

Abstract
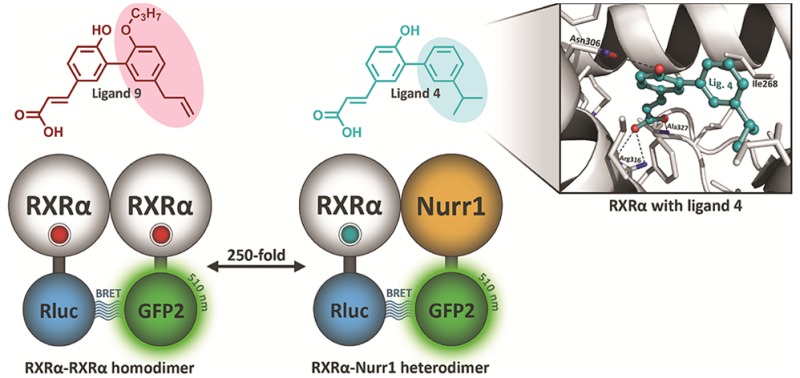
Retinoid X receptors (RXRs) play key roles in many physiological processes in both the periphery and central nervous system. In addition, RXRs form heterodimers with other nuclear receptors to exert their physiological effects. The nuclear receptor related 1 protein (NURR1) is particularly interesting because of its role in promoting differentiation and survival of dopamine neurons. However, only a small number of RXR-heterodimer selective modulators are available, with limited chemical diversity. This work describes the synthesis, biochemical evaluation, and structural elucidation of a novel series of RXR ligands with strongly biased interactions with RXRα–NURR1 heterodimers. Targeted modifications to the small molecule biaryl scaffold caused local RXRα side-chain disturbances and displacement of secondary structural elements upon ligand binding. This resulted in the repositioning of protein helices in the heterodimer interface of RXRα, alterations in homo- versus heterodimer formation, and modulation of activation function 2 (AF2). The data provide a rationale for the design of RXR ligands consisting of a highly conserved hydrophilic region, strongly contributing to the ligand affinity, and a variable hydrophobic region, which efficiently probes the effects of structural changes at the level of the ligand on co-regulator recruitment or the RXRα–NURR1 dimerization interface.
Keywords: Nuclear receptors, heterodimerization, ligand binding domain, retinoid X receptor, nuclear receptor related 1
Introduction
The retinoid X receptor (RXR) plays a key role as a transcriptional regulator through formation of heterodimers with other nuclear receptor partners. Activation of RXR heterodimers exerts neuroprotective effects in animal models of neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease and multiple sclerosis.1−4 The activity of RXRs is influenced by a class of compounds related to the naturally occurring 9-cis-13,14-dihydroretinoic acid.5 The L-shaped ligand binding pocket is unique to RXRs, and structural information derived from X-ray crystallographic data of the RXR ligand binding domains in the apo or holo state6,7 has aided the design of specific ligands for this receptor.8 Comprehensive overviews of the structure, biology, and therapeutic implications of targeting RXRs with small molecule ligands are available.8−12 However, the chemical diversity of these ligands is limited by the structural constraints placed by the RXR ligand binding pocket and the availability of synthetic methodologies to access designed ligands.13 Controlled RXR heterodimerization and RXR partial agonism are contemporary biomedical challenges,11,14 both of which could in principle be addressed via exploration of an appropriate structural class.
