

Abstract
Kaposi’s sarcoma (KS) is the most common AIDS-associated malignancy etiologically caused by Kaposi’s sarcoma-associated herpesvirus (KSHV). KS is a highly disseminated and vascularized tumor comprised of poorly differentiated spindle-shaped endothelial cells. KSHV encodes 12 pre-microRNAs (pre-miRNAs) that yield 25 mature miRNAs, but their roles in KSHV-induced tumor dissemination and angiogenesis remain largely unknown. KSHV-encoded miR-K12-6 (miR-K6) can produce two mature miRNAs, miR-K6-3p and miR-K6-5p. Recently, we have shown that miR-K6-3p promoted cell migration and angiogenesis by directly targeting SH3 domain binding glutamate-rich protein (SH3BGR) (PLoS Pathog. 2016;12(4):e1005605). Here, by using mass spectrometry, bioinformatics analysis and luciferase reporter assay, we showed that miR-K6-5p directly targeted the coding sequence (CDS) of CD82 molecule (CD82), a metastasis suppressor. Ectopic expression of miR-K6-5p specifically inhibited the expression of endogenous CD82 and strongly promoted endothelial cells invasion in vitro and angiogenesis in vivo. Overexpression of CD82 significantly inhibited cell invasion and angiogenesis induced by miR-K6-5p. Mechanistically, CD82 directly interacted with c-Met to inhibit its activation. MiR-K6-5p directly repressed CD82, relieving its inhibition on c-Met activation and inducing cell invasion and angiogenesis. Deletion of miR-K6 from KSHV genome abrogated KSHV suppression of CD82 resulting in compromised KSHV activation of c-Met pathway, and KSHV-induced invasion and angiogenesis. In conclusion, these results show that by inhibiting CD82, KSHV miR-K6-5p promotes cell invasion and angiogenesis by activating the c-Met pathway. Our findings illustrate that KSHV miRNAs may play an essential role in the dissemination and angiogenesis of KSHV-induced malignancies.
Keywords: KSHV miRNAs, Cell Invasion, Angiogenesis, CD82, c-Met
Introduction
Kaposi’s sarcoma-associated herpesvirus (KSHV) is an oncogenic virus involved in the development of several human malignancies, including Kaposi’s sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman’s disease (MCD)1. KS is a highly angiogenic vascular tumor expressing mixed cellular markers2. It is characterized by abnormally dense and irregular blood vessels, extravasated erythrocytes with hemosiderin deposits, and vast inflammatory infiltration, as well as an abundance of inflammatory cytokines (ICs), growth factors (GFs), angiogenic factors and invasive factors3. These cytokines and factors, including vascular endothelial growth factor (VEGF), interleukin 6 (IL-6), tumor necrosis factor α (TNF-α), angiogenin and matrix metalloproteinases (MMPs), might promote the angiogenic phenotype in KS lesions4–6 and contribute to the dissemination of KS involving visceral organs7, 8.
Like other herpesviruses, KSHV life cycle consists two phases: latency and lytic replication. During latency, only a few viral genes are expressed, including latency-associated nuclear antigen [LANA, open reading frame (ORF) 73], viral cyclin (vCyclin, ORF72), and viral FLIP (vFLIP, ORF71); and these genes are essential for KSHV-induced tumorigenesis. Besides these latent proteins, KSHV genome also encodes 12 precursor miRNAs (pre-miRs) that are further processed into 25 mature miRNAs, all of which are highly expressed during viral latency and in KS tumors1, 9–14.
MicroRNAs (miRNAs) are small non-coding RNAs that post-transcriptionally regulate gene expression predominantly via binding to the 3′UTRs of mRNAs, leading to translational inhibition15. The KSHV miRNAs regulate viral lytic reactivation, apoptosis, cell cycle, immune evasion, epigenetic regulation, cellular transformation, and angiogenesis by directly regulating viral and/or host genes16–44. For example, miR-K5, miR-K7-5p, miR-K9-5p, miR-K3 and miR-K4, have been reported to maintain latency by directly or indirectly targeting KSHV lytic switch protein (RTA)16, 18, 20, 21. MiR-K1 maintains NF-κB-dependent viral latency by targeting IκBα30. In addition, miR-K9 targets IRAK1 (interleukin-1 receptor-associated kinase 1) to mediate TLR/IL-1R signaling, resulting in reduced inflammation; and miR-K5 also regulates TLR/IL-1R signaling by targeting MYD88 (myeloid differentiation primary response protein 88)25. Moreover, miR-K3 and miR-K7 inhibit the expression of C/EBPβ (CCAAT/enhancer-binding protein β), a transcriptional repressor of IL-6 and IL-10, to promote cell growth of KSHV-infected cells and angiogenesis33, 45. Our recent study showed that miR-K3 induced cell migration, invasion and angiogenesis and enhanced viral latency by directly targeting G protein-coupled receptor (GPCR) kinase 2 (GRK2), modulating CXCR2/AKT signaling pathway46, 47. We also found that miR-K6-3p contributed to cell migration and angiogenesis by directly targeting SH3 domain binding glutamate-rich protein (SH3BGR), leading to the activation of STAT3 pathway48. Even though several functions of KSHV miRNAs have been revealed, the roles of these miRNAs in the development of KSHV-related malignancies, especially in KSHV-induced tumor dissemination and angiogenesis, remain unknown.
CD82 was identified as a metastasis suppressor 20 years ago49, and is now widely recognized as a biomarker in predicting metastatic potential of solid malignant tumors50. In addition to suppressing tumor metastasis, CD82 was also reported to affect tumor growth51, 52 and regulate various cellular functions, including cell adhesion, movement, senescence, proliferation, and survival53–60. Despite the fact that CD82 inhibits cell migration and cancer invasion, its role in the pathogenesis of KS remains unclear.
KSHV miR-K6 is processed into two mature miRNAs, miR-K6-3p and miR-K6-5p, which are highly expressed in KSHV latently infected B cells61 and KS tumors62. MiR-K6-3p has been shown to promote endothelial cell migration and angiogenesis, implying that miR-K6-5p might have a role in the development of KS tumors. In this study, we aimed to examine the role of miR-K6-5p on cell mobility and angiogenesis. We found that miR-K6-5p induced endothelial cell invasion and angiogenesis by directly targeting the CDS of CD82. In addition, activation of c-Met pathway was required for miR-K6-5p-induced endothelial cell invasion and angiogenesis, which was suppressed by CD82. Our findings provide further insights into the molecular mechanism by which KSHV miRNAs contribute to the pathogenesis of KSHV-associated malignancies.
Results
Ectopic Expression of miR-K6-5p Promotes Endothelial Cell Invasion and Angiogenesis
We previously reported that KSHV miR-K6-3p contributed to cell migration and angiogenesis by regulating the SH3BGR/STAT3 pathway48. Considering that miR-K6-5p was derived from the same precursor miRNA with miR-K6-3p, we wondered whether miR-K6-5p could also contribute to KSHV-induced tumor dissemination and angiogenesis. To address this issue, we first transduced HUVECs with lentivirus expressing miR-K6-5p (Figure 1a). Luciferase reporter assay showed that miR-K6-5p significantly inhibited the activity of pGL3-miR-K6-5p sensor reporter, indicating that the miR-K6-5p expression construct was functional in HUVECs (Figure 1b). Then, we conducted transwell migration and Matrigel invasion assays to determine whether miR-K6-5p had any effect on cell motility. As shown in Figures 1c and 1d, ectopic expression of miR-K6-5p in endothelial cells markedly enhanced cell invasion when compared to cells expressing the vector control. However, we did not observe any effect of miR-K6-5p on cell migration (Supplementary Figure S1). To assess the effect of miR-K6-5p on angiogenesis, we performed a tubule formation assay and found that ectopic expression of miR-K6-5p dramatically increased tube formation in HUVECs compared to control cells (Figures 1e and f). Moreover, expression of miR-K6-5p increased the levels of MMP10 and VEGFA mRNA transcripts (Figure 1g), which are known to promote cell invasion and angiogenesis.
Figure 1. Ectopic expression of miR-K6-5p promotes endothelial cells invasion and angiogenesis in vitro.
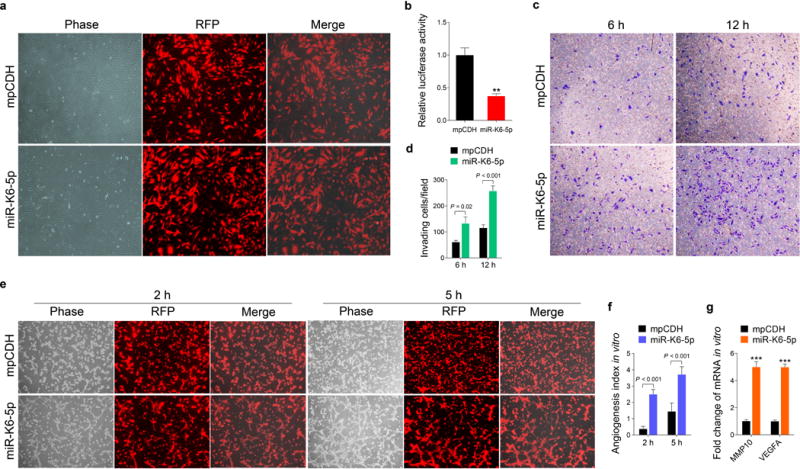
(a). HUVECs were transduced with lentivirus empty vector (mpCDH; top) and lentivirus-miR-K6-5p (miR-K6-5p; bottom), and representative images were taken under the light microscope (Phase) and fluorescent microscope (RFP) (Original magnification, ×100).
(b). Luciferase activity was detected in lentivirus empty vector (mpCDH) or lentivirus-miR-K6-5p (miR-K6-5p) transduced HUVECs followed by transfection with the pGL3-Control (Control) or the pGL3-miR-K6-5p sensor reporter (Sensor). ** P < 0.01 for Student’s t-test versus mpCDH group.
(c). Matrigel invasion assay with HUVECs transduced with lentivirus empty vector (mpCDH) or lentivirus-miR-K6-5p (miR-K6-5p). The representative images were captured at 6 h and 12 h post seeding (original magnification, ×100).
(d). The quantified results of Matrigel invasion assay in (c). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed.
(e). Microtubule formation assay for HUVECs transduced with lentivirus empty vector (mpCDH; top) or lentivirus-miR-K6-5p (miR-K6-5p; bottom). The representative images were captured under the light microscope (Phase) and fluorescent microscope (RFP) at 2 and 5 h post seeding (original magnification, ×100).
(f). The quantified results of microtubule formation assay in (e). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed.
(g). The mRNA expression levels of MMP10 and VEGFA in HUVECs treated as in (a) were determined by RT-qPCR. The quantified results represent the mean ± SD. Three independent experiments, each with four technical replicates, were performed. *** P < 0.001 for Student’s t-test versus mpCDH group.
We further determined the effect of miR-K6-5p on KSHV-induced angiogenesis in an in vivo chick chorioallantoic membrane (CAM) angiogenesis model. Expression of miR-K6-5p increased the angiogenesis index (Figures 2a and b). We also performed an in vivo Matrigel plug angiogenesis assay to confirm these observations. Hemoglobin of the plugs was strikingly increased with miR-K6-5p overexpression (Figures 2c and d and Supplementary Figure S2). Hematoxylin and eosin (H&E) staining showed extensive dense neovascularization and erythrocyte infiltration in the miR-K6-5p plugs (Figures 2e). We then examined the levels of smooth muscle actin (SMA), a marker of the vascular and lymphatic endothelial cells, and VEGFA and b-FGF, both are pro-angiogenic factors by immunohistochemistry. As shown, there were more SAM-, VEGFA- and b-FGF-positive cells in plugs induced by miR-K6-5p than those of control plugs (Figures 2e and f). Consistent with these results, the levels of MMP10 and VEGFA mRNAs were significantly elevated in plugs of miR-K6-5p-transduced HUVECs (Figure 2g). These results indicated that miR-K6-5p promoted endothelial cell invasion and angiogenesis.
Figure 2. Ectopic expression of miR-K6-5p promotes endothelial cell angiogenesis in vivo.
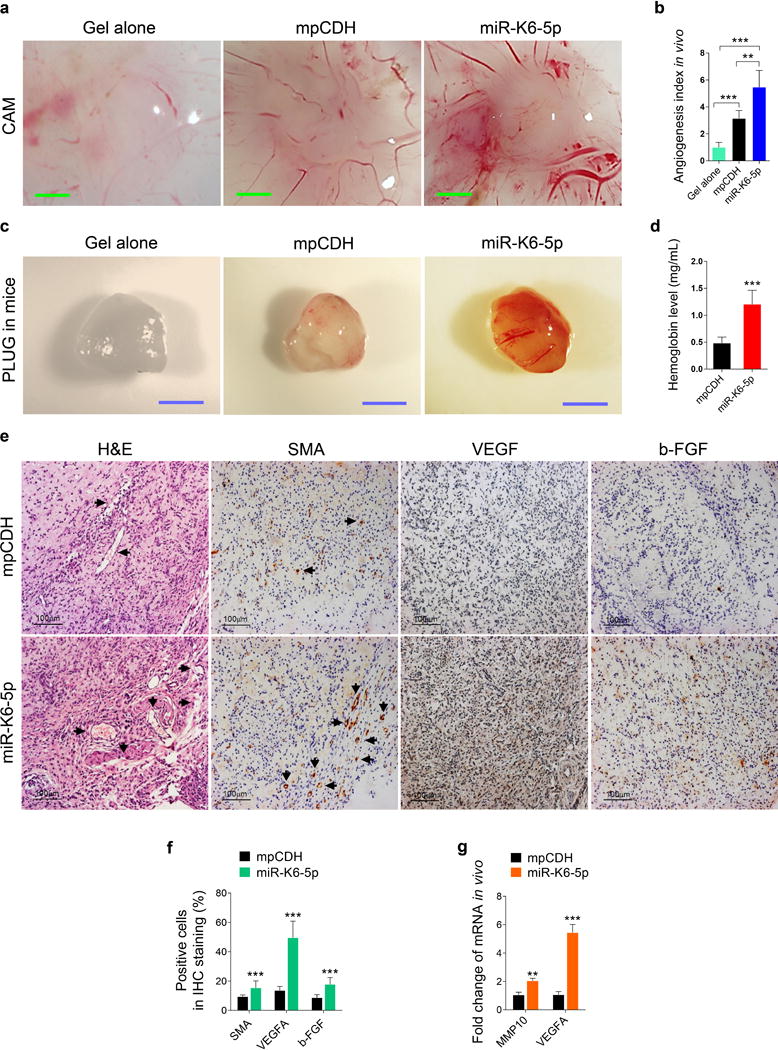
(a). HUVECs transduced with lentivirus empty vector (mpCDH) or lentivirus-miR-K6-5p (miR-K6-5p) were examined for their proangiogenic effects in a CAM model as described in the “Materials and Methods” section. Representative photographs of angiogenesis are shown.
