906. 10.1038/nprot.2007.261 [DOI] [PubMed] [Google Scholar]
- 17. Cox JUR, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome—wide protein quantification. Nature Biotechnology. 2008;26:1367–1372. 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
- 18. Lanfermeijer FC, Venema K, Palmgren MG. Purification of a Histidine-Tagged Plant Plasma Membrane H+-ATPase Expressed in Yeast. Protein Expression and Purification. 1998;12(1):29–37. 10.1006/prep.1997.0788 [DOI] [PubMed] [Google Scholar]
- 19. Rouser G, Fkeischer S, Yamamoto A. Two dimensional thin layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids. 1970;5(5):494–496. 10.1007/BF02531316 [DOI] [PubMed] [Google Scholar]
- 20. Gao M, Cui HR, Loe DW, Grant CE, Almquist KC, Cole SPC, et al. Comparison of the Functional Characteristics of the Nucleotide Binding Domains of Multidrug Resistance Protein 1. Journal of Biological Chemistry. 2000;275(17):13098–13108. 10.1074/jbc.275.17.13098 [DOI] [PubMed] [Google Scholar]
- 21. Özvegy C, Váradi A, Sarkadi B. Characterization of Drug Transport, ATP Hydrolysis, and Nucleotide Trapping by the Human ABCG2 Multidrug Transporter MODULATION OF SUBSTRATE SPECIFICITY BY A POINT MUTATION. Journal of Biological Chemistry. 2002;277(50):47980–47990. 10.1074/jbc.M207857200 [DOI] [PubMed] [Google Scholar]
- 22. Urbatsch IL, Al-Shawi MK, Senior AE. Characterization of the ATPase Activity of Purified Chinese Hamster P-glycoprotein. Biochemistry. 1994;33(23):7069–7076. 10.1021/bi00189a008 [DOI] [PubMed] [Google Scholar]
- 23. Mao Q, Leslie EM, Deeley RG, Cole SPC. ATPase activity of purified and reconstituted multidrug resistance protein MRP1 from drug-selected H69AR cells. Biochimica et Biophysica Acta (BBA)—Biomembranes. 1999;1461(1):69–82. 10.1016/S0005-2736(99)00150-9 [DOI] [PubMed] [Google Scholar]
- 24. Hirayama H, Kimura Y, Kioka N, Matsuo M, Ueda K. ATPase activity of human ABCG1 is stimulated by cholesterol and sphingomyelin. Journal of Lipid Research. 2013;54(2):496–502. 10.1194/jlr.M033209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Urbatsch IL, Sankaran B, Weber J, Senior AE. P-glycoprotein Is Stably Inhibited by Vanadate-induced Trapping of Nucleotide at a Single Catalytic Site. Journal of Biological Chemistry. 1995;270(33):19383–19390. 10.1074/jbc.270.33.19383 [DOI] [PubMed] [Google Scholar]
- 26. Benabdelhak H, Schmitt L, Horn C, Jumel K, Blight MA, Holland IB. Positive co-operative activity and dimerization of the isolated ABC ATPase domain of HlyB from Escherichia coli. Biochemical Journal. 2005;386:489–495. 10.1042/BJ20041282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lamping E, Monk BC, Niimi K, Holmes AR, Tsao S, Tanabe K, et al. Characterization of Three Classes of Membrane Proteins Involved in Fungal Azole Resistance by Functional Hyperexpression in Saccharomyces cerevisiae. Eukaryotic Cell. 2007;6(7):1150–1165. 10.1128/EC.00091-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cai J, Daoud R, Alqawi O, Georges E, Pelletier J, Gros P. Nucleotide Binding and Nucleotide Hydrolysis Properties of the ABC Transporter MRP6 (ABCC6). Biochemistry. 2002;41(25):8058–8067. 10.1021/bi012082p [DOI] [PubMed] [Google Scholar]
- 29. Tanaka AR, Abe-Dohmae S, Ohnishi T, Aoki R, Morinaga G, Okuhira Ki, et al. Effects of Mutations of ABCA1 in the First Extracellular Domain on Subcellular Trafficking and ATP Binding/Hydrolysis. Journal of Biological Chemistry. 2003;278(10):8815–8819. 10.1074/jbc.M206885200 [DOI] [PubMed] [Google Scholar]