Three RXR subtypes are known, RXRα (subject of this study), RXRβ, and RXRγ (NR2B1, NR2B2, and NR2B3), and all three interact similarly with many coregulator proteins, and with several nuclear receptors to form heterodimers. Ligand binding to RXR can induce the transcriptional activity of some of its heterodimeric partner receptors (NURR1, NGFIB, FXR, LXR, CAR, and PPAR).15 Thus, RXR-selective ligands that only activate specific RXR heterodimers may have greater therapeutic potential, because they would be expected to cause fewer side effects compared to ligands that cause general activation of RXR–NR heterodimers.5,9,11 In this respect, RXR–PPAR and RXR–LXR heterodimers have gained a lot of attention, as the clearance of Aβ through apoE in Alzheimer’s mouse models is believed to be facilitated by the activation of these heterodimers.16,17 For example, the ligand LG101506 was identified as the most potent of a series of selective RXR–PPAR heterodimer activators,18,19 whereas it did not activate the RXR-LXR heterodimer. The potential of this selective RXR modulator as a treatment for type 2 diabetes with reduced side effects was shown in mice. Activation of RXR–nuclear receptor heterodimers with RXR receptor ligands also provides an important strategy for activating orphan nuclear receptors which themselves do not readily bind ligands (e.g., NURR1).
The nuclear receptor related 1 protein or NURR1 (also referred to as NR4A2) controls the development, function, and survival of dopaminergic neurons.20−22 NURR1 knockout mice have reduced dopaminergic neurons and show perinatal lethality.23,24 Loss of function NURR1 genetic polymorphisms in patients are linked to familial Parkinson’s disease.25,26 The crystal structure of the LBD of NURR1 reveals a “closed” ligand binding pocket, with the C-terminal helix 12 in a canonical fold analogous to agonist bound ligand binding domains,27 which may not easily allow access to small molecule ligands.28 Therefore, modulation of NURR1 activity via RXRs would constitute a viable entry point for NURR1 activitation.4,29−31 A limited number of RXR–NURR1 heterodimer activators have been reported with promising selectivity over other RXR-heterodimer pairs.4,32,33 Notwithstanding these important advances in developing selective RXR-heterodimer modulators, there is a demand for a broader portfolio of RXR–NURR1 modulators, both for use as research tools, to address open question such as regarding the resulting effect on RXR signaling via other heterodimers and the potential presence of physiological ligands with similar profiles, and as potential drugs.22
Here we report the synthesis, biophysical evaluation and structural elucidation of a new series of RXRα ligands with a strong bias toward promoting RXRα–NURR1 heterodimer versus RXRα–RXRα homodimer formation. The work makes use of the knowledge derived from studies on the natural product34 honokiol derived RXRα agonists described previously.35 The biaryl scaffold of this series is straightforward to derivatize, which in principle enables a rapid pharmacophore mapping of the RXRα ligand binding pocket. In this present study, ligands 1–5 (Figure 1) were designed to probe the hydrophobic region of the RXRα ligand binding pocket, while keeping the polar interactions intact, to access the flexibility and local displacements of amino acid side-chains of the ligand binding domain. Ortho-substituted ligands 6 and 7 were synthesized to investigate how constraining the conformation of the biaryl system would affect the activity of these ligands. Finally, a canonical side-chain extension36 of the biaryl scaffold in the direction of the RXRα helix 12 produced compounds 9 and 10, for the purpose to investigate for possible RXRα partial and full antagonism. This compact set of chemical probes was subsequently evaluated as RXRα ligands, using a fluorescence-based polarization assay and cellular mammalian two-hybrid assay. In addition, their effectiveness in modulating RXRα–NURR1 heterodimers over RXRα–RXRα homodimers was tested using cell-based bioluminescence resonance energy transfer (BRET2) assays.2,33 To corroborate these results and to elucidate the binding mode and conformational changes in the protein induced by these compounds, the X-ray structures of five of these novel ligands bound to RXRα were solved.
Figure 1.

Designed biaryl-based RXR ligands 1–10 and established potent RXR agonist LG100268.