(b). The quantification of results in (a). The number of blood vessels was normalized to that of Matrigel alone. Data represent mean ± SD, each group with at least five tumors. Three independent experiments were performed. ** P < 0.01 for Student’s t-test versus mpCDH group.
(c). HUVECs treated as in (a) were examined for their proangiogenic effects in Matrigel plug assay in nude mice as described in the “Materials and Methods” section. Representative photographs of angiogenesis in the nude mice are shown.
(d). The hemoglobin level of the Matrigel plugs treated as in (c) was determined with hemoglobin content calculated based on the standard curve. Data represent mean ± SD, each group with at least five tumors. Three independent experiments were performed. *** P < 0.001 for Student’s t-test versus mpCDH group.
(e). Hematoxylin and eosin (H&E) staining analysis of histologic features (original magnification, ×400) and immunohistochemical (IHC) staining analysis of the expression of SMA (original magnification, ×400), VEGFA (original magnification, ×400) and b-FGF (original magnification, ×400) in plugs induced by HUVECs transduced with mpCDH or miR-K6-5p. Black arrows point to neovascularization and hemorrihagic foci in H&E staining sections and the SMA in IHC staining sections, respectively.
(f). Quantification of results in (e). *** P < 0.001 for chi-square test versus mpCDH group.
(g). The mRNA expression of MMP10 and VEGFA in the Matrigel plugs treated as in (c) were determined by RT-qPCR. The quantified results represent the mean ± SD. Three independent experiments, each with four technical replicates, were performed. ** P < 0.01 and *** P < 0.001 for Student’s t-test versus mpCDH group.
miR-K6-5p Induces Cell Invasion and Angiogenesis by directly Targeting the CDS of CD82 Gene
To identify the targets that mediate miR-K6-5p-induced cell invasion and angiogenesis, we examined proteins differentially expressed in miR-K6-5p- and mpCDH-transduced HUVECs by mass spectrometry analysis. Among the downregulated cellular proteins (Table 1), five proteins had miR-K6-5p putative binding sites in their 3′UTR or CDS based on the results of bioinformatics analysis with programs including TargetScan, RNAhybrid, Findtar, and Pita (Table 2). Luciferase reporter assay indicated that miR-K6-5p suppressed luciferase activity of both CD82 and B4GALT5 CDS reporter (Figure 3a). However, miR-K6-5p can only reduce CD82 protein expression (Figure 3b), which was chosen for further validation. Moreover, the mRNA level of CD82 was markedly down-regulated in miR-K6-5p-expressing HUVECs compared to cells expressing the control vector (Figure 3c). In KSHV-infected HUVECs, both protein and mRNA levels of CD82 were also dramatically reduced compared to the mock-infected control (Figures 3d and e). Consistent with these observations, there were less CD82-postive cells in KS lesion compared to the normal skin tissue as shown by IHC staining (Figures 3f and g).
Table 1.
Cellular proteins downregulated >1.33 folds in HUVECs infected with miR-K6-5p.
| Protein name | Fold | Protein name | Fold | Protein name | Fold | Protein name | Fold |
|---|---|---|---|---|---|---|---|
| FAM213A | −3.62±0.12 | ADAM10 | −2.18±0.07 | FBXL6 | −1.84±0.01 | USP33 | −1.50±0.06 |
| C19orf43 | −3.26±0.09 | CD47 | −2.16±0.15 | COX6A1 | −1.83±0.15 | GANAB | −1.49±0.08 |
| NRD1 | −2.91±0.21 | COX20 | −2.13±0.10 | L1CAM | −1.81±0.05 | LEPRE1 | −1.49±0.07 |
| PSME4 | −2.87±0.15 | MPZL1 | −2.10±0.14 | AASS | −1.79±0.13 | JAK1 | −1.49±0.04 |
| RETSAT | −2.77±0.19 | PDCL3 | −2.08±0.13 | SPCS1 | −1.74±0.07 | EIF4A2 | −1.49±0.06 |
| INTS9 | −2.67±0.19 | BRIX1 | −2.05±0.19 | SPAG9 | −1.69±0.09 | PPIC | −1.47±0.03 |
| ZFPL1 | −2.66±0.17 | INTS5 | −2.03±0.12 | PFKL | −1.65±0.11 | CHD1 | −1.45±0.06 |
| CNOT3 | −2.65±0.06 | WDR36 | −2.00±0.22 | VPS29 | −1.64±0.07 | ECE1 | −1.43±0.05 |
| SLC7A6 | −2.59±0.06 | NO66 | −1.99±0.18 | LUC7L | −1.63±0.06 | LGALS1 | −1.42±0.01 |
| DNMT1 | −2.52±0.23 | C8orf33 | −1.97±0.21 | NDUFAF1 | −1.63±0.12 | ADAM9 | −1.40±0.03 |
| RAB5C | −2.48±0.26 | RIT1 | −1.96±0.10 | DHCR24 | −1.61±0.12 | NMES1 | −1.38±0.01 |
| KIAA0754 | −2.46±0.20 | KATNAL2 | −1.90±0.10 | CCNB2 | −1.60±0.06 | RFTN1 | −1.38±0.02 |
| ATP6AP2 | −2.35±0.19 | TRIM23 | −1.90±0.09 | P4HA2 | −1.56±0.05 | PLOD2 | −1.37±0.01 |
| TCF3 | −2.30±0.05 | CD82 | −1.89±0.02 | PLOD1 | −1.55±0.06 | CALR | −1.36±0.02 |
| RNMT | −2.30±0.09 | DCTN1 | −1.88±0.19 | CD320 | −1.54±0.03 | APP | −1.34±0.01 |
| RALGAPB | −2.23±0.16 | ARPC3 | −1.88±0.17 | B4GALT5 | −1.53±0.05 | ||
| C14orf166 | −2.23±0.26 | ECT2 | −1.84±0.19 | CFDP1 | −1.52±0.05 |
Table 2.
Fold changes for selected proteins identified in miR-K6-5p-transduced HUVECs using TMT quantification.
| Protein name | Fold change
|
Function | Target prediction
|
|||||
|---|---|---|---|---|---|---|---|---|
| Rep1 | Rep2 | Rep3 | Seed | Site | △G | Recommendation | ||
| TCF3 | 0.37 | 0.45 | 0.43 | Stem cell self renewal | 8:0:0 | 3′UTR | −14.2 | excellent |
| CD82 | 0.54 | 0.50 | 0.55 | Tumor metastasis | 8:1:0 | CDS | −20.02 | excellent |
| B4GALT5 | 0.71 | 0.60 | 0.65 | Glioma-initiating cell self renewal | 8:1:0 | CDS | −20.84 | excellent |
| 8:1:0 | 3′UTR | −18.44 | excellent | |||||
| APP | 0.76 | 0.74 | 0.74 | Alzheimer’s disease | 8:1:0 | 3′UTR | −11.9 | OK |
| GANAB | 0.76 | 0.61 | 0.64 | Lung tumor | 8:1:0 | 3′UTR | −18.7 | excellent |
Figure 3. CD82 expression is downregulated in miR-K6-5p-expressing HUVECs and KS lesion samples.
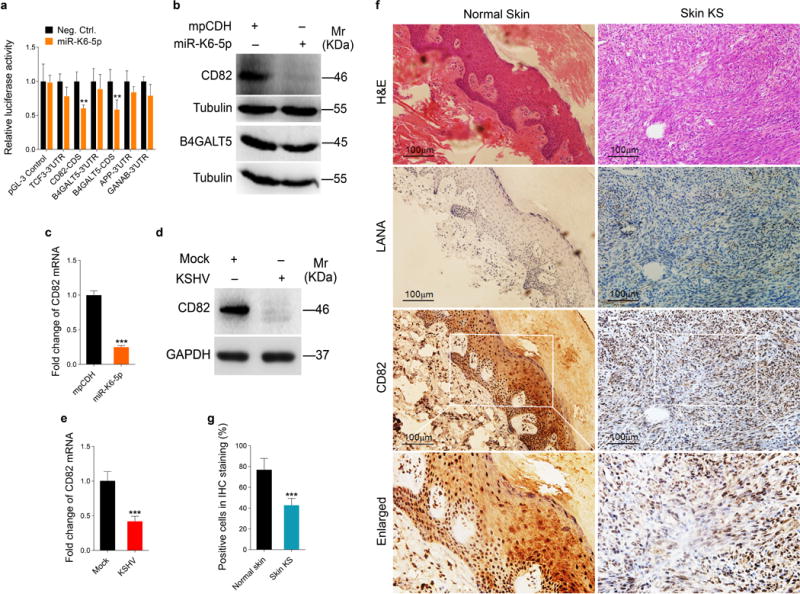
(a). Luciferase activity was detected in HEK293T cells co-transfected by a mimic of miR-K6-5p (miR-K6-5p) or a negative control nucleotide (Neg. Ctrl.) together with pGL3-Control (pGL3-Control), pGL3-TCF3 3′UTR luciferase reporter (TCF3-3′UTR), pGL3-CD82 CDS luciferase reporter (CD82-CDS), pGL3-B4GALT5 3′UTR luciferase reporter (B4GALT5-3′UTR), pGL3-B4GALT5 CDS luciferase reporter (B4GATL5-CDS), pGL3-APP 3′UTR luciferase reporter (APP-3′UTR) or pGL3-GANAB 3′UTR luciferase reporter (GANAB-3′UTR) for 24 h. The relative reporter activity level of the Neg. Ctrl. group was set as “1” for comparison. ** P < 0.01 for Student’s t-test versus Neg. Ctrl. group.
(b). The expression of CD82 and B4GALT5 protein in HUVECs transduced with lentivirus empty vector (mpCDH) or lentivirus-miR-K6-5p (miR-K6-5p) was detected by Western-blotting. Results shown were from a representative experiment of three independent experiments with similar results.
(c). The mRNA level of CD82 in HUVECs transduced with lentivirus empty vector (mpCDH) or lentivirus-miR-K6-5p (miR-K6-5p) was examined by qPCR. The quantified results represent the mean ± SD. Three independent experiments, each with three technical replicates, were performed. *** P < 0.001 for Student’s t-test versus mpCDH group.
(d). Western-blotting analysis of CD82 protein in KSHV-infected HUVECs (KSHV) as described in the ‘Materials and Methods’ section or in HUVECs treated with PBS as the negative control (Mock). Results shown were from a representative experiment of three independent experiments with similar results.
(e). qPCR analysis for CD82 mRNA in KSHV-infected HUVECs (KSHV) as described in the ‘Materials and Methods’ section or in HUVECs treated with PBS as the negative control (Mock). The quantified results represent the mean ± SD. Three independent experiments, each with four technical replicates, were performed. *** P < 0.001 for Student’s t-test versus Mock group.
(f). Hematoxylin and eosin (H&E) staining of KS lesion (right) and normal skin (left) to show histologic features (top panel; original magnification, ×200) and immunohistochemical (IHC) staining of KSHV LANA and CD82 (middle and bottom panels, respectively; original magnification, ×200).
(g). Quantification of results in (f). *** P < 0.001 for chi-square test versus Normal skin group.
As shown in Figure 4a, miR-K6-5p inhibited the reporter activity of CD82 CDS in a dose-dependent fashion. RT-qPCR and Western blotting confirmed that miR-K6-5p attenuated the expression of CD82 in a dose-dependent manner (Figures 4b and c). To evaluate the direct effect of miR-K6-5p on CD82, we performed cotransfection of miR-K6-5p mimics along with a CD82 expression plasmid and a pEGFP-N2 plasmid into HUVECs and HEK293T cells, respectively. Western blotting indicated that miR-K6-5p strongly inhibited the expression of exogenous CD82 in a dose-dependent manner but not that of EGFP, which was used as a control for transfection efficiency (Figure 4d).
Figure 4. The CD82 CDS is directly targeted by miR-K6-5p.