- 30. Janvilisri T, Venter H, Shahi S, Reuter G, Balakrishnan L, Veen HWv. Sterol Transport by the Human Breast Cancer Resistance Protein (ABCG2) Expressed in Lactococcus lactis. Journal of Biological Chemistry. 2003;278(23):20645–20651. 10.1074/jbc.M301358200 [DOI] [PubMed] [Google Scholar]
- 31. Xu P, Okkeri J, Hanisch S, Hu RY, Xu Q, Pomorski TG, et al. Identification of a novel mouse P4-ATPase family member highly expressed during spermatogenesis. Journal of Cell Science. 2009;122(16):2866–2876. 10.1242/jcs.047423 [DOI] [PubMed] [Google Scholar]
- 32. Jensen NB, Strucko T, Kildegaard KR, David F, Maury J, Mortensen UH, et al. EasyClone: method for iterative chromosomal integration of multiple genes Saccharomyces cerevisiae. FEMS Yeast Research. 2014;14(2):238–248. 10.1111/1567-1364.12118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Albertsen L, Chen Y, Bach LS, Rattleff S, Maury J, Brix S, et al. Diversion of Flux toward Sesquiterpene Production in Saccharomyces cerevisiae by Fusion of Host and Heterologous Enzymes. Applied and Environmental Microbiology. 2011;77(3):1033–1040. 10.1128/AEM.01361-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mead DJ, Gardner DCJ, Oliver SG. The yeast 2 μ plasmid: strategies for the survival of a selfish DNA. Molecular and General Genetics MGG. 1986;205(3):417–421. 10.1007/BF00338076 [DOI] [PubMed] [Google Scholar]
- 35. Sun H, Molday RS, Nathans J. Retinal Stimulates ATP Hydrolysis by Purified and Reconstituted ABCR, the Photoreceptor-specific ATP-binding Cassette Transporter Responsible for Stargardt Disease. Journal of Biological Chemistry. 1999;274(12):8269–8281. 10.1074/jbc.274.12.8269 [DOI] [PubMed] [Google Scholar]
- 36. Ketchum CJ, Schmidt WK, Rajendrakumar GV, Michaelis S, Maloney PC. The Yeast a-factor Transporter Ste6p, a Member of the ABC Superfamily, Couples ATP Hydrolysis to Pheromone Export. Journal of Biological Chemistry. 2001;276(31):29007–29011. 10.1074/jbc.M100810200 [DOI] [PubMed] [Google Scholar]
- 37. Takahashi K, Kimura Y, Kioka N, Matsuo M, Ueda K. Purification and ATPase activity of human ABCA1. The Journal of Biological Chemistry. 2006;281(16):10760–10768. 10.1074/jbc.M513783200 [DOI] [PubMed] [Google Scholar]
- 38. Gorbulev S, Abele R, Tampé R. Allosteric crosstalk between peptide-binding, transport, and ATP hydrolysis of the ABC transporter TAP. Proceedings of the National Academy of Sciences. 2001;98(7):3732–3737. 10.1073/pnas.061467898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prasad R, Goffeau A. Yeast ATP-Binding Cassette Transporters Conferring Multidrug Resistance. Annual Review of Microbiology. 2012;66(1):39–63. 10.1146/annurev-micro-092611-150111 [DOI] [PubMed] [Google Scholar]
- 40. Lamping E, Baret PV, Holmes AR, Monk BC, Goffeau A, Cannon RD. Fungal PDR transporters: phylogeny, topology, motifs and function. Fungal genetics and biology: FG & B. 2010;47(2):127 10.1016/j.fgb.2009.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Z, Stalcup LD, Harvey BJ, Weber J, Chloupkova M, Dumont ME, et al. Purification and ATP Hydrolysis of the Putative Cholesterol Transporters ABCG5 and ABCG8. Biochemistry. 2006;45(32):9929–9939. 10.1021/bi0608055 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
All ATPase data are within the paper and in the Supporting Information. Flow cytometry files are available under Figshare: DOI 10.6084/m9.figshare.5259457 and DOI 10.6084/m9.figshare.5259454.