Results and Discussion
Synthesis
The synthesis and cocrystal structure of ligand 1 were described previously.35 The allyl side chain of 1 partly occupies the lipophilic pocket in the ligand binding domain of RXRα, analogously to the tetramethyl-cyclohexene unit found in typical RXR ligands, leading to closure of the ligand binding pocket via repositioning of helix 12 in an agonist conformation and subsequent increased binding toward coactivators. The high binding affinity and low molecular weight of 1 makes it an ideal scaffold to explore modifications targeting the lipophilic pocket (Table 1). Ligands 2–5 were therefore synthesized bearing structural variations in the hydrophobic side chain. The synthesis of compounds 2–5 relied on efficient palladium-catalyzed cross coupling reactions (Scheme 1). The cinnamic acid derivative 11 was treated with thionyl chloride in methanol to obtain the methyl cinnamate derivative 12. Four boronic acids or esters were then reacted with 12 using Suzuki coupling (Buchwald-modified) to provide intermediates 13–16 in excellent yields (78–99%).35,37 The biaryls 13–16 were thereafter demethylated using boron tribromide and hydrolyzed using sodium hydroxide, yielding ligands 2–5 in reasonable yields with high purities after a preparative HPLC purification. Molecules 6 and 7 were designed and synthesized to access the contorted conformation necessary for the biaryl ligands to fit within the ligand binding pocket of RXRα.8 To enable a more efficient synthesis, the methyl protection of the phenol (11) was replaced by a methoxymethyl acetal (MOM) group (17). Subsequent Miyaura borylation enabled the key palladium cross coupling with 19 or 20, accessible in a single step in moderate yields (51–57%). The biaryl products were then treated with dilute hydrochloric acid (3 M) to deprotect the phenol in quantitative yields. Finally, the methyl ester was efficiently hydrolyzed using sodium hydroxide, yielding ligands 6 and 7.
Table 1. Summary of Fluorescence Polarization (FP) and Mammalian Two-Hybrid (M2H) Data for the RXRα Agonistsa.
| compd | FP/EC50 (nM) | M2H (luciferase)/EC50 (nM) |
|---|---|---|
| LG100268 | 150 ± 40 | 5.1 ± 2.0 |
| 1 (R = CH2CH=CH2) | 260 ± 110 | 6.3 ± 4.0 |
| 2 (R = Ph) | 140 ± 23 | 85 ± 9 |
| 3 (R = Bn) | 142 ± 9 | 92 ± 36 |
| 4 (R = iPr) | 89 ± 7 | 5.8 ± 1.8 |
| 5 (R = nPr) | 170 ± 80 | 18 ± 10 |
| 6 | 9900 ± 2500 | >2500 |
| 7 | 1020 ± 60 | 14 600 ± 1800 |
EC50 values for LG100268 and ligands 1–7. See experimental section for details of the assays. The 20- to 30-fold difference between the FP and M2H data for the more potent compounds is a common phenomemon,35,40 which can be explained by intrinsic differences between the two different assay formats, in particular, the different protein and peptide concentrations used.
Scheme 1. Synthesis of Novel Biaryl RXRα Ligands.

The syntheses of 1 and 8 have been previously reported.35 Conditions: (a) thionyl chloride, MeOH, 0 °C; (b) arylboronic acid or arylboronic ester, Pd2(dba)3, SPhos, KF, dioxane/H2O (10:1 v/v), 110 °C; (c) BBr3, CH2Cl2 −78 °C; (d) NaOH, dioxane/MeOH (14:5 v/v), 40 °C; (e) bis(pinacolato)diboron, Pd(OAc)2, XPhos, KOAc, dioxane 110 °C; (f) 19 or 20, Pd2(dba)3, SPhos, KF, dioxane/H2O (6:1 v/v), 110 °C; (g) 23 or 24, Pd2(dba)3, SPhos, KF, dioxane/H2O (6:1 v/v), 110 °C; (h) HCl, THF, room temperature; (i) NaOH, dioxane/MeOH (14:5 v/v).
