

Abstract
Deuterodifluoromethyl ketones and sulfones were assembled in three synthetic steps from methyl ketones and sulfones, respectively. The key synthetic transformation is the deuteration of the difluorocarbanion generated by the release of trifluoroacetate from highly α-fluorinated gem-diols. High levels of deuterium on the “CF2D” group were routinely observed. This strategy is mild and versatile and it can be applied to both ketones and sulfones without additional concerns of over- or under-fluorination. Additional examples address issues of over-deuteration when compounds with other acidic protons are subjected to the reaction conditions. This process not only demonstrates a new method to install a “CF2D” group but also extends the scope of trifluoroacetate release to sulfones.
Keywords: fluorine, enolate, deuteration, ketones, base
Graphical Abstract

Fluorinated organic compounds are particularly attractive to the pharmaceutical industry, because fluorination may increase potency, modulate basicity, or enhance resistance to metabolic transformations.1,2 Accordingly, an increasing number of newly approved pharmaceuticals display a fluorine on their structure. Another more recent strategy to address drug metabolism is the incorporation of deuterium in the place of hydrogen atom.3,4 Although this process is substantially less explored compared to fluorination in medicinal chemistry, deuteration is commonly used in organic chemistry to label compounds for mechanistic or kinetic studies. The convergence of these two fields has yielded some examples of organic molecules that display both fluorine and deuterium;5–7 however, this area is widely under-developed and there are few synthetic methods to access these unique structures.8
The two existing “CF2D” reagents have been derived from trifluoromethylation reagents, however a deuterium replaces a fluorine atom. The most recognizable example is trimethyl(deuterodifluoromethyl)silane 1, derived from the Ruppert-Prakash reagent, but the reagent 1 has only been reported to transfer deuterium and not the “CF2D” group (Scheme 1).9 The other reagent, sulfoximine 2, has been prepared twice, but the levels of deuterium incorporation on the “CF2D” group too low to enable its use.10 The other published compounds arise from quenching difluorinated anions with D2O or CD3OD, yet none of these reports are primarily directed to the synthesis of deuterofluorocarbons.11–19 The only exception is the synthesis of 19-deutero-19,19-difluoroandrost-4-3,17-dione (3) which was conducted to study the mechanism of inactivation of aromatase.5 Herein, we report that deuterodifluoromethyl ketones and sulfones can be readily accessed via deuteration of the difluoroenolate generated from highly α-fluorinated gem-diols.
Scheme 1.

Notable compounds displaying a “CF2D” group.
In 2011, we reported that the base-mediated fragmentation of highly α-fluorinated gem-diols produces difluoroenolates that readily participate in aldol reactions (Scheme 2).20 This mild yet powerful process releases trifluoroacetate and has enabled the reaction of difluoroenolates with halogenation reagents,21,22 imines,23 trifluoromethyl ketones,24 and disulfides.25 Also, the difluoroenolates can be treated with water to produce difluoromethyl ketones without concerns of over- or under-fluorinated by-products.26 Based on these prior data, we hypothesized that a difluoroenolate, formed following the release of trifluoroacetate, could be effectively trapped with deuterium. We envisioned that this method could assemble “CF2D” groups adjacent to ketones with high levels of deuterium incorporation and furthermore, we aimed to conduct side-by-side comparisons between the pronated and deuterated products (see below).
Scheme 2.

Reactions of difluoroenolates formed by the release of trifluoroacetate from highly α-fluorinated gem-diols.