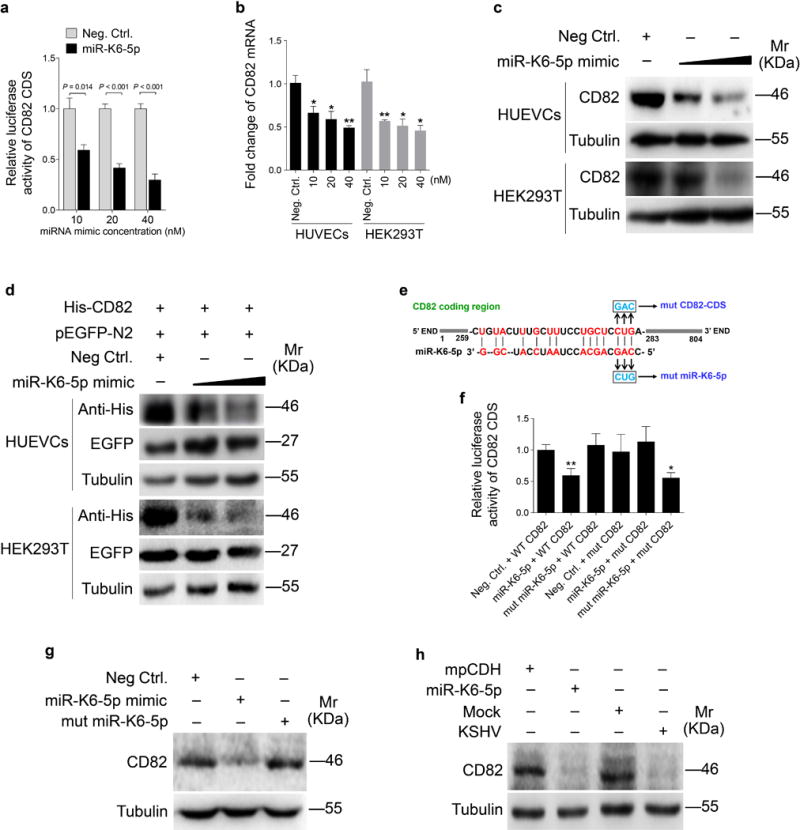
(a). Luciferase assay with HEK293T cells co-transfected with the pGL3-CD82 CDS reporter together with an increasing amount (10, 20, and 40 nM) of negative control nucleotide (Neg. Ctrl.) or a mimic of miR-K6-5p (miR-K6-5p) for 24 h.
(b). The mRNA level of CD82 in HUVECs and HEK293T transfected with an increasing amount (10, 20, and 40 nM) mimic of miR-K6-5p or a negative control for 48 h was examined by qPCR. The quantified results represent the mean ± SD. Three independent experiments, each with four technical replicates, were performed. * P < 0.05 and ** P < 0.01 for Student’s t-test versus Neg. Ctrl. group.
(c). HUVECs and HEK293T cells transfected with an increasing amount (20 and 40 nM) mimic of miR-K6-5p or negative control for 48 h. The transfected cells were collected and Western-blotting was performed with the indicated antibodies. Results shown were from a representative experiment of three independent experiments with similar results.
(d). miR-K6-5p inhibited the expression of exogenous CD82 protein by targeting its CDS. Western-blotting was performed with HUVECs and HEK293T cells co-transfected by pHAGE-CD82, together with pEGFP and increasing amounts (20 and 40 nM) mimic of miR-K6-5p.
(e). Schematic illustration of the putative seed sequences of miR-K6-5p complementary with CD82 CDS and mutagenesis of the putative binding sites in the CDS of CD82.
(f). Effect of mutation of the putative binding site on the CD82 CDS reporter. After co-transfection of CD82 wild type CDS (WT CD82) or the mutant CD82 CDS reporter construct (mut CD82) together with a negative control nucleotide of miRNA (Neg. Ctrl.), a mimic of miR-K6-5p (miR-K6-5p) or a mutant mimic of miR-K6-5p (mut miR-K6-5p) for 24 h in HEK293T cells, cells were collected and assayed for luciferase activity. * P < 0.05 and ** P < 0.01 for Student’s t-test versus Neg. Ctrl. group.
(g). Mutant miR-K6-5p failed to target endogenous CD82 in HUVECs. A miRNA negative control nucleotide (Neg. Ctrl.), a mimic of miR-K6-5p (miR-K6-5p mimic; 40 nM) or a mutant mimic of miR-K6-5p (mut miR-K6-5p) lacking the seed sequences were transfected into HUVECs for 48 h, respectively. The transfected cells were collected and Western-blotting was performed with the indicated antibodies. Results shown were from a representative experiment of three independent experiments with similar results.
(h). Inhibition of CD82 expression by miR-K6-5p following transfection with a mimic (40 nM) and by KSHV following infection. Results shown were from a representative experiment of three independent experiments with similar results.
To further confirm that CD82 is the direct target of miR-K6-5p, we performed mutagenesis with the CD82 CDS reporter and miR-K6-5p (Figure 4e). Mutation of the putative binding site abolished the inhibitory effect of miR-K6-5p on CD82 CDS reporter activity; however, the mutant mimic lacking the seed sequence failed to inhibit the CD82 CDS reporter activity (Figure 4f). The mutant mimic designed to match the CD82 CDS mutation (Figure 4e) exhibited a strong inhibitory effect on the mutant CDS reporter (Figure 4f). Consistent with these results, mimic of miR-K6-5p suppressed the expression of endogenous CD82 in HUVECs while the mutant mimic of miR-K6-5p lacking the seed sequence did not (Figure 4g). The expression level of CD82 in HUVECs transfected with miR-K6-5p mimic was similar to that of KSHV infection (Figure 4h). These results indicated that miR-K6-5p directly targeted the CD82 CDS.
To examine the role of CD82 in miR-K6-5p-induced cell invasion and angiogenesis, miR-K6-5p-expressing HUVECs were transduced with lentivirus-CD82 and further analyzed for invasion and angiogenesis activities. Overexpression of CD82 significantly abolished miR-K6-5p-induced invasion at 6 and 12 h post-seeding (Figures 5a and b). Meanwhile, overexpression of CD82 blocked tube formation of HUEVCs induced by miR-K6-5p (Figures 5c and d). Consistently, overexpression of CD82 decreased the expression of MMP10 and VEGFA on mRNA levels (Figure 5e). Western blotting confirmed the suppression of endogenous CD82 by miR-K6-5p (Lane 3 vs lane 1 in Figure 5f). Transduction with lentivirus-CD82 increased CD82 expression (Lanes 2 and 4 in Figure 5f). Furthermore, CAM and Matrigel plug assays showed that overexpression of CD82 inhibited miR-K6-5p-induced angiogenesis in vivo (Figures 5g–j and Supplementary Figure S3). Consistent with these observations, overexpression of CD82 reduced the expression of MMP10 and VEGFA transcripts in miR-K6-5p-induced plugs (Figure 5k).
Figure 5. Overexpression of CD82 inhibits miR-K6-5p-induced cell invasion and angiogenesis in vitro and in vivo.
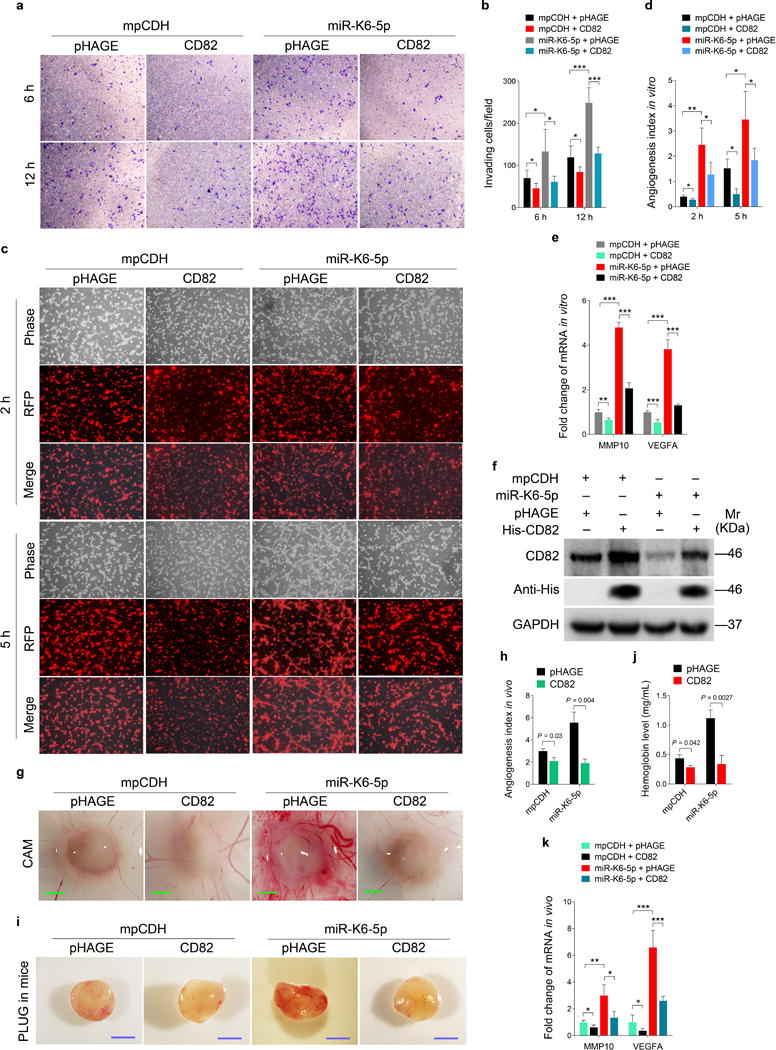
(a). Matrigel invasion assay for HUVECs transduced with lentivirus-mediated miR-K6-5p (miR-K6-5p) or empty vector (mpCDH), which were subsequently co-transduced with lentivirus-CD82 (CD82) or its control pHAGE (pHAGE), respectively. The representative images were captured at 6 h and 12 h post seeding (original magnification, ×100).
(b). Quantification of results in (a). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05 and *** P < 0.001 for Student’s t-test.
(c). Microtubule formation assay for HUVECs treated as in (a). The representative images were captured at 2 h and 5 h post seeding (original magnification, ×100).
(d). Quantification of results in (c). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05 and ** P < 0.01 for Student’s t-test.
(e). The mRNA expression levels of MMP10 and VEGFA in HUVECs treated as in (a) were determined by qPCR. The quantified results represent the mean ± SD. Three independent experiments, each with four technical replicates, were performed. ** P < 0.01 and *** P < 0.001 for Student’s t-test.
(f). Western blotting was performed in HUVECs treated as in (a) with the indicated antibodies. The antibody against His-tag was used to detect the exogenous expression of CD82. Results shown were from a representative experiment of three independent experiments with similar results.
(g). HUVECs treated as in (a) were examined for their proangiogenic effects in CAM model as described in the “Materials and Methods” section. Representative photographs of angiogenesis are shown.
(h). Quantification of results in (g). The number of blood vessels was normalized to that of Matrigel alone. Data represent mean ± SD, each group with at least five tumors. Three independent experiments were performed and similar results were obtained.
(i). HUVECs treated as in (a) were examined for their proangiogenic effects in Matrigel plug assay in nude mice as described in the “Materials and Methods” section. Representative photographs of angiogenesis in the nude mice are shown.
(j). The hemoglobin level of the Matrigel plugs treated as in (I) was determined with hemoglobin content calculated based on the standard curve. Data represent mean ± SD, each group with six tumors (n=6). Three independent experiments were performed and similar results were obtained.
(k). The mRNA expression levels of MMP10 and VEGFA in the Matrigel plugs treated as in (I) were determined by qPCR. The quantified results represent the mean ± SD. Three independent experiments, each with four technical replicates, were performed. * P < 0.05, ** P < 0.01 and *** P < 0.001 for Student’s t-test.
Suppression of CD82 Activates the c-Met Signaling, Which is Required for miR-K6-5p-Induced Cell Invasion and Angiogenesis
It has been reported that CD82 inhibited the HGF receptor c-Met by attenuating the crosstalk between c-Met and integrin, and integrin-dependent crosstalk between c-Met and Src, leading to reduction of invasion and growth of cells63, 64. CD82 and c-Met were also reported to co-localize at the cell periphery and form a complex65. Given these findings, we reasoned that c-Met may also be involved in CD82 mediated miR-K6-5p-induced cell invasion and angiogenesis. Indeed, KSHV latently infected HUVECs had increased the level of c-Met phosphorylation66. Importantly, expression of miR-K6-5p alone in endothelial cells was sufficient to increase c-Met phosphorylation (Supplementary Figure S4a). Overexpression of CD82 in miR-K6-5p-expressing HUVECs inhibited c-Met activation (Supplementary Figure S4b), indicating that CD82 down-regulated c-Met activity in endothelial cells. We examined the physiological interaction between CD82 and c-Met in the current system. Co-immunoprecipitation assay indeed showed that CD82 interacted with c-Met in endothelial cells (Supplementary Figures S4c–f). Overexpression of CD82 increased the amount of CD82-immunoprecipiated c-Met and reduced the level of activated c-Met (Supplementary Figures S4c and d). Similarly, interaction of CD82 and c-Met was also observed in c-Met-transfected HUVECs (Supplementary Figures S4e and f). Consistently, overexpression of CD82 reduced the level of activated c-Met. Under this condition, we could detect colocalization of CD82 with c-Met by confocal microscope (Supplementary Figure S4g). In addition, knockdown of CD82 with shRNAs was sufficient to increase the phosphorylated c-Met level (Supplementary Figures S4g and 5). These data indicated that CD82 directly interacted with c-Met to inhibit its activation, and miR-K6-5p directed repression of CD82 to relieve its inhibition of c-Met phosphorylation, resulting in higher level of c-Met activation.
Next, we explored whether c-Met activation mediated miR-K6-5p-induced cell invasion and angiogenesis. As expected, transduction of a mixture of shRNAs to knock down c-Met expression (shc-Met; Figure 6a and Supplementary Figure S6) reduced miR-K6-5p-induced invasion and tube formation (Figures 6b and c), as well as levels of MMP10 and VEGFA transcripts (Figure 6d). Matrigel plug assay showed that knock down of c-Met diminished miR-K6-5p-induced angiogenesis in vivo (Figure 6e), and blocked miR-K6-5p induction of MMP10, and VEGFA (Figure 6f). We further used a selective c-Met inhibitor PF-2341066 to confirm the role of c-Met activation in miR-K6-5p-induced cell invasion and angiogenesis. PF-2341066 not only decreased the level of phosphorylated c-Met (Figure 6g) but also inhibited cell invasion and tube formation (Figures 6h and i) in HUVECs transduced with miR-K6-5p. Collectively, these results suggest that activation of the c-Met pathway mediated miR-K6-5p-induced cell invasion and angiogenesis.
Figure 6. Activation of c-Met, which is negatively regulated by CD82, contributes to miR-K6-5p-induced endothelial cell invasion and angiogenesis.