In our efforts to selectively drive RXRα homodimers, and not RXRα–NURR1 heterodimers, toward an antagonistic conformation, we applied the previously validated strategy by Nahoum and co-workers for inducing RXR antagonism.36 Compound 8 (Figure 1) was previously described by us and found to lack significant RXRα activity, because of the additional polar phenolic functionality, which points toward a lipophilic environment.35 X-ray crystallographic data have demonstrated that alkylation of the appropriate phenol displaces the position of helix 12 toward a (partial) antagonistic fold, influencing the position of L436, which plays a determining role in the communication with helix 12.36,38 Therefore, using the biaryl scaffold, agonist 8 was modified with two different length alkoxy chains. The length of the alkoxy chain was hypothesized to be directly correlated with the displacement of helix 12 and therefore its antagonist properties.36 The antagonists 9 and 10 were synthesized using intermediate 18 for the Suzuki couplings, using the ligands introduced by Buchwald,35,37 with either 23 or 24. Intermediates 23 and 24 were each made in two steps in excellent yields via sp2–sp3 Pd-catalyzed cross couplings on the bromide group to introduce the allyl-substituted group. Finally, intermediates 25 and 26 were treated with hydrochloric acid in THF for the deprotection of the MOM-group and subsequently with sodium hydroxide for the hydrolysis of the methyl ester to provide the antagonists 9 and 10.
Pharmacological Evaluation
The activity of the ligands on RXRα was initially profiled using a fluorescence-based coactivator recruitment polarization (FP) assay and then in a more biologically relevant mammalian two-hybrid (M2H) assay. The FP and M2H assays revealed an EC50(FP) = 260 nM and EC50(M2H) = 6.3 nM for 1, which compares favorably to the established, but more bulky, full agonist LG100268: EC50(FP) = 150 nM and EC50(M2H) = 5.1 nM (Table 1). The differences in measured EC50 affinities between the FP assay and the M2H assay are a common phenomenon because of intrinsic differences between the two assay formats, the protein concentrations, and the coregulator peptide.35,39 Besides 1, ligands 2–5 also displayed full agonism in both assays with the measured affinities (EC50) in the nanomolar range (Table 1). Replacing the allyl group with the closely related i-propyl (4) or n-propyl (5) did not strongly affect the ligand affinity for RXRα. The aromatic phenyl (2) and benzyl (3) substituents displayed a 10-fold decrease in potency in the M2H cell-based assay compared to the smaller propyl substituents. Nevertheless, also these ligands still activate RXRα with nanomolar potencies. The RXRα binding of 1–5 is thus dominated by the hydrophilic portion of the biaryl ligands, i.e., the hydroxyl-cinnamic acid moiety. The hydrophobic substituents tune the ligand affinity but are not crucial for high ligand affinity. As such, ligand modifications at this part of the molecule could provide an entry to affect the homo- vs heterodimer preference of RXRα.
The terphenyl ligand 2, provides an interesting platform to study the importance and effects of the rotation around the two phenyl–phenyl bonds. Addition of a single methyl group at the central phenyl (6 and 7) was thought to direct the rotation to preferred orientations in complex with RXRα. Ligands 6 and 7 both displayed full agonism in the FP assay and the M2H assay, albeit with potencies in the 1–10 μM range (Table 1). Comparison with the nanomolar affinities observed for ligand 2, demonstrates that the addition of the single methyl groups strongly impacts affinity for RXRα. This very strict SAR is typical for this class of biphenyls, as in our previous studies the placement of a hydroxyl functionality, such as in 8, similarly impacted affinity by changes over 100-fold.35 In the case of 6 and 7, the decrease in affinity might be caused by a suboptimal conformational match of the ligand for binding to the protein in the conformation befitting the binding pocket (vide infra).