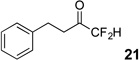
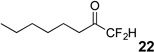
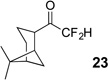
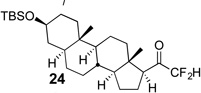
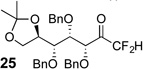
The requisite highly α-fluorinated gem-diols 4–14 were synthesized as described in the literature (Table 1).20–22 Specifically, this class of compound can be access from methyl ketones by trifluoroacetylation followed by difluorination. Next, we treated substrates 4–14 in a 3:1 mixture of H2O and THF with Et3N (4 equiv.) and received the difluoromethyl ketones 15–25 in good to excellent yields (i.e. 71–99%). Phenyl- and heteroaromatic substituted ketones are compatible with the process, but LiBr was added to the reaction mixture in the case of the thiophene 7. Alkyl substituted and α,β-unsaturated ketones participate in the process as well. Also, these data correlate well with similar studies conducted by Pattison and coworkers using CH3CN as solvent,26 however, the mixture of THF and water are more adaptable to the subsequent deuteration studies. Notable, examples of complex difluoromethyl ketones are the steroid- and glucose-derived compounds 24 and 25, respectively. Although no epimerization of the α-stereogenic center of the steroid derivative 24 was observed, the basic conditions did promote epimerization of the α-benzyloxy substituent on 25. Clearly, the center on the latter compound is more acidic than the former; however, the subtle differences among the fluorinated ketones 21–25 will play a more prominent role in creating deuterated analogues. Specifically, processes to prevent over-deuteration as well as under deuteration must be identified.
Table 1.
Preparation of difluoromethyl ketones 15–25.
 | |||
|---|---|---|---|
| Entry | Substrate | Major Product | Yielda |
| 1 |  |
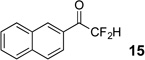 |
96% |
| 2 |  |
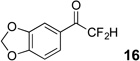 |
88% |
| 3 | 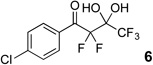 |
 |
71% |
| 4 | 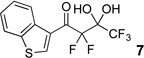 |
 |
92%b |
| 5 | 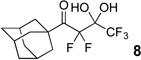 |
 |
96% |
| 6 | 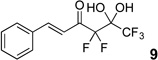 |
 |
78%b |
| 7 | 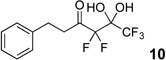 |
 |
99% |
| 8 | 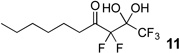 |
 |
88% |
| 9 | 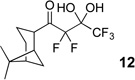 |
 |
94% |
| 10 | 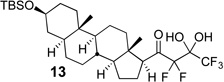 |
 |
94% |
| 11 | 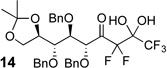 |
 |
74% (d.r. = 5:3) |
Isolated yields.
LiBr was added.
The deactivation of carbanion formation from α-fluoro substituents was shown in 1976 by Hine.27 Specifically, the rate of deuterium exchange was considerably slower in methyl difluoroacetate than methyl acetate when compared in methanold with sodium methoxide. Subsequent computation studies28 and synthetic studies29 have further supported that fluorine substitution can have a dramatic effect on acidity of neighboring protons. For example, NaH-mediated deprotonation of 1-cyclohexyl-2,2-difluoro-1-ethanone followed by trapping with TMSCl fails to generate the difluoroenolate as reported by Liu and Zhou (Scheme 3).29 To demonstrate that difluoromethyl ketones cannot be used to assemble the “CF2D” group, difluoromethyl ketone 21 was treated with Et3N in a mixture of 3:1 D2O and THF. Across 24 hours, deuteration occurred slowly at the CH2 group adjacent to ketone, but the “CF2H” group was untouched as observed 19F NMR. The undesired deuterated product 26 was isolated in 83% yield.
Scheme 3.

Examples of deactivated carbanion formation from α-fluorosubstituents.
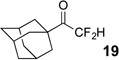
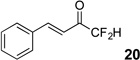
Treatment of the pentafluorinated gem-diols with D2O in the place of water in the presence of base provided the desired deuterodifluoromethyl ketones 27–35 (Table 2). Levels of deuterium incorporation were typically 97–99% as determined by 19F NMR. This method is a powerful process to access the “CF2D” groups which is devoid of under- or over-fluorination as well as low levels of deuteration. It is fully compatible with aryl, heteroaryl, alkenyl, and alkyl substituted ketones. The case of the steroid-based derivative 34 shows that the “CF2D” group can be installed on complex structures; however, the formation of glucose derivative 35 demonstrates that the other α-position is subjected to deuteration under these conditions. Specifically, analyses of HRMS, 1H, 13C, and 19F NMR confirmed that no additional deuterium atoms were present on steroid 34, yet the presence of the additional deuterium was readily observed for glucose 35. These data prompted additional studies to address the potential of over-deuteration.