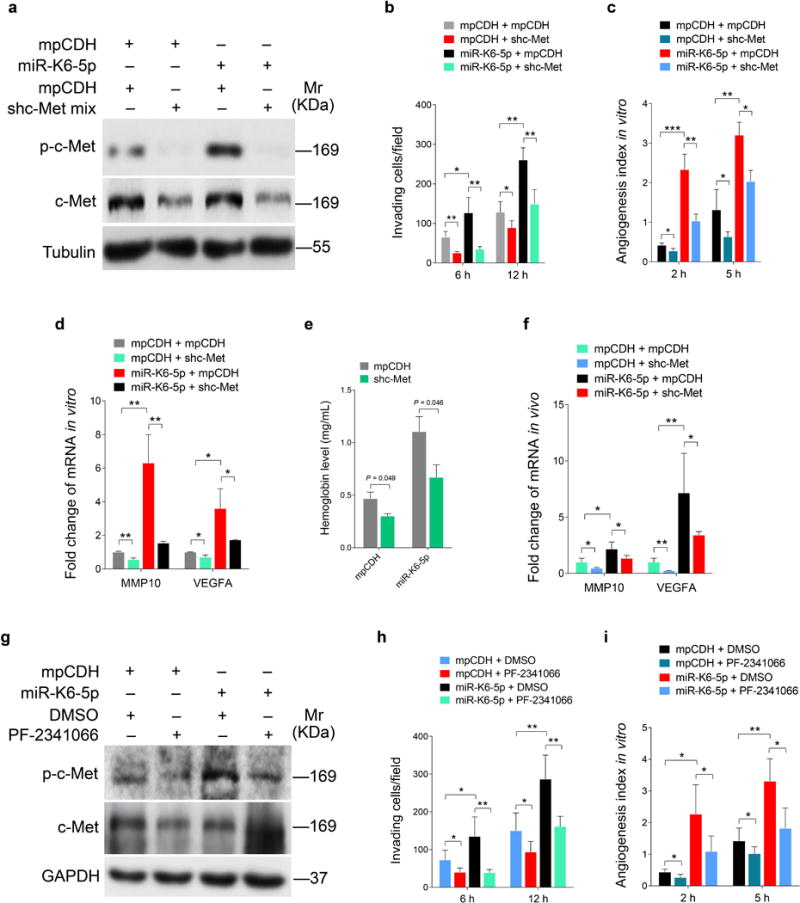
(a). Western-blotting analysis of phosphorylated c-Met in HUVECs transduced with lentivirus-mediated empty vector (mpCDH) or miR-K6-5p (miR-K6-5p), and further transduced with lentivirus-mediated a mixture of short hairpin RNAs targeting c-Met (shc-Met). Results shown were from a representative experiment of three independent experiments with similar results.
(b). Matrigel invasion assay for HUVECs treated as in (a). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05 and ** P < 0.01 for Student’s t-test.
(c). Microtubule formation assay for HUVECs treated as in (a). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05, ** P < 0.01 and *** P < 0.001 for Student’s t-test.
(d). The mRNA expression levels of MMP10 and VEGFA in HUVECs treated as in (a) were determined by qPCR. The quantified results represent the mean ± SD. Three independent experiments, each with three technical replicates, were performed. * P < 0.05 and ** P < 0.01 for Student’s t-test.
(e). HUVECs treated as in (a) were examined for their proangiogenic effects in Matrigel plug assay in nude mice as described in the “Materials and Methods” section. The hemoglobin levels of the Matrigel plugs were determined with hemoglobin content calculated based on the standard curve. Data represent mean ± SD, each group with at least six tumors. Three independent experiments were performed and similar results were obtained.
(f). The mRNA expression levels of MMP10 and VEGFA in the Matrigel plugs treated as in (e) were determined by qPCR. The quantified results represent the mean ± SD. Three independent experiments, each with four technical replicates, were performed. * P < 0.05 and ** P < 0.01 for Student’s t-test.
(g). Western-blotting analysis of phosphorylated c-Met and total c-Met in HUVECs transduced with lentivirus-mediated empty vector (mpCDH) or miR-K6-5p (miR-K6-5p) and further treated with the c-Met inhibitor, PF-2341066 (PF-2341066) or its control (DMSO). Results shown were from a representative experiment of three independent experiments with similar results.
(h). Matrigel invasion assay for HUVECs treated as in (g). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05 and ** P < 0.01 for Student’s t-test.
(i). Microtubule formation assay for HUVECs treated as in (g). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05 and ** P < 0.01 for Student’s t-test.
Deletion of miR-K6 from KSHV Genome Attenuates KSHV-induced Cell Invasion and Angiogenesis
In our previous study, we have already shown that deleted miR-K6 from KSHV genome inhibited KSHV-induced cell migration and tube formation48. Here, we found that deleted miR-K6 also decreased the levels of cell invasion (Figures 7a and b). Importantly, cells infected by the mutant virus increased CD82 expression and reduced c-Met phosphorylation compared to cells infected by the WT virus (Figure 7c). As we previously showed, cells infected by the mutant virus had decreased level of VEGFA. Compared to cells infected by the WT virus, those cells infected by the mutant virus also had a decreased level of MMP10 mRNA but an increased level of CD82 mRNA (Figure 7d). Furthermore, inhibition of miR-K6-5p by transfecting a specific inhibitor into WT KSHV-infected HUVECs reduced cell invasiveness and the extent of angiogenesis (Supplementary Figure S7).
Figure 7. Deletion of miR-K6 from KSHV genome attenuates KSHV induction of endothelial cell invasion and angiogenesis.
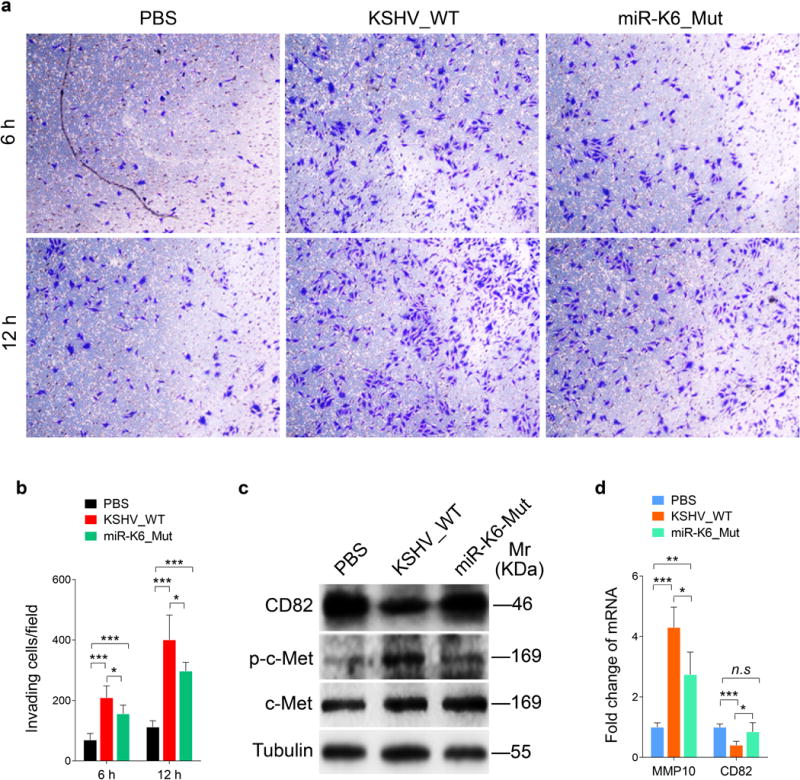
(a). Matrigel invasion assay for HUVECs were treated with PBS (PBS), infected with BAC16 KSHV wide type virus (KSHV_WT) or BAC16 KSHV miR-K6 deletion mutant virus (miR-K6_Mut). The representative images were captured at 6 h and 12 h post seeding (original magnification, ×100).
(b). Quantification of the results in (a). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05 and *** P < 0.001 for Student’s t-test.
(c). Western-blotting analysis of expression of CD82, phosphorylated c-Met and total c-Met in HUVECs treated as in (a) with the indicated antibodies. Results shown were from a representative experiment of three independent experiments with similar results.
(d). The mRNA levels of MMP10 and CD82 in HUVECs treated as in (a) were determined by RT-qPCR. The quantified results represent the mean ± SD. Three independent experiments, each with four technical replicates, were performed. * P < 0.05, ** P < 0.01 and *** P < 0.001 for Student’s t-test. n.s, not significant.
Interestingly, knockdown of CD82 with a mixture of shRNAs (shCD82) in mutant cells was sufficient to increase the level of activated c-Met (Figure 8a and Supplementary Figure S5), and partially increased cell invasiveness and the extent of angiogenesis (Figures 8b and c). Consistent with these results, overexpression of c-Met in miR-K6_mut-infected cells increased cell invasion and angiogenesis (Figures 8d–f). Overexpression of miR-K6-5p in the mutant cells also increased in cell invasion and angiogenesis (Supplementary Figure S8).
Figure 8. CD82/c-Met signaling partially mediates miR-K6-induced endothelial cell invasion and angiogenesis.
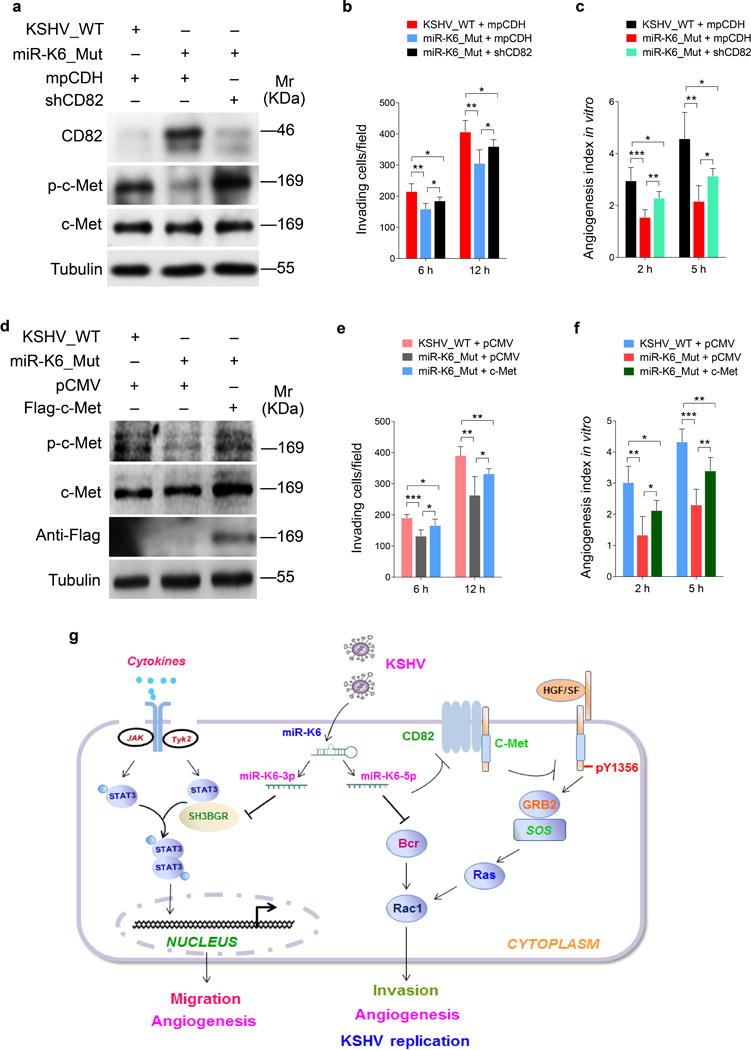
(a). Western blotting analysis of expression of CD82, phosphorylated c-Met and total c-Met in HUVECs infected with BAC16 KSHV wide type virus (KSHV_WT) or BAC16 KSHV miR-K6 deletion mutant virus (miR-K6_Mut) and further transduced with lentiviruses expressing a mixture of short hairpin RNAs targeting CD82 (shCD82). Results shown were from a representative experiment of three independent experiments with similar results.
(b). Matrigel invasion assay for HUVECs treated as in (a). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05 and ** P < 0.01 for Student’s t-test.
(c). Microtubule formation assay for HUVECs treated as in (a). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05, ** P < 0.01 and *** P < 0.001 for Student’s t-test.
(d). Western-blotting analysis of expression of phosphorylated c-Met and total c-Met in HUVECs infected with BAC16 KSHV wide type virus (KSHV_WT) or BAC16 KSHV miR-K6 deletion mutant virus (miR-K6_Mut) and further transfected with pCMV3-Flag-c-Met construct (c-Met) or its control pCMV3-C-Flag (pCMV). The antibody against Flag-tag was used to detect the exogenous expression of c-Met. Results shown were from a representative experiment of three independent experiments with similar results.
(e). Matrigel invasion assay for HUVECs treated as in (d). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05 and ** P < 0.01 for Student’s t-test.
(f). Microtubule formation assay for HUVECs treated as in (d). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05, ** P < 0.01, and *** P < 0.001 for Student’s t-test.
(g). Schematic illustration of the mechanism by which miR-K6-5p promotes endothelial cell invasion and angiogenesis. In normal endothelial cells, CD82 binds to c-Met resulting in inhibition of c-Met activation. During KSHV infection, miR-K6-5p targets and inhibits CD82 expression releasing c-Met from CD82 inhibition, leading to the activation of c-Met pathway, and ultimately promotes endothelial cell invasion and angiogenesis.
Taken together, these results indicated that in the context of KSHV infection, miR-K6-5p promoted cell invasion and angiogenesis by targeting CD82 to activate the c-Met pathway.