We previously studied ligand 8, which demonstrated full agonism, but with only moderate affinity for RXRα in FP as well as M2H assays.35 The design of 9 and 10 prompted us to study these compounds in a competition format to profile their antagonist characteristics. RXRα was therefore stimulated with the full agonist LG100268 and the subsequent impact of ligands 9 and 10 on coregulator recruitment was studied via fluorescence polarization studies and on transcription via M2H assays. The addition of ligands 9 or 10 to the agonist-stimulated RXRα resulted in decreased fluorescence polarization, indicating lowered coactivator recruitment via displacement of the agonist and stabilization of an inactive RXRα conformation. Ligand 10 demonstrated full antagonism, while ligand 9 showed partial antagonism (Figure 3a). Consistent with its partial antagonist activity, ligand 9 also displayed partial agonism in an agonist assay format. The measured affinities of 9 (IC50(FP) = 48.5 ± 4.6 μM) and 10 (IC50(FP) = 46.9 ± 5.9 μM) at the RXRα receptor were approximately 25-fold lower than that of the known antagonist UVI3003: IC50(FP) = 1.8 ± 0.6 μM. Compounds 9 and 10 were also RXRα antagonists in the cell-based M2H assay. M2H competition experiments showed a clear decrease in luciferase expression upon addition of either ligand in the 10–40 μM range after stimulation with agonist LG100268 (Figure 3b).
Figure 3.

Ligands 9 and 10 are antagonists at RXRα. (a) Fluorescence polarization data showing antagonist activity for 9 and 10. Ligand 9 displays partial antagonism, while 10 acts as a full antagonist in a competition assay against LG100268 (50 nM). (b) Cellular antagonist activities of 9 and 10 measured in an M2H luciferase competition assay against LG100268 (10 nM). (c) X-ray cocrystal structures of ligand 9 (purple, PDB: 5MKJ, 2.5 Å resolution) bound in the ligand binding pocket of RXRα in ribbon representation with the TIF2 derived coregulator peptide. Ligand 9 is shown with final 2Fo – Fc electron density maps (contoured at 1σ). Note that part of RXRα helix 11 is omitted to allow visualization of the ligand. (d) Zoom-in on an overlay of the ligand binding pockets of RXRα cocrystallized with ligands 1 (blue), 4 (gray), and 9 (purple). Relevant helices and amino acids and their displacements are shown. Compared to 4, ligand 1 induces an outward expansion of helices 6 and 7. Next to this, ligand 9 induces an additional outward displacement of leucines 436 and 455 on helices 11 and 12.
The potency and efficacy of the biaryl ligands to induce RXRα–RXRα homodimer and RXRα–NURR1 heterodimer conformational changes was determined using cellular BRET2 assays2,33 (Table 2). Agonist ligands 1–5 all displayed remarkably strong potencies (single digit and sub-nanomolar EC50’s) and high efficacies, comparable to the chemically optimized agonist LG100268 (Figure 1). Ligands 1–5 all feature preferential affinity for RXRα–NURR1 heterodimers. Ligand 4 displayed an encouraging 25-fold higher potency at RXRα–NURR1 over RXRα–RXRα, with a pEC50 of 9.1. It should be noted that in these same assays both the well-studied RXR ligand bexarotene and the recently developed dihydrobenzofuran-based ligands only showed 2–7-fold selectivity.2,33 In contrast, the methylated terphenyl ligands 6 and 7 showed higher potency at RXRα–RXRα homodimers. The addition of the single methyl group to 2ortho to the biphenyl bonds, resulting in 6 and 7, thus leads to a reversal in homo- vs heterodimer affinity.
Table 2. Pharmacological Evaluation of Hetero- and Homodimerization Using BRET2 Assaysa.