Table 2.
Preparation of deuterodifluoromethyl ketones 27–35.
 | ||||
|---|---|---|---|---|
| Entry | Substrate | Major Product | Yielda | % Db |
| 1 | 4 | 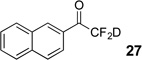 |
89% | 98% |
| 2 | 5 | 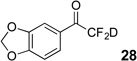 |
86% | 98% |
| 3 | 6 | 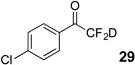 |
47% | 96% |
| 4 | 7 | 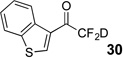 |
89% | 97% |
| 5 | 8 |  |
90% | 98% |
| 6 | 9 | 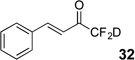 |
26%c | 99% |
| 7 | 12 | 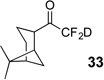 |
90% | 99% |
| 8 | 13 | 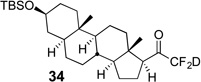 |
81% | 99% |
| 9 | 14 | 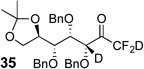 |
76% | 98% (d.r. = 4:3) |
Isolated yields.
Percent deuterium incorporation were determined by 19F NMR, see Supporting Information for details.
The major product was the dimer and it was isolated in 69% yield, see Supporting Information for details.
Over-deuteration of difluoromethyl ketones can be readily addressed due to the differences in acidity of the two α-carbons. For example, treatment of the gem-diol 10 with D2O and Et3N provides the product 36 with complete deuteration of all three α-protons (Scheme 4). Subsequently, if the trideuterated compound 36 is subjected to Et3N and H2O, only two of the deuteriums are exchanged and the “CF2D” remains intact on 37. A similar result occurred during the deuteration of 11 to give 38.
Scheme 4.

Additional deuterium incorporation studies on 10 and 11.
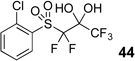
Difluoromethyl sulfones are an important class of fluorinated building blocks that have been championed by Prakash.30,31 Despite the widespread use of these reagents, the deuterated analogues have not been established. In order extend the scope of this strategy and produce another group of “CF2D” compounds, we synthesized the requisite highly α-fluorinated gem-diols from the methylphenylsulfones 39–42 (Table 3). The two-step trifluoroacetylation and difluorination protocol20 provided the gem-diols 43–46.
Table 3.
Preparation of fluorosulfone-based gem-diols 43–46.
 | |||
|---|---|---|---|
| Entry | Substrate | Major Product | Yielda |
| 1 |  |
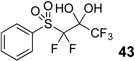 |
87% |
| 2 | 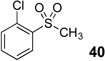 |
 |
93% |
| 3 |  |
 |
82% |
| 4 |  |
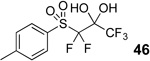 |
71% |
Isolated yields.
The base-promoted release of trifluoroacetate in the presence of H2O or D2O provided the difluoromethyl sulfones 47–50 or deuterodifluoromethyl sulfones 51–54, respectively (Table 4). Again, deuteration occurred in very high levels (i.e., >96% by 19F NMR) and yields were very good. Also, trifluoroacetate release still occurs exclusively, regardless of the exchange of the ketone group with a sulfone group.
Table 4.