Discussion
CD82, a protein of 267 amino acids, is a type III transmembrane protein belonging to the tetraspanin superfamily, which is widely distributed in normal tissues50. CD82 contains four transmembrane domains, an N and a C cytoplasmic domain, a large extracellular loop (LEL), a small extracellular and an intercellular loop53. The transmembrane domains contain three highly conserved polar residues, an Asn, a Gln, and a Glu polar residue; they interact with each other and with transmembrane domains of other tetraspanins67. CD82 was initially identified as a metastasis suppressor of prostate cancer, and later reported to suppress metastasis in a variety of solid malignant tumors, such as colon, gastric, hepatocarcinoma, thyroid and breast cancers68. However, whether CD82 expression is involved in the development and progression of KS has not been investigated. In this study, we found that CD82 expression in KS tumors was lower than in normal skin tissues. We also showed that KSHV infection reduced CD82 expression. In addition, KSHV miR-K6-5p directly targeted CD82 to suppress its expression. In the context of KSHV infection, miR-K6-5p reduced the expression of metastasis suppressor CD82, and induced cell invasion and angiogenesis, which might contribute to the development and progression of KS.
c-Met, encoded by proto-oncogene Met, is a receptor tyrosine kinase mainly expressed in epithelial and endothelial cells69. c-Met is overexpressed in a variety of human tumors, such as thyroid, gastric, pancreatic, breast, prostate, cervical and endometrial cancers70–76. c-Met is the only known high-affinity receptor for hepatocyte growth factor (HGF), also known as scatter factor (SF)77, 78. Upon binding with HGF, c-Met activates a wide range of pathways, including those involved in regulating tumor invasion, metastasis and angiogenesis79. A previous study reported that KSHV activates the HGF/c-Met pathway in vitro and in vivo. A c-Met inhibitor induced apoptosis of KSHV-positive PEL cells and effectively suppressed PEL tumor progression in vivo by inducing cell cycle arrest and DNA damage66. These reports imply that the HGF/c-Met pathway might be a potential therapeutic target for KSHV-induced tumors. In this report, we found that KSHV latently infected HUVECs had increased level of c-Met phosphorylation. Moreover, we demonstrated that ectopic expression of miR-K6-5p alone in HUVECs was sufficient to induce c-Met signaling while deletion of miR-K6 from the KSHV genome significantly decreased the phosphorylation level of c-Met in KSHV-infected endothelial cells. Thus, miR-K6-5p promotes cell invasion and angiogenesis in part by inducing aberrant c-Met signaling during KSHV infection. Our study is the first report to show that the activation of c-Met pathway is important for the pathogenesis of KS, and reveal a novel role of c-Met signaling in the metastasis and angiogenesis of KSHV-associated tumors.
CD82 has been demonstrated to suppress integrin-induced invasion by inhibiting integrin-dependent crosstalk with c-Met receptor and Src kinases63. It is also reported that CD82 inhibits HGF-stimulated phosphorylation of c-Met at Y1313 and Y1365 without affecting c-Met expression80. CD82 expression selectively attenuated c-Met signaling via phosphatidylinositol 3-kinase/Akt signaling but had no effect on the activity of the MAPK pathway80. CD82 inhibited HGF-dependent c-Met activity by forming a complex with GM264, and CD82 and c-Met could form a complex to attenuate c-Met signaling by inhibiting adapter protein binding to the c-Met tyrosine residue65. These findings suggests that in normal cells, CD82, which presents in highly quantities, binds to c-Met, leading to reduced c-Met activation, hence inhibiting the progression of human tumors. In this study, we showed that KSHV miR-K6-5p suppressed CD82 expression to release c-Met leading to its activation, therefore contributing to KSHV-induced cell invasion and angiogenesis.
Several studies have identified functions of KSHV pre-miR-K6. For example, miR-K6-5p silenced the cellular transcription factor MAF (musculoaponeurotic fibrosarcoma oncogene homolog) to induce endothelial cell reprogramming, and thereby contributed to KSHV-induced oncogenesis81. miR-K6-5p also increased angiogenesis in vitro by targeting breakpoint cluster region (Bcr)39. On the other hand, miR-K6-3p, derived from the same precursor miRNA with miR-K6-5p, inhibited TGF-β activity by directly suppressing the expression of tumor suppressor thrombospondin 1 (THBS1) to regulate cell adhesion, migration, and angiogenesis35. In addition, miR-K6-3p contributed to KSHV-induced angiogenesis by regulating SH3BGR/STAT3 pathway48. In the current study, we showed that either inhibition of CD82 or overexpression of c-Met did not totally reverse KSHV-induced cell invasion and angiogenesis. These results were consistent with the previous studies, indicating that besides the CD82/c-Met pathway, pre-miR-K6 might target other cellular genes to induce cell invasion and angiogenesis.
Activation of c-Met results in the recruitment of effector proteins such as growth-factor-receptor-bound protein 2 (Grb2)82. Grb2 binding is linked to c-Met downstream signaling pathways, including key components of oncogenic signaling Ras, Rac1 and p21-activated kinase (PAK) involved in cell proliferation, cytoskeletal rearrangement and motility83. miR-K6-5p has been shown to increase Rac1 activity by repressing Bcr expression39. In the current study, by directly inhibiting CD82 expression, miR-K6-5p might also contribute to activation of Rac1-PAK pathway by enhancing c-Met activation (Figure 8g).
Interestingly, we found that ectopic expression of miR-K6-5p alone did not affect the activity of cell migration (Supplementary Figure S1), and in our previous study, miR-K6-3p was shown to have no effect on cell invasion48. However, deletion of pre-miR-K6 from the KSHV genome abolished miR-K6-5p signaling, which was sufficient to suppress KSHV-induced cell migration and invasion. Therefore, by encoding two synergistic mature miRNAs with different functions, pre-miR-K6 might enhance the dissemination and invasion of KSHV-associated tumors.
It was initially believed that functional mammalian miRNA recognition sequences might be located preferentially in the 3′UTR of target mRNAs. However, more and more miRNA target sites within coding regions have been found in recent studies84. In this study, we have also found that miR-K6-5p binding site is located in the coding region of CD82 rather than 3′UTR.
In summary, we have revealed that miR-K6-5p directly targets the CD82/c-Met pathway to promote endothelial cell invasion and angiogenesis (Figure 8g). The current findings further extend the regulatory networks of KSHV miRNAs and their multiple targets. The essential roles of KSHV miRNAs in KSHV-induced tumor dissemination and angiogenesis illustrate that these miRNAs and their regulated proteins and pathways could serve as novel therapeutic targets of KSHV-associated cancers.
Materials and Methods
Cell Culture
Primary human umbilical vein endothelial cells (HUVECs) were isolated from human umbilical cords by digesting the interior of the umbilical vein with collagenase (Sigma, St. Louis, MO, USA) and cultured in complete EBM-2 culture media (LONZA, Allendale, NJ, USA)85. HUVECs were used between passage 3 and 6. ISLK-BAC16 cells and iSLK-BAC16△miR-K6 cells were maintained as previously described86. HEK293T were obtained from American Type Culture Collection (ATCC, Rockville, MD) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) +10% FBS. All cells were cultured at 37°C in a humidified, 5% CO2 atmosphere.
Plasmids
The construct pGL3-CD82 CDS and the mutant CD82 CDS construct were generated by cloning the full length and mutant CD82 CDS sequences into the downstream of the luciferase sequence in the pGL3-Control plasmid (Promega, Shanghai, China), respectively. Human CD82 gene with a His tag at the C-terminus (pHAGE-CD82) was generated using the cDNA of HUVECs as PCR templates and inserted into the lentiviral transferring plasmid pHAGE as previously described87, 88. The miRNA (miR-K6-5p) expressing plasmid was constructed by two steps: First, the miR-30 precursor stem-loops plus RFP coding sequences were amplified from the pTRIPZ plasmid (Open Biosystems, AL, USA) and inserted into another lentiviral plasmid pCDH-CMV-MCS-EF1-copGFP (System Bioscience, CA, USA) to create a new lentiviral plasmid, which has GFP and RFP two signals and was designed as modified pCDH (mpCDH)46. Second, the precursor stem-loops of miR-K6-5p was amplified using primers (forward) 5′-CAG AAG GCT CGA GAA GGT ATA TTG CTG TTG ACA GTG AGC G-3′ and (reverse) 5′-CTA AAG TAG CCC CTT GAA TTC CGA GGC AGT AGG CA-3′ and cloned into mpCDH. Meanwhile, the mpCDH plasmid was used for the short hairpin RNA (shRNA) expressing lentiviral vector. ShRNA complementary sequences to CD82 and c-Met were listed in Table 3. The pCMV3-Flag-c-Met construct containing c-Met cDNA was purchased from Sino Biological Inc. (Beijing, China). The plasmid pEGFP-N2 contained the enhanced green fluorescent protein (eGFP) gene fused to the cytomegalovirus IE gene promoter was from CLONTECH Laboratories (Palo Alto, CA, USA). In this study, pHAGE was used as a control of pHAGE-CD82 while a modified pCDH (mpCDH for short) was used as a control for the miR-K6-5p expression construct and all the shRNA expression constructs.
Table 3.
The sequences of the shRNAs
| Gene | shRNA No. | Sequence of shRNA (5′ to 3′) |
|---|---|---|
| c-Met | sh1 | CUGUCAGAGGAUACUGCACTT |
| sh2 | UCUUCUUCCCAGUGAUAACCATT | |
| CD82 | sh1 | AAAGGCUGUUGUCCUCUUCTT |
| sh2 | AAUGAGGAUCAGGAGCAGGTT |
Antibodies, Western-Blotting, Transfection and Reagents
An anti-KSHV LANA rat monoclonal antibody (MAb) was purchased from Advanced Biotechnologies Inc. (Columbia, MD, USA)89. The anti-phospho-c-Met (Y1356) rabbit polyclonal antibody (PAb) was obtained from Abcam (Cambridge, MA, USA). The anti-c-Met rabbit MAb and anti-Flag M2 rabbit MAb were obtained from Cell Signaling Technologies (Beverly, MA, USA). The anti-CD82 rabbit PAb, anti-b-FGF goat PAb, anti-GAPDH mouse MAb, anti-α-Tubulin mouse MAb, and horseradish peroxidase (HRP)-conjugated goat anti-mouse or anti-rabbit IgG were all purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-His mouse MAb, anti-GFP rabbit antibody, and anti-mouse immunoglobulin G (IgG) were from Beyotime Institute of Biotechnology (Nantong, Jiangsu, China). Western-blotting analysis was performed as previously described90, 91. Transfection of HUVECs was performed with the Effectence transfection reagent (Qiagen, Valencia, CA, USA) while transfections in other cells were performed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. PF-2341066, a c-Met inhibitor, was purchased from Selleck Chemicals (Shanghai, China).
Generation of WT or Mutant miR-K6 Recombinant Viruses
KSHV BAC16 virus and miR-K6 mutant viruses were generated as described in the previous studies48, 92.
Lentiviral Transduction
To obtain the recombinant lentiviruses, HEK293T cells were transfected with lentiviral plasmids, packaging vector psPAX2 and envelope vector pMD2.G as previously described93. The viruses were collected from the culture supernatant 48 h after transfection.
Transwell Migration and Matrigel Invasion Assays
Transwell migration and Matrigel invasion assays were performed as previously described46, 48. Briefly, 1.2 × 105 HUVECs were seeded in top of the chamber after the chamber was coated with 40 μl matrigel mixed with 20 μl basic medium. After 6 h and 12 h incubation at 37°C, cells were fixed with 70% methanol and stained with 0.5% crystal violet solution. The cells at the bottom of the membrane were counted by two observers in a double-blind manner and the average cell number per field for each well was calculated. For each experiment, at least three replicate wells were used and the representative images were taken from five randomly selected fields of each well.
RT-qPCR
Total RNA was isolated from cells by Trizol reagent (Invitrogen, Carlsbad, CA, USA). cDNA was synthesized from equal total RNA using Promega Reverse-Transcription Kit (Promega, Madison, WI) according to the manufacturer’s instructions. The sequences of specific primers of RT-qPCR for several genes were listed in Table 4. qPCR was performed using SYBR Premix Ex Taq Kit (TaKaRa Biotechnology Co. Ltd., Dalian, China) according to the manufacturer’s instructions.
Table 4.
The sequences of specific primers for qPCR
| Target | Application | Primer |
|---|---|---|
| MMP10 | RT-qPCR | F: 5′- GGACCTGGGCTTTATGGAGATAT -3′ |
| R: 5′- CCCAGGGAGTGGCCAAGT -3′ | ||
| VEGFA | RT-qPCR | F: 5′-CTTGCCTTGCTGCTCTACCT -3′ |
| R: 5′-GATTCTGCCCTCCTCCTTCT -3′ | ||
| CD82 | RT-qPCR | F: 5′- GCTCATTCGAGACTACAACAGC -3′ |
| R: 5′- GTGACCTCAGGGCGATTCA -3′ | ||
| β-actin | RT-qPCR | F: 5′- TTGCCGACAGGATGCAGAAGGA -3′ |
| R: 5′- AGGTGGACAGCGAGGCCAGGAT -3′ |
Protein Extraction, TMT Protein Labeling, Mass Spectrometry, and Data Analysis
Mass spectrometry analysis was done as previously described48. Peptide digests were analyzed by nanoscalereversed phase liquid chromatography (Easy-nLC, Thermo Fisher Scientific Inc.) coupled online with an LTQ-OrbitrapVelos mass spectrometer (Thermo Fisher Scientific Inc.). Spectra were searched using Maxquant (version 1.2.2.5), and results were filtered to 1% FDR at the unique peptide level using the COMPASS software suite.
Immunohistochemistry (IHC)
The clinical section of the research was reviewed and approved by the Institutional Ethics Committee of the First Affiliated Hospital of Nanjing Medical University (Nanjing, China; Study protocol # 2015-SR-116). Written informed consent was obtained from all participants, and all samples were anonymized. All participants were adults. The KS clinical tissue specimens and the normal skin tissue specimens were provided by the First Affiliated Hospital of Nanjing Medical University for hematoxylin and eosin (H&E) and immunohistochemistry (IHC) staining. Sections of formalin-fixed and paraffn-embedded samples were stained with the indicated antibodies as previously described46, 93. The expression level was estimated by calculating the percentage of positive cells.