| compd | RXRα–NURR1 |
RXRα–RXRα |
NURR1–RXRα vs RXRα–RXRα selectivity | ||
|---|---|---|---|---|---|
| pEC50 (SD) | %Eff (SD) | pEC50 (SD) | %Eff (SD) | fold | |
| LG100268 | 9.3 (0.3) | 100 (7) | 8.2 (0.2) | 100 (11) | 13 |
| 1 | 8.6 (0.2) | 129 (14) | 7.5 (0.1) | 269 (32) | 13 |
| 2 | 8.5 (0.2) | 148 (15) | 7.5 (0.1) | 283 (15) | 10 |
| 3 | 8.6 (0.2) | 178 (18) | 7.7 (0.1) | 286 (44) | 8 |
| 4 | 9.1 (0.4) | 129 (4) | 7.7 (0.1) | 291 (28) | 25 |
| 5 | 8.3 (0.2) | 141 (9) | 7.0 (0.0) | 286 (37) | 20 |
| 6 | 6.1 (0.2) | 330 (38) | 6.8 (0.2) | 236 (11) | 0.2 |
| 7 | 7.1 (0.4) | 123 (12) | 7.5 (0.1) | 175 (24) | 0.4 |
| 9 | 6.8 (0.2) | 41 (25) | 7.7 (0.2) | 66 (6) | 0.1 |
| 10 | <5.0 | 32 (19) | <5.0 | 42 (18) | |
The potency and efficacy of the antagonistic biaryl ligands 9 and 10 to selectively induce RXRα–RXRα homodimers over RXRα–NURR1 heterodimers toward antagonistic conformational changes was also determined. Partial antagonist 9 shows a profile similar to 6 and 7, but with further biased interactions toward RXRα homodimers over RXRα–NURR1 heterodimers by a factor of 10. Additionally, ligand 9 featured a lower efficacy consistent with its partial antagonist character. The full antagonist 10 did not show appreciable activity in the BRET2 assays.
Structural Evaluation
The cocrystallization of ligands 1, 3, and 4 with RXRα showed the canonical interactions of the carboxylate group of the ligands with Arg316, the backbone nitrogen of Ala327, and a conserved water molecule (Figure 2a). The free hydroxyl group on the ligands makes a hydrogen bond with Asn306. This hydrogen bonding network is conserved for all the ligands and directs the positioning of the hydrophobic part of the molecules. The hydrophobic component of 1, 3, and 4 occupies the lipophilic region of the ligand binding pocket. In this region, ligand-dependent RXRα amino acid reorientations can be observed. Especially ligand 4 (i-propyl substitution) repositions Ile324, Val332, Ser336, and Val342 compared to ligands 1 and 3, creating a smaller ligand binding pocket (Figure 2a, zoom and Supporting Information Figure S60). This tighter packing of helices is less pronounced for the region around Ile 324, but mostly affects helices 6 and 7, showing amino acid displacements up to 2.8 Å, and the end of RXRα helix 11, and with that the loop between helix 11 and 12. The carboxy-terminal part of helix 11 has been identified to play a pivotal role in the dimerization of RXRs,42−44 such as via polar contacts between the C-terminal carboxylic acid of PPAR Helix 12 and lysine 431 of RXR helix 10/11.45 NURR1 features an atypical, longer, helix 1227 which, following a modeled RXR-NURR1 heterodimer46 and published RXR-PPAR crystal structures,45,47 probably points toward the RXRα LBD, notably RXRα helices 7 and 11. RXRα–NURR1 heterodimerization thus implies repositioning of RXRα structural elements in this region to accommodate binding of the NURR1 helix 12. Ligand 4 shows the strongest bias toward RXRα–NURR1 heterodimerization (Table 2). The repositioning of RXRα helices 7 and 11 by the compact ligand structure might therefore explain its strong selectivity for heterodimerization.
Figure 2.

Crystal structures of RXRα with agonists 1, 3, 4, 6, and 7. (a) Overlay of the X-ray cocrystal structures of ligands 1 (blue, PDB: 4OC7),353 (orange, PDB: 5MJ5, 1.9 Å resolution), and 4 (gray, PDB: 5MKU, 1.8 Å resolution) bound in the ligand binding pocket of RXRα in ribbon representation with the TIF2 derived coregulator peptide. Zoom-in on the ligand binding pocket of RXRα with the amino acids represented as sticks showing the interactions and displacements. (b) Overlay of the X-ray cocrystal structures of ligands 6 (light green, PDB: 5MMW, 2.7 Å resolution) and 7 (dark green, PDB: 5MK4, 2.0 Å resolution) bound to the ligand binding pocket of RXRα in ribbon representation with the TIF2 derived coregulator peptide. Zoom-in on the ligand binding pocket of RXR with the amino acid represented as sticks showing the interactions between the ligands and the protein. (c) Final 2Fo – Fc electron density maps (contoured at 1σ) of ligands 1, 3, 4, 6, and 7.