Preparation of difluoromethyl sulfones 47–50 and deuterodifluoromethyl sulfones 51–54.
 | |||||
|---|---|---|---|---|---|
| Entry | Substrate | Conditions | Major Product | Yielda | % Db |
| 1 | 43 | Et3N, H2O | 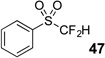 |
87% | na |
| 2 | 44 | Et3N, H2O | 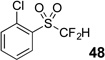 |
quant. | na |
| 3 | 45 | Et3N, H2O | 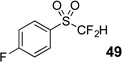 |
89% | na |
| 4 | 46 | Et3N, H2O | 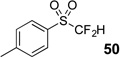 |
85% | na |
| 5 | 43 | Et3N, D2O | 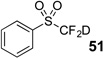 |
74% | 97% |
| 6 | 44 | Et3N, D2O |  |
96% | 96% |
| 7 | 45 | Et3N, D2O | 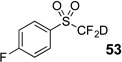 |
80% | 98% |
| 8 | 46 | Et3N, D2O | 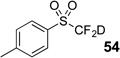 |
91% | 98% |
Isolated yields.
Percent deuterium incorporation were determined by 19F NMR, see Supporting Information for details.
In conclusion, deuterodifluoromethyl ketones and sulfones were assembled with high levels of deuterium on the “CF2D” group. This strategy is mild and versatile as it can be applied to both ketones and sulfones without concerns of over- or under-fluorination. Additional examples were provided to address issues of over-deuteration when compounds with other acidic protons are subjected to the reaction conditions. This process not only demonstrates a new method to install a “CF2D” group but also extends the scope of trifluoroacetate release to sulfones.
Supplementary Material
Highlights.
Deuterodifluoromethyl ketones and sulfones are prepared in three synthetic steps.
High levels of deuterium on the “CF2D” group are observed.
Over- and under-fluorinated by-products are not isolated using this process.
Acknowledgments
Funding for this work was provided by the University of Mississippi, the American Association of Colleges of Pharmacy as a New Pharmacy Faculty Research Award, the National Institute on Aging (R21AG039718), and the National Institute of General Medical Sciences (P20GM104932). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the National Institutes of Health (NIH). The authors acknowledge the Mass Spectrometry and Proteomics Facility of the University of Notre Dame for acquisition of high-resolution mass spectrometry data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Muller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 2.Hagmann WK. J. Med. Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 3.Mullard A. Nat. Rev. Drug Discov. 2016;15:219–221. doi: 10.1038/nrd.2016.63. [DOI] [PubMed] [Google Scholar]
- 4.Timmins GS. Expert Opin. Ther. Pat. 2014;24:1067–1075. doi: 10.1517/13543776.2014.943184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Furth PS, Robinson CH. Biochemistry. 1989;28:1254–1259. doi: 10.1021/bi00429a045. [DOI] [PubMed] [Google Scholar]
- 6.Giovanelli E, Leroux S, Moisan L, Carreyre H, Thuéry P, Buisson D-A, Meddour A, Coustard J-M, Thibaudeau S, Rousseau B, Nicolas M, Hellier P, Doris E. Org. Lett. 2011;13:4116–4119. doi: 10.1021/ol201637m. [DOI] [PubMed] [Google Scholar]