Chicken Chorioallantoic Membrane (CAM) Assay
CAM assay was performed as previously described87, 88, 94. Briefly, cells were mixed with High Concentration Matrigel (BD Biosciences), and inoculated onto the CAM of 9-day-old embryos through the false gas chamber hole. After implantation for 4 days, the CAM was harvested and examined under stereomicroscope. The blood vessels were counted by two observers in a double-blind manner. Assays for each treatment were carried out using 8-day-old embryos.
Matrigel Plug Assay for Angiogenesis in Nude Mice
The animal experiments were approved by the Institution of Animal Care and Use Committee of Nanjing Medical University (Animal protocol # NJMU/IACUC_2013-8-18-01). All animal care and use protocols were performed in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals approved by the State Council of People’s Republic of China. Matrigel plug assay was performed as previously described87, 94. In brief, 3–4-week-old male athymic BALB/c nu/nu mice (Nanjing Biomedical Research Institute of Nanjing University, Nanjing, China) were maintained under pathogen-free conditions. Cell aliquots (1 × 107 cells in 0.2 ml of serum-free medium) were mixed with 0.4 ml of High Concentration Matrigel (BD Biosciences, Bedford, MA, USA), and the mixture was immediately injected subcutaneously into the right flanks of nude mice. The mice were killed 10 days after the injection, and the Matrigel plugs were removed from the mice. The hemoglobin content of the Matrigel was determined using Drabkin’s reagent kit (Sigma-Aldrich) according to the manufacturer’s instructions. The final hemoglobin concentration was calculated from a standard calibration curve after spectrophotometric analysis at 540 nm. For all animal studies, animals were randomly chosen. Concealed allocation and blinding of outcome assessment were used. No statistical method was used to predetermine sample sizes.
Microtubule Formation Assay
The microtubule formation assay was performed on micro-slide Angiogenesis ibiTreat (ibidi, Martinsried, Germany) coated with 10 μl of Matrigel (BD Biosciences) as previously described48. Tubule formation was quantified by counting the number of branching points and measuring the total length of the capillary tubes by two observers in a double-blind manner in at least five images using NIH Image software.
Co-immunoprecipitation (Co-IP)
Immunoprecipitation was performed using a standard protocol as previously described48. Briefly, cells were collected, rinsed, and lysed. Lysates were clarified, and the supernatants were subjected to immunoprecipitation using specified antibody. After extensive washing with the lysis buffer, the precipitated material was separated by SDS-PAGE and analyzed by immunoblotting.
Confocal Microscopy
The immunofluorescence staining was carried out as previously described87. In brief, cells were cultured on glass bottom cell culture dishes, fixed and washed. Mouse MAb to CD82 (Abcam, Cambridge, MA, USA) and rabbit PAb to phospho-c-Met (Y1356) were labeled with secondary Alexa Fluro 555-conjugated anti-rabbit and Alexa Fluro 488-conjugated anti-mouse antibodies (Invitrogen), respectively. The cells were counterstained with DAPI. Images were observed and recorded with a Zeiss Axiovert 200M epifluorescence microscope (Carl Zeiss Inc., Freistaat Thuüringen, Germany).
Statistical Analysis
Quantitative data were presented as mean ± SD. Statistical analysis was based on chi-square test for Figure 2f and 3g, and two-side Student’s t-test for the remaining figures. P < 0.05 was considered statistically significant. All the experiments were repeated three times, unless otherwise stated.
Supplementary Material
Figure S1. miR-K6-5p doesn’t induce cell migration.
Transwell migration for HUVECs transduced with lentivirus empty vector (mpCDH) or lentivirus-miR-K6-5p (miR-K6-5p). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. n.s., not significant.
Figure S2. miR-K6-5p promotes angiogenesis in vivo.
HUVECs treated as in (Figure 2a) were examined for their proangiogenic effects in Matrigel plug assay in nude mice as described in the “Materials and Methods” section. Five images of angiogenesis in the nude mice in one experiment are shown. The images in the green box are shown in Figure 2c as representative photographs.
Figure S3. Overexpression of CD82 inhibits miR-K6-5p-induced angiogenesis in vivo.
HUVECs treated as in (Figure 5a) were examined for their proangiogenic effects in Matrigel plug assay in nude mice as described in the “Materials and Methods” section. Six images of angiogenesis in the nude mice in one experiment are shown. The images in the green box are shown in Figure 5i as representative photographs.
Figure S4. The interaction between CD82 and c-Met in endothelial cells.
(a). Western-blotting analysis of CD82, phosphorylated c-Met and c-Met in HUVECs transduced with lentivirus empty vector (mpCDH) or lentivirus-miR-K6-5p (miR-K6-5p), and HUVECs treated with PBS (Mock) or infected with KSHV (KSHV), respectively. Results shown were from a representative experiment of three independent experiments with similar results.
(b). Western-blotting analysis of phosphorylated c-Met in HUVECs transduced with lentivirus-mediating empty vector (mpCDH) or miR-K6-5p (miR-K6-5p), which were subsequently co-transduced with lentivirus CD82 (CD82) or its control pHAGE (pHAGE), respectively. The antibody against His-tag was used to detect the exogenous expression of CD82. Results shown were from a representative experiment of three independent experiments with similar results.
(c). The physiological interaction between CD82 and c-Met. HUVECs were transduced with lentivirus-CD82 (CD82) or its control pHAGE (pHAGE) and subjected to co-immunoprecipitation with the antibody against CD82 (IP: CD82) or c-Met (IP: c-Met) followed by Western-blotting using the indicated antibodies.
(d). Lentivirus-CD82 construct-transduced HUVECs subjected to co-immunoprecipitation with the antibody against immunoglobulin G (IgG) or His (His-CD82) followed by Western-blotting using indicated antibodies. Results shown were from a representative experiment of three independent experiments with similar results.
(e). HUVECs transfected with pCMV3-Flag-c-Met construct (Flag-c-Met) or its control pCMV3-C-Flag (pCMV) were subjected to co-immunoprecipitation with the antibody against CD82 (IP: CD82) or c-Met (IP: c-Met) followed by Western-blotting using indicated antibodies.
(f). pCMV3-Flag-c-Met construct-transfected HUVECs subjected to co-immunoprecipitation with the antibody against immunoglobulin G (IgG) or Flag (Flag-c-Met) followed by Western-blotting using indicated antibodies. Results shown were from a representative experiment of three independent experiments with similar results.
(g). Confocal microscopy for localization of CD82 and phosphorylated c-Met. HUVECs transduced with lentivirus CD82 (CD82) and its control pHAGE (pHAGE), or lentivirus-mediated a mixture of short hairpin RNAs targeting CD82 (shCD82) and its empty vector (mpCDH) were stained for CD82 (green) and p-c-Met (red) with indicated antibodies. 4′, 6′-diamidino-2-phenylindole (DAPI) (blue) stains nuclei.
Figure S5. Screening and identification of lentivirus-mediated short hairpin RNA targeting CD82.
Western-blotting was performed in HUVECs transduced with lentivirus-mediated No.1 (sh1CD82), No. 2 (sh2CD82), and a mixture of No. 1 and 2 together (shCD82) of short hairpin RNAs targeting CD82 or the control (mpCDH) with the indicated antibodies. Results shown were from a representative experiment of three independent experiments with similar results.
Figure S6. Screening and identification of lentivirus-mediated short hairpin RNA targeting c-Met.
Western blotting was performed in HUVECs transduced with lentivirus-mediated No.1 (sh1c-Met), No. 2 (sh2c-Met), and a mixture of No. 1 and 2 together (shc-Met) of short hairpin RNAs targeting c-Met or the control (mpCDH) with the indicated antibodies. Results shown were from a representative experiment of three independent experiments with similar results.
Figure S7. Knockdown of miR-K6-5p from KSHV genome attenuates KSHV induction of endothelial cell invasion and angiogenesis.
(a). Matrigel invasion assay for HUVECs treated with PBS (PBS), infected with BAC16 KSHV wide type virus (KSHV_WT) and further transfected with miR-K6-5p inhibitor (miR-K6-5p inhibitor) or its control (Neg. Ctrl. of inhibitor). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05, ** P < 0.01, and *** P < 0.001 for Student’s t-test.
(b). Microtubule formation assay for HUVECs treated as in (a). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05, ** P < 0.01, and *** P < 0.001 for Student’s t-test.
Figure S8. MiR-K6-5p is necessary for KSHV-induced endothelial cell invasion and angiogenesis.
(a). Matrigel invasion assay for HUVECs infected with BAC16 KSHV wild type virus (KSHV_WT) or BAC16 KSHV miR-K6 deletion mutant virus (miR-K6_Mut) and further transduced with lentivirus empty vector (mpCDH) or lentivirus-miR-K6-5p (miR-K6-5p). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed.* P < 0.05, ** P < 0.01, and *** P < 0.001 for Student’s t-test.
(b). Microtubule formation assay for HUVECs treated as in (a). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05, ** P < 0.01, and *** P < 0.001 for Student’s t-test.
Acknowledgments
This work was supported by grants from National Natural Science Foundation of China (81761128003, 81371824, 81401662, and 81571984), grants from NIH (R01CA213275, R01CA177377 and R01CA132637), and the Natural Science Youth Foundation of Jiangsu Province (BK20140908).
Footnotes
Author Contributions: Conceived and designed the experiments: CL. Performed the experiments: WL MH CW CF. Provided the reagents: CW HL BJR RR SJG. Analyzed the data: JX YS FW QY. Wrote the paper: WL JQ SJG CL.
Competing Interests: The authors have declared that no competing interests exist.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Mesri EA, Cesarman E, Boshoff C. Kaposi’s sarcoma and its associated herpesvirus. Nature reviews Cancer. 2010;10:707–719. doi: 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dourmishev LA, Dourmishev AL, Palmeri D, Schwartz RA, Lukac DM. Molecular genetics of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiology and molecular biology reviews : MMBR. 2003;67:175–212. doi: 10.1128/MMBR.67.2.175-212.2003. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheung TW. AIDS-related cancer in the era of highly active antiretroviral therapy (HAART): a model of the interplay of the immune system, virus, and cancer. “On the offensive–the Trojan Horse is being destroyed”–Part A: Kaposi’s sarcoma. Cancer investigation. 2004;22:774–786. doi: 10.1081/cnv-200032788. [DOI] [PubMed] [Google Scholar]
- 4.Sadagopan S, Sharma-Walia N, Veettil MV, Bottero V, Levine R, Vart RJ, et al. Kaposi’s sarcoma-associated herpesvirus upregulates angiogenin during infection of human dermal microvascular endothelial cells, which induces 45S rRNA synthesis, antiapoptosis, cell proliferation, migration, and angiogenesis. Journal of virology. 2009;83:3342–3364. doi: 10.1128/JVI.02052-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ensoli B, Barillari G, Gallo RC. Cytokines and growth factors in the pathogenesis of AIDS-associated Kaposi’s sarcoma. Immunol Rev. 1992;127:147–155. doi: 10.1111/j.1600-065x.1992.tb01412.x. [DOI] [PubMed] [Google Scholar]
- 6.Pantanowitz L, Dezube BJ, Hernandez-Barrantes S, Tahan SR, Dabbous MK. Matrix metalloproteinases in the progression and regression of Kaposi’s sarcoma. Journal of cutaneous pathology. 2006;33:793–798. doi: 10.1111/j.1600-0560.2006.00567.x. [DOI] [PubMed] [Google Scholar]
- 7.Rosano L, Spinella F, Di Castro V, Nicotra MR, Albini A, Natali PG, et al. Endothelin receptor blockade inhibits molecular effectors of Kaposi’s sarcoma cell invasion and tumor growth in vivo. The American journal of pathology. 2003;163:753–762. doi: 10.1016/S0002-9440(10)63702-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blankaert D, Simonart T, Van Vooren JP, Parent D, Liesnard C, Farber CM, et al. Constitutive release of metalloproteinase-9 (92-kd type IV collagenase) by Kaposi’s sarcoma cells. Journal of acquired immune deficiency syndromes and human retrovirology : official publication of the International Retrovirology Association. 1998;18:203–209. doi: 10.1097/00042560-199807010-00002. [DOI] [PubMed] [Google Scholar]
- 9.Cullen BR. Viruses and microRNAs: RISCy interactions with serious consequences. Genes & development. 2011;25:1881–1894. doi: 10.1101/gad.17352611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5570–5575. doi: 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Samols MA, Hu J, Skalsky RL, Renne R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. Journal of virology. 2005;79:9301–9305. doi: 10.1128/JVI.79.14.9301-9305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grundhoff A, Sullivan CS, Ganem D. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. Rna. 2006;12:733–750. doi: 10.1261/rna.2326106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marshall V, Parks T, Bagni R, Wang CD, Samols MA, Hu J, et al. Conservation of virally encoded microRNAs in Kaposi sarcoma–associated herpesvirus in primary effusion lymphoma cell lines and in patients with Kaposi sarcoma or multicentric Castleman disease. The Journal of infectious diseases. 2007;195:645–659. doi: 10.1086/511434. [DOI] [PubMed] [Google Scholar]