Terphenyl ligands 6 and 7 were provided with a methyl functionality at the ortho-position at either of the two biphenyl bonds. Compared to nonmethylated 2, ligands 6 and 7 featured decreased affinity for RXRα and bias toward RXRα–RXRα homodimerization. The X-ray cocrystallization of ligands 6 and 7 with RXRα showed the canonical NR fold, bound to a co-regulator peptide (Figure 2b). The biphenyl core scaffold of 6 and 7 was more out-of-plane rotated in comparison to the other agonists. Ligands 6 and 7 fit the canonical L-shaped ligand binding pocket via the same hydrogen bonding network, but compared to, for example, ligand 4 induce significant changes in the positioning of several amino acids and helices 6, 7, and 11 and their connecting loops, which form the lipophilic region of the binding pocket (Figure 2b). More specifically, compared to ligand 4, the side chain residues of Val332, Ser336 and Val342 are displaced by 1.0 to 2.2 Å when either ligand 6 or 7 are bound (Figures S60–S62). The ortho-methyl substituent on ligand 6 causes the phenyl ring to rotate more out of plane than the other ligands due to steric clash between the methyl and phenyl groups (Figure S61). This specific out-of-plane orientation of the third phenyl group is apparently unfavorable for the binding of ligand 6 as illustrated by the 10-fold lower RXRα activity compared to 7 (Table 1). The additional ortho-methyl substituent on ligand 7 points into the direction of helix 11, displacing Leu436 (Figure S61). This steric interaction is unfavorable in the context of the phenyl substitution pattern, as highlighted by the strong drop in RXRα activity of 7 compared to ligand 2. Combined, the methyl substituents on 6 and 7 result in both cases in repositioning of RXRα structural elements, which may correlate with a lower bias toward RXRα–NURR1 heterodimerization as observed in the BRET2 assay.
To gain structural information about the effects of the additional alkoxy group of 9 and 10, the X-ray structure of the RXRα ligand binding domain complexed with ligand 9 and the co-regulator peptide TIF2 was solved. The protein was crystallized in the agonist conformation, stabilized by interactions with the coregulator peptide, and reflected the partial (ant)agonist character of 9. Comparison of this structure with that of agonist 1 and its analogs (vide supra) revealed, as expected,36 reorientations of amino acid residues related to helix 12 positioning (Figure 3c,d). The most dominant reorientation affected Leu436 in helix 11 induced by the n-propoxy chain, which rotated around 3.0 Å toward helix 12, notably L455. Leu436 has been described as a key residue for the communication between the ligand and helix 12 and the correlated activation function-2 (AF2).9,36,48 The repulsive interaction between the L436 and L455 will lower the association strength between helix 12 and the ligand binding domain, shifting the equilibrium of conformations of the receptor in solution toward an antagonist conformation as demonstrated by the FP and M2H data (Figure 3a,b). The effect might be proportional to the chain length and provides a reasonable rationale for the full antagonism of ligand 10.
The position of the biaryl scaffold of 9 in the RXRα ligand binding pocket is shifted compared to full agonists like 1 and 4 (Figure 3d). This results in concomitant shifts of RXRα helices 6 and 7, especially when compared to ligand 4 and analogous to ligands 6 and 7. Similar to ligand 7, the double substitution pattern on the biaryl scaffold allows partial agonist 9 to simultaneously address two parts of the ligand binding pocket, via helices 6 and 7 and via helices 11 and 12. The combined addressing of the different parts of the RXRα pocket via the hydrophobic substitution pattern leads to amino acid shifts that correlate with preferential affinity for RXRα–RXRα homodimers (Table 2).