- 7.Kerekes AD, Esposite SJ, Doll RJ, Tagat JR, Yu T, Xiao Y, Zhang Y, Prelusky DB, Tevar S, Gray K, Terracina GA, Lee S, Jones J, Liu M, Basso AD, Smith EB. J. Med. Chem. 2011;54:201–210. doi: 10.1021/jm1010995. [DOI] [PubMed] [Google Scholar]
- 8.Richardson DP, Bauer GS, Bravo AA, Drzyzga M, Motazedi T, Pelegri-O’Day EM, Poudyal S, Suess DLM, Thoman JW., Jr J. Fluorine Chem. 2015;180:208–215. [Google Scholar]
- 9.Fier PS, Hartwig JF. J. Am. Chem. Soc. 2012;134:5524–5527. doi: 10.1021/ja301013h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang W, Wang F, Hu J. Org. Lett. 2009;11:2109–2112. doi: 10.1021/ol900567c. [DOI] [PubMed] [Google Scholar]
- 11.Surya Prakash GK, Hu J, Olah GA. J. Fluorine Chem. 2001;112:355–360. [Google Scholar]
- 12.Griffith GA, Hillier IH, Percy JM, Roig R, Vincent MA. J. Org. Chem. 2006;71:8250–8255. doi: 10.1021/jo061450y. [DOI] [PubMed] [Google Scholar]
- 13.Aliouane L, Rigaud B, Pfund E, Jean L, Renard P-Y, Lequeux T. Tetrahedron Lett. 2011;52:3681–3685. [Google Scholar]
- 14.Hu M, Wang F, Zhao Y, He Z, Zhang W, Hu J. J. Fluorine Chem. 2012;135:45–58. [Google Scholar]
- 15.Thomoson CS, Dolbier WR. J. Org. Chem. 2013;78:8904–8908. doi: 10.1021/jo401392f. [DOI] [PubMed] [Google Scholar]
- 16.Aikawa K, Maruyama K, Honda K, Mikami K. Org. Lett. 2015;17:4882–4885. doi: 10.1021/acs.orglett.5b02438. [DOI] [PubMed] [Google Scholar]
- 17.Dang H, Whittaker AM, Lalic G. Chem. Sci. 2016;7:505–509. doi: 10.1039/c5sc03415a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ismalaj E, Le Bars D, Billard T. Angew. Chem. Int. Ed. 2016;55:4790–4793. doi: 10.1002/anie.201601280. [DOI] [PubMed] [Google Scholar]
- 19.Lin Q-Y, Xu X-H, Zhang K, Qing F-L. Angew. Chem. Int. Ed. 2016;55:1479–1483. doi: 10.1002/anie.201509282. [DOI] [PubMed] [Google Scholar]
- 20.Han C, Kim EH, Colby DA. J. Am. Chem. Soc. 2011;133:5802–5805. doi: 10.1021/ja202213f. [DOI] [PubMed] [Google Scholar]
- 21.John JP, Colby DA. J. Org. Chem. 2011;76:9163–9168. doi: 10.1021/jo2017179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hazlitt RA, John JP, Tran Q-L, Colby DA. Tetrahedron Letters. 2016;57:1906–1908. doi: 10.1016/j.tetlet.2016.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie C, Wu L, Mei H, Soloshonok VA, Han J, Pan Y. Tetrahedron Lett. 2014;55:5908–5910. [Google Scholar]
- 24.Zhang P, Wolf C. J. Org. Chem. 2012;77:8840–8844. doi: 10.1021/jo3017583. [DOI] [PubMed] [Google Scholar]
- 25.Lin Y-M, Yi W-B, Shen W-Z, Lu G-P. Org. Lett. 2016;18:592–595. doi: 10.1021/acs.orglett.5b03654. [DOI] [PubMed] [Google Scholar]
- 26.Leng DJ, Black CM, Pattison G. Org. Biomol. Chem. 2016;14:1531–1535. doi: 10.1039/c5ob02468d. [DOI] [PubMed] [Google Scholar]
- 27.Hine J, Mahone LG, Liotta CL. J. Am. Chem. Soc. 1967;89:5911–5920. [Google Scholar]
- 28.Castejon HJ, Wiberg KB. J. Org. Chem. 1998;63:3937–3942. [Google Scholar]
- 29.Liu Y-L, Zhou J. Chem. Commun. 2012;48:1919–1921. doi: 10.1039/c2cc17140f. [DOI] [PubMed] [Google Scholar]
- 30.Prakash GKS, Ni C, Wang F, Hu J, Olah GA. Angew. Chem. Int. Ed. 2011;50:2559–2563. doi: 10.1002/anie.201007594. [DOI] [PubMed] [Google Scholar]
- 31.Prakash GKS, Hu J. Acc. Chem. Res. 2007;40:921–930. doi: 10.1021/ar700149s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.