- 14.O’Hara AJ, Chugh P, Wang L, Netto EM, Luz E, Harrington WJ, et al. Pre-micro RNA signatures delineate stages of endothelial cell transformation in Kaposi sarcoma. PLoS pathogens. 2009;5:e1000389. doi: 10.1371/journal.ppat.1000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 16.Bellare P, Ganem D. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell host & microbe. 2009;6:570–575. doi: 10.1016/j.chom.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lei X, Bai Z, Ye F, Xie J, Kim CG, Huang Y, et al. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nature cell biology. 2010;12:193–199. doi: 10.1038/ncb2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu F, Stedman W, Yousef M, Renne R, Lieberman PM. Epigenetic regulation of Kaposi’s sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. Journal of virology. 2010;84:2697–2706. doi: 10.1128/JVI.01997-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang D, Gao Y, Lin X, He Z, Zhao Q, Deng Q, et al. A human herpesvirus miRNA attenuates interferon signaling and contributes to maintenance of viral latency by targeting IKKepsilon. Cell research. 2011;21:793–806. doi: 10.1038/cr.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin X, Liang D, He Z, Deng Q, Robertson ES, Lan K. miR-K12-7-5p encoded by Kaposi’s sarcoma-associated herpesvirus stabilizes the latent state by targeting viral ORF50/RTA. PloS one. 2011;6:e16224. doi: 10.1371/journal.pone.0016224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu CC, Li Z, Chu CY, Feng J, Feng J, Sun R, et al. MicroRNAs encoded by Kaposi’s sarcoma-associated herpesvirus regulate viral life cycle. EMBO reports. 2010;11:784–790. doi: 10.1038/embor.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bai Z, Huang Y, Li W, Zhu Y, Jung JU, Lu C, et al. Genomewide Mapping and Screening of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) 3′ Untranslated Regions Identify Bicistronic and Polycistronic Viral Transcripts as Frequent Targets of KSHV MicroRNAs. Journal of virology. 2014;88:377–392. doi: 10.1128/JVI.02689-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plaisance-Bonstaff K, Choi HS, Beals T, Krueger BJ, Boss IW, Gay LA, et al. KSHV miRNAs decrease expression of lytic genes in latently infected PEL and endothelial cells by targeting host transcription factors. Viruses. 2014;6:4005–4023. doi: 10.3390/v6104005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abend JR, Uldrick T, Ziegelbauer JM. Regulation of tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi’s sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. Journal of virology. 2010;84:12139–12151. doi: 10.1128/JVI.00884-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abend JR, Ramalingam D, Kieffer-Kwon P, Uldrick TS, Yarchoan R, Ziegelbauer JM. Kaposi’s sarcoma-associated herpesvirus microRNAs target IRAK1 and MYD88, two components of the toll-like receptor/interleukin-1R signaling cascade, to reduce inflammatory-cytokine expression. Journal of virology. 2012;86:11663–11674. doi: 10.1128/JVI.01147-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boss IW, Nadeau PE, Abbott JR, Yang Y, Mergia A, Renne R. A Kaposi’s sarcoma-associated herpesvirus-encoded ortholog of microRNA miR-155 induces human splenic B-cell expansion in NOD/LtSz-scid IL2Rgammanull mice. Journal of virology. 2011;85:9877–9886. doi: 10.1128/JVI.05558-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dahlke C, Maul K, Christalla T, Walz N, Schult P, Stocking C, et al. A microRNA encoded by Kaposi sarcoma-associated herpesvirus promotes B-cell expansion in vivo. PloS one. 2012;7:e49435. doi: 10.1371/journal.pone.0049435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, Chi JT, et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature. 2007;450:1096–1099. doi: 10.1038/nature05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hansen A, Henderson S, Lagos D, Nikitenko L, Coulter E, Roberts S, et al. KSHV-encoded miRNAs target MAF to induce endothelial cell reprogramming. Genes & development. 2010;24:195–205. doi: 10.1101/gad.553410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lei X, Zhu Y, Jones T, Bai Z, Huang Y, Gao SJ. A Kaposi’s sarcoma-associated herpesvirus microRNA and its variants target the transforming growth factor beta pathway to promote cell survival. Journal of virology. 2012;86:11698–11711. doi: 10.1128/JVI.06855-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Sun R, Lin X, Liang D, Deng Q, Lan K. Kaposi’s Sarcoma-Associated Herpesvirus-Encoded MicroRNA miR-K12-11 Attenuates Transforming Growth Factor Beta Signaling through Suppression of SMAD5. Journal of virology. 2012;86:1372–1381. doi: 10.1128/JVI.06245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell host & microbe. 2009;5:376–385. doi: 10.1016/j.chom.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 33.Qin Z, Freitas E, Sullivan R, Mohan S, Bacelieri R, Branch D, et al. Upregulation of xCT by KSHV-encoded microRNAs facilitates KSHV dissemination and persistence in an environment of oxidative stress. PLoS pathogens. 2010;6:e1000742. doi: 10.1371/journal.ppat.1000742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qin Z, Kearney P, Plaisance K, Parsons CH. Pivotal advance: Kaposi’s sarcoma-associated herpesvirus (KSHV)-encoded microRNA specifically induce IL-6 and IL-10 secretion by macrophages and monocytes. Journal of leukocyte biology. 2010;87:25–34. doi: 10.1189/jlb.0409251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samols MA, Skalsky RL, Maldonado AM, Riva A, Lopez MC, Baker HV, et al. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS pathogens. 2007;3:e65. doi: 10.1371/journal.ppat.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skalsky RL, Samols MA, Plaisance KB, Boss IW, Riva A, Lopez MC, et al. Kaposi’s sarcoma-associated herpesvirus encodes an ortholog of miR-155. Journal of virology. 2007;81:12836–12845. doi: 10.1128/JVI.01804-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suffert G, Malterer G, Hausser J, Viiliainen J, Fender A, Contrant M, et al. Kaposi’s sarcoma herpesvirus microRNAs target caspase 3 and regulate apoptosis. PLoS pathogens. 2011;7:e1002405. doi: 10.1371/journal.ppat.1002405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ziegelbauer JM, Sullivan CS, Ganem D. Tandem array-based expression screens identify host mRNA targets of virus-encoded microRNAs. Nature genetics. 2009;41:130–134. doi: 10.1038/ng.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramalingam D, Happel C, Ziegelbauer JM. Kaposi’s Sarcoma-Associated Herpesvirus MicroRNAs Repress Breakpoint Cluster Region Protein Expression, Enhance Rac1 Activity, and Increase In Vitro Angiogenesis. Journal of virology. 2015;89:4249–4261. doi: 10.1128/JVI.03687-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kang JG, Majerciak V, Uldrick TS, Wang X, Kruhlak M, Yarchoan R, et al. Kaposi’s sarcoma-associated herpesviral IL-6 and human IL-6 open reading frames contain miRNA binding sites and are subject to cellular miRNA regulation. The Journal of pathology. 2011;225:378–389. doi: 10.1002/path.2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kieffer-Kwon P, Happel C, Uldrick TS, Ramalingam D, Ziegelbauer JM. KSHV MicroRNAs Repress Tropomyosin 1 and Increase Anchorage-Independent Growth and Endothelial Tube Formation. PloS one. 2015;10:e0135560. doi: 10.1371/journal.pone.0135560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moody R, Zhu Y, Huang Y, Cui X, Jones T, Bedolla R, et al. KSHV MicroRNAs Mediate Cellular Transformation and Tumorigenesis by Redundantly Targeting Cell Growth and Survival Pathways. PLoS pathogens. 2013;9:e1003857. doi: 10.1371/journal.ppat.1003857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo Y, Li W, Qin J, Lu C, Fan W. Kaposi’s sarcoma-associated herpesvirus (KSHV)-encoded microRNAs promote matrix metalloproteinases (MMPs) expression and pro-angiogenic cytokine secretion in endothelial cells. Journal of medical virology. 2017 doi: 10.1002/jmv.24773. [DOI] [PubMed] [Google Scholar]
- 44.Qin J, Li W, Gao SJ, Lu C. KSHV microRNAs: Tricks of the Devil. Trends in microbiology. 2017 doi: 10.1016/j.tim.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qin Z, Jakymiw A, Findlay V, Parsons C. KSHV-Encoded MicroRNAs: Lessons for Viral Cancer Pathogenesis and Emerging Concepts. International journal of cell biology. 2012;2012:603961. doi: 10.1155/2012/603961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu M, Wang C, Li W, Lu W, Bai Z, Qin D, et al. A KSHV microRNA Directly Targets G Protein-Coupled Receptor Kinase 2 to Promote the Migration and Invasion of Endothelial Cells by Inducing CXCR2 and Activating AKT Signaling. PLoS pathogens. 2015;11:e1005171. doi: 10.1371/journal.ppat.1005171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li W, Jia X, Shen C, Zhang M, Xu J, Shang Y, et al. A KSHV microRNA enhances viral latency and induces angiogenesis by targeting GRK2 to activate the CXCR2/AKT pathway. Oncotarget. 2016;7:32286–32305. doi: 10.18632/oncotarget.8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li W, Yan Q, Ding X, Shen C, Hu M, Zhu Y, et al. The SH3BGR/STAT3 Pathway Regulates Cell Migration and Angiogenesis Induced by a Gammaherpesvirus MicroRNA. PLoS pathogens. 2016;12:e1005605. doi: 10.1371/journal.ppat.1005605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dong JT, Lamb PW, Rinker-Schaeffer CW, Vukanovic J, Ichikawa T, Isaacs JT, et al. KAI1, a metastasis suppressor gene for prostate cancer on human chromosome 11p11.2. Science. 1995;268:884–886. doi: 10.1126/science.7754374. [DOI] [PubMed] [Google Scholar]
- 50.Zoller M. Tetraspanins: push and pull in suppressing and promoting metastasis. Nature reviews Cancer. 2009;9:40–55. doi: 10.1038/nrc2543. [DOI] [PubMed] [Google Scholar]
- 51.Yang X, Wei LL, Tang C, Slack R, Mueller S, Lippman ME. Overexpression of KAI1 suppresses in vitro invasiveness and in vivo metastasis in breast cancer cells. Cancer research. 2001;61:5284–5288. [PubMed] [Google Scholar]
- 52.Nishioka C, Ikezoe T, Furihata M, Yang J, Serada S, Naka T, et al. CD34(+)/CD38(-) acute myelogenous leukemia cells aberrantly express CD82 which regulates adhesion and survival of leukemia stem cells. International journal of cancer Journal international du cancer. 2013;132:2006–2019. doi: 10.1002/ijc.27904. [DOI] [PubMed] [Google Scholar]
- 53.Liu WM, Zhang XA. KAI1/CD82, a tumor metastasis suppressor. Cancer letters. 2006;240:183–194. doi: 10.1016/j.canlet.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 54.Tonoli H, Barrett JC. CD82 metastasis suppressor gene: a potential target for new therapeutics? Trends in molecular medicine. 2005;11:563–570. doi: 10.1016/j.molmed.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 55.Wei Q, Zhang F, Richardson MM, Roy NH, Rodgers W, Liu Y, et al. CD82 restrains pathological angiogenesis by altering lipid raft clustering and CD44 trafficking in endothelial cells. Circulation. 2014;130:1493–1504. doi: 10.1161/CIRCULATIONAHA.114.011096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bandyopadhyay S, Zhan R, Chaudhuri A, Watabe M, Pai SK, Hirota S, et al. Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nature medicine. 2006;12:933–938. doi: 10.1038/nm1444. [DOI] [PubMed] [Google Scholar]
- 57.Ruseva Z, Geiger PX, Hutzler P, Kotzsch M, Luber B, Schmitt M, et al. Tumor suppressor KAI1 affects integrin alphavbeta3-mediated ovarian cancer cell adhesion, motility, and proliferation. Experimental cell research. 2009;315:1759–1771. doi: 10.1016/j.yexcr.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 58.Guo XZ, Xu JH, Liu MP, Kleeff J, Ho CK, Ren LN, et al. KAI1 inhibits anchorage-dependent and -independent pancreatic cancer cell growth. Oncology reports. 2005;14:59–63. [PubMed] [Google Scholar]
- 59.Zismanov V, Lishner M, Tartakover-Matalon S, Radnay J, Shapiro H, Drucker L. Tetraspanin-induced death of myeloma cell lines is autophagic and involves increased UPR signalling. British journal of cancer. 2009;101:1402–1409. doi: 10.1038/sj.bjc.6605291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tohami T, Drucker L, Shapiro H, Radnay J, Lishner M. Overexpression of tetraspanins affects multiple myeloma cell survival and invasive potential. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2007;21:691–699. doi: 10.1096/fj.06-6610com. [DOI] [PubMed] [Google Scholar]
- 61.Gottwein E, Corcoran DL, Mukherjee N, Skalsky RL, Hafner M, Nusbaum JD, et al. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell host & microbe. 2011;10:515–526. doi: 10.1016/j.chom.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Catrina Ene AM, Borze I, Guled M, Costache M, Leen G, Sajin M, et al. MicroRNA expression profiles in Kaposi’s sarcoma. Pathology oncology research : POR. 2014;20:153–159. doi: 10.1007/s12253-013-9678-1. [DOI] [PubMed] [Google Scholar]
- 63.Sridhar SC, Miranti CK. Tetraspanin KAI1/CD82 suppresses invasion by inhibiting integrin-dependent crosstalk with c-Met receptor and Src kinases. Oncogene. 2006;25:2367–2378. doi: 10.1038/sj.onc.1209269. [DOI] [PubMed] [Google Scholar]
- 64.Todeschini AR, Dos Santos JN, Handa K, Hakomori SI. Ganglioside GM2-tetraspanin CD82 complex inhibits met and its cross-talk with integrins, providing a basis for control of cell motility through glycosynapse. The Journal of biological chemistry. 2007;282:8123–8133. doi: 10.1074/jbc.M611407200. [DOI] [PubMed] [Google Scholar]
- 65.Takahashi M, Sugiura T, Abe M, Ishii K, Shirasuna K. Regulation of c-Met signaling by the tetraspanin KAI-1/CD82 affects cancer cell migration. International journal of cancer Journal international du cancer. 2007;121:1919–1929. doi: 10.1002/ijc.22887. [DOI] [PubMed] [Google Scholar]