Conclusions
Despite the fact that RXR receptors play major roles in many biological processes through heterodimerization with other nuclear receptors, only a small number of small molecule RXR-heterodimer selective modulators are available, with limited chemical diversity and biophysical properties. This study has delivered a compact and focused selection of RXRα–NURR1 agonists based on a versatile biaryl scaffold, structurally different than previously reported molecules.10 In earlier work using chiral dihydrobenzofuran acids, we demonstrated a 3-fold biased interaction with RXRα–NURR1.33 The biaryl scaffold presented here provides a 25-fold selectivity bias for RXRα–NURR1 in the case of analogue 4, and a >100-fold switch in homo- vs heterodimer selectivity when comparing analogs 4 and 9 in Table 2. The structural elucidation of five of these novel RXRα ligands, in our view, provides a first rationale toward understanding how to generate RXRα–NURR1 heterodimer selective ligands. Key ligand–protein interactions and correlated side-chain displacements were identified, modulating both selective dimerization and coregulator recruitment. Interactions of the ligand with key amino acid chains such as Ile324, Val332, Ser336, and Val342 on helices 6 and 7 tune the size of the ligand binding pocket. These compact ligands bind RXRα in a manner that allows movement of helix 7 and 11 to generate a compact ligand binding pocket conformation which arguable is more suited for heterodimerization with NURR1, potentially by enabling the accommodation of the long NURR1 helix 12. Interactions of substituents on the biaryl scaffold with RXRα amino acids involved in formation of the AF2, such as Leu436, induce helix 12 repositioning and translate into lower ligand affinities or, alternatively, into (partial) antagonist properties. These interactions with helices 11 and 12 are matched by the expansion of the RXRα ligand binding pocket via helices 6 and 7, leading to a selectivity of the biaryl scaffold for RXRα–RXRα heterodimers.
This novel series of ligands allows addressing a wide range of RXRα receptor conformations and associated functional outcomes via substitution patterns on the same biaryl scaffold, expanding the current RXR modulator repertoire with agonist as well as antagonist ligands. The data provide a rationale for the design of RXR ligands comprised of a unique hydrophilic region with a conserved hydrogen bonding network contributing to the binding affinity, and a hydrophobic region to probe the other parts of the receptor influencing dimerization properties or coregulator recruitment. These findings justify further exploration of the ligand-controlled homo- vs heterodimerization of RXR and its interaction partners, for activation of the NURR1:RXRα heterodimer as monotherapy for Parkinson’s disease,2,4 for delineating the resulting physiological effects on other RXR heterodimers, and also for potentially revealing conserved mechanisms for other nuclear receptors.
Methods
Synthesis
All the solvents employed were commercially available and used without purification unless stated otherwise. Water was purified using a Millipore purification train. All the reagents are commercially available and used without purification. All the NMR data were recorded on a Varian Gemini 400 MHz NMR, a Bruker Cryomagnet 400 MHz, a Bruker UltraShield Magnet 400 MHz, or a Varian 200 MHz (400 or 200 MHz for 1H NMR and 100 or 50 MHz for 13C NMR). Proton experiments are reported in parts per million (ppm) downfield of TMS. All 13C spectra were reported in ppm relative to residual chloroform (77 ppm). Analytical LC-MS was performed on a C4, Jupiter SuC4300A, 150 × 2.00 mm column with a gradient 5%–100% acetonitrile in H2O supplemented with 0.1% v/v formic acid (FA) in 15 min. Silica column chromatography was performed manually using silica with particle size 60–200 μm. Preparative HP-LC was performed on a Gemini S4 110A 150 × 21.20 mm column using H2O and acetonitrile