- 66.Dai L, Trillo-Tinoco J, Cao Y, Bonstaff K, Doyle L, Del Valle L, et al. Targeting HGF/c-MET induces cell cycle arrest, DNA damage, and apoptosis for primary effusion lymphoma. Blood. 2015;126:2821–2831. doi: 10.1182/blood-2015-07-658823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bari R, Zhang YH, Zhang F, Wang NX, Stipp CS, Zheng JJ, et al. Transmembrane interactions are needed for KAI1/CD82-mediated suppression of cancer invasion and metastasis. The American journal of pathology. 2009;174:647–660. doi: 10.2353/ajpath.2009.080685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feng J, Huang C, Wren JD, Wang DW, Yan J, Zhang J, et al. Tetraspanin CD82: a suppressor of solid tumors and a modulator of membrane heterogeneity. Cancer metastasis reviews. 2015;34:619–633. doi: 10.1007/s10555-015-9585-x. [DOI] [PubMed] [Google Scholar]
- 69.Hanna JA, Bordeaux J, Rimm DL, Agarwal S. The function, proteolytic processing, and histopathology of Met in cancer. Advances in cancer research. 2009;103:1–23. doi: 10.1016/S0065-230X(09)03001-2. [DOI] [PubMed] [Google Scholar]
- 70.Koo BS, Kim JM, Seo ST, Yoon YH, Kwon KR, Kim SH, et al. Upregulation of HGF and c-MET is associated with subclinical central lymph node metastasis in papillary thyroid microcarcinoma. Annals of surgical oncology. 2014;21:2310–2317. doi: 10.1245/s10434-014-3553-5. [DOI] [PubMed] [Google Scholar]
- 71.Fuse N, Kuboki Y, Kuwata T, Nishina T, Kadowaki S, Shinozaki E, et al. Prognostic impact of HER2, EGFR, and c-MET status on overall survival of advanced gastric cancer patients. Gastric cancer : official journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association. 2016;19:183–191. doi: 10.1007/s10120-015-0471-6. [DOI] [PubMed] [Google Scholar]
- 72.Neuzillet C, Couvelard A, Tijeras-Raballand A, de Mestier L, de Gramont A, Bedossa P, et al. High c-Met expression in stage I-II pancreatic adenocarcinoma: proposal for an immunostaining scoring method and correlation with poor prognosis. Histopathology. 2015;67:664–676. doi: 10.1111/his.12691. [DOI] [PubMed] [Google Scholar]
- 73.Yan S, Jiao X, Zou H, Li K. Prognostic significance of c-Met in breast cancer: a meta-analysis of 6010 cases. Diagnostic pathology. 2015;10:62. doi: 10.1186/s13000-015-0296-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.16996116Han Y, Luo Y, Zhao J, Li M, Jiang Y. Overexpression of c-Met increases the tumor invasion of human prostate LNCaP cancer cells in vitro and in vivo. Oncology letters. 2014;8:1618–1624. doi: 10.3892/ol.2014.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leo C, Horn LC, Einenkel J, Hentschel B, Hockel M. Tumor hypoxia and expression of c-met in cervical cancer. Gynecologic oncology. 2007;104:181–185. doi: 10.1016/j.ygyno.2006.07.040. [DOI] [PubMed] [Google Scholar]
- 76.Yoshida S, Harada T, Iwabe T, Taniguchi F, Fujii A, Sakamoto Y, et al. Induction of hepatocyte growth factor in stromal cells by tumor-derived basic fibroblast growth factor enhances growth and invasion of endometrial cancer. The Journal of clinical endocrinology and metabolism. 2002;87:2376–2383. doi: 10.1210/jcem.87.5.8483. [DOI] [PubMed] [Google Scholar]
- 77.Naldini L, Weidner KM, Vigna E, Gaudino G, Bardelli A, Ponzetto C, et al. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the MET receptor. The EMBO journal. 1991;10:2867–2878. doi: 10.1002/j.1460-2075.1991.tb07836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 79.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nature reviews Molecular cell biology. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 80.Li Y, Huang X, Zhang J, Li Y, Ma K. Synergistic inhibition of cell migration by tetraspanin CD82 and gangliosides occurs via the EGFR or cMet-activated Pl3K/Akt signalling pathway. The international journal of biochemistry & cell biology. 2013;45:2349–2358. doi: 10.1016/j.biocel.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 81.17500590Hansen A, Henderson S, Lagos D, Nikitenko L, Coulter E, Roberts S, et al. KSHV-encoded miRNAs target MAF to induce endothelial cell reprogramming. Genes & development. 2010;24:195–205. doi: 10.1101/gad.553410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Atabey N, Gao Y, Yao ZJ, Breckenridge D, Soon L, Soriano JV, et al. Potent blockade of hepatocyte growth factor-stimulated cell motility, matrix invasion and branching morphogenesis by antagonists of Grb2 Src homology 2 domain interactions. The Journal of biological chemistry. 2001;276:14308–14314. doi: 10.1074/jbc.M010202200. [DOI] [PubMed] [Google Scholar]
- 83.Royal I, Lamarche-Vane N, Lamorte L, Kaibuchi K, Park M. Activation of cdc42, rac, PAK, and rho-kinase in response to hepatocyte growth factor differentially regulates epithelial cell colony spreading and dissociation. Molecular biology of the cell. 2000;11:1709–1725. doi: 10.1091/mbc.11.5.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lin HR, Ganem D. Viral microRNA target allows insight into the role of translation in governing microRNA target accessibility. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5148–5153. doi: 10.1073/pnas.1102033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Poon M, Zhang X, Dunsky KG, Taubman MB, Harpel PC. Apolipoprotein(a) induces monocyte chemotactic activity in human vascular endothelial cells. Circulation. 1997;96:2514–2519. doi: 10.1161/01.cir.96.8.2514. [DOI] [PubMed] [Google Scholar]
- 86.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, et al. Construction and manipulation of a new Kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome clone. Journal of virology. 2012;86:9708–9720. doi: 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhu X, Guo Y, Yao S, Yan Q, Xue M, Hao T, et al. Synergy between Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6 and HIV-1 Nef protein in promotion of angiogenesis and oncogenesis: role of the AKT signaling pathway. Oncogene. 2014;33:1986–1996. doi: 10.1038/onc.2013.136. [DOI] [PubMed] [Google Scholar]
- 88.Zhou F, Xue M, Qin D, Zhu X, Wang C, Zhu J, et al. HIV-1 Tat Promotes Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) vIL-6-Induced Angiogenesis and Tumorigenesis by Regulating PI3K/PTEN/AKT/GSK-3beta Signaling Pathway. PloS one. 2013;8:e53145. doi: 10.1371/journal.pone.0053145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yan Q, Ma X, Shen C, Cao X, Feng N, Qin D, et al. Inhibition of Kaposi’s Sarcoma-Associated Herpesvirus Lytic Replication by HIV-1 Nef and Cellular MicroRNA hsa-miR-1258. Journal of virology. 2014;88:4987–5000. doi: 10.1128/JVI.00025-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zeng Y, Zhang X, Huang Z, Cheng L, Yao S, Qin D, et al. Intracellular Tat of human immunodeficiency virus type 1 activates lytic cycle replication of Kaposi’s sarcoma-associated herpesvirus: role of JAK/STAT signaling. Journal of virology. 2007;81:2401–2417. doi: 10.1128/JVI.02024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Qin D, Zeng Y, Qian C, Huang Z, Lv Z, Cheng L, et al. Induction of lytic cycle replication of Kaposi’s sarcoma-associated herpesvirus by herpes simplex virus type 1: involvement of IL-10 and IL-4. Cellular microbiology. 2008;10:713–728. doi: 10.1111/j.1462-5822.2007.01079.x. [DOI] [PubMed] [Google Scholar]
- 92.Jain V, Plaisance-Bonstaff K, Sangani R, Lanier C, Dolce A, Hu J, et al. A Toolbox for Herpesvirus miRNA Research: Construction of a Complete Set of KSHV miRNA Deletion Mutants. Viruses. 2016;8 doi: 10.3390/v8020054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xue M, Yao S, Hu M, Li W, Hao T, Zhou F, et al. HIV-1 Nef and KSHV oncogene K1 synergistically promote angiogenesis by inducing cellular miR-718 to regulate the PTEN/AKT/mTOR signaling pathway. Nucleic acids research. 2014;42:9862–9879. doi: 10.1093/nar/gku583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yao S, Hu M, Hao T, Li W, Xue X, Xue M, et al. MiRNA-891a-5p mediates HIV-1 Tat and KSHV Orf-K1 synergistic induction of angiogenesis by activating NF-kappaB signaling. Nucleic acids research. 2015;43:9362–9378. doi: 10.1093/nar/gkv988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. miR-K6-5p doesn’t induce cell migration.
Transwell migration for HUVECs transduced with lentivirus empty vector (mpCDH) or lentivirus-miR-K6-5p (miR-K6-5p). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. n.s., not significant.
Figure S2. miR-K6-5p promotes angiogenesis in vivo.
HUVECs treated as in (Figure 2a) were examined for their proangiogenic effects in Matrigel plug assay in nude mice as described in the “Materials and Methods” section. Five images of angiogenesis in the nude mice in one experiment are shown. The images in the green box are shown in Figure 2c as representative photographs.
Figure S3. Overexpression of CD82 inhibits miR-K6-5p-induced angiogenesis in vivo.
HUVECs treated as in (Figure 5a) were examined for their proangiogenic effects in Matrigel plug assay in nude mice as described in the “Materials and Methods” section. Six images of angiogenesis in the nude mice in one experiment are shown. The images in the green box are shown in Figure 5i as representative photographs.
Figure S4. The interaction between CD82 and c-Met in endothelial cells.
(a). Western-blotting analysis of CD82, phosphorylated c-Met and c-Met in HUVECs transduced with lentivirus empty vector (mpCDH) or lentivirus-miR-K6-5p (miR-K6-5p), and HUVECs treated with PBS (Mock) or infected with KSHV (KSHV), respectively. Results shown were from a representative experiment of three independent experiments with similar results.
(b). Western-blotting analysis of phosphorylated c-Met in HUVECs transduced with lentivirus-mediating empty vector (mpCDH) or miR-K6-5p (miR-K6-5p), which were subsequently co-transduced with lentivirus CD82 (CD82) or its control pHAGE (pHAGE), respectively. The antibody against His-tag was used to detect the exogenous expression of CD82. Results shown were from a representative experiment of three independent experiments with similar results.
(c). The physiological interaction between CD82 and c-Met. HUVECs were transduced with lentivirus-CD82 (CD82) or its control pHAGE (pHAGE) and subjected to co-immunoprecipitation with the antibody against CD82 (IP: CD82) or c-Met (IP: c-Met) followed by Western-blotting using the indicated antibodies.
(d). Lentivirus-CD82 construct-transduced HUVECs subjected to co-immunoprecipitation with the antibody against immunoglobulin G (IgG) or His (His-CD82) followed by Western-blotting using indicated antibodies. Results shown were from a representative experiment of three independent experiments with similar results.
(e). HUVECs transfected with pCMV3-Flag-c-Met construct (Flag-c-Met) or its control pCMV3-C-Flag (pCMV) were subjected to co-immunoprecipitation with the antibody against CD82 (IP: CD82) or c-Met (IP: c-Met) followed by Western-blotting using indicated antibodies.
(f). pCMV3-Flag-c-Met construct-transfected HUVECs subjected to co-immunoprecipitation with the antibody against immunoglobulin G (IgG) or Flag (Flag-c-Met) followed by Western-blotting using indicated antibodies. Results shown were from a representative experiment of three independent experiments with similar results.
(g). Confocal microscopy for localization of CD82 and phosphorylated c-Met. HUVECs transduced with lentivirus CD82 (CD82) and its control pHAGE (pHAGE), or lentivirus-mediated a mixture of short hairpin RNAs targeting CD82 (shCD82) and its empty vector (mpCDH) were stained for CD82 (green) and p-c-Met (red) with indicated antibodies. 4′, 6′-diamidino-2-phenylindole (DAPI) (blue) stains nuclei.
Figure S5. Screening and identification of lentivirus-mediated short hairpin RNA targeting CD82.
Western-blotting was performed in HUVECs transduced with lentivirus-mediated No.1 (sh1CD82), No. 2 (sh2CD82), and a mixture of No. 1 and 2 together (shCD82) of short hairpin RNAs targeting CD82 or the control (mpCDH) with the indicated antibodies. Results shown were from a representative experiment of three independent experiments with similar results.
Figure S6. Screening and identification of lentivirus-mediated short hairpin RNA targeting c-Met.
Western blotting was performed in HUVECs transduced with lentivirus-mediated No.1 (sh1c-Met), No. 2 (sh2c-Met), and a mixture of No. 1 and 2 together (shc-Met) of short hairpin RNAs targeting c-Met or the control (mpCDH) with the indicated antibodies. Results shown were from a representative experiment of three independent experiments with similar results.
Figure S7. Knockdown of miR-K6-5p from KSHV genome attenuates KSHV induction of endothelial cell invasion and angiogenesis.
(a). Matrigel invasion assay for HUVECs treated with PBS (PBS), infected with BAC16 KSHV wide type virus (KSHV_WT) and further transfected with miR-K6-5p inhibitor (miR-K6-5p inhibitor) or its control (Neg. Ctrl. of inhibitor). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05, ** P < 0.01, and *** P < 0.001 for Student’s t-test.
(b). Microtubule formation assay for HUVECs treated as in (a). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05, ** P < 0.01, and *** P < 0.001 for Student’s t-test.
Figure S8. MiR-K6-5p is necessary for KSHV-induced endothelial cell invasion and angiogenesis.
(a). Matrigel invasion assay for HUVECs infected with BAC16 KSHV wild type virus (KSHV_WT) or BAC16 KSHV miR-K6 deletion mutant virus (miR-K6_Mut) and further transduced with lentivirus empty vector (mpCDH) or lentivirus-miR-K6-5p (miR-K6-5p). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed.* P < 0.05, ** P < 0.01, and *** P < 0.001 for Student’s t-test.
(b). Microtubule formation assay for HUVECs treated as in (a). The quantified results represent the mean ± SD. Three independent experiments, each with five technical replicates, were performed. * P < 0.05, ** P < 0.01, and *** P < 0.001 for Student’s t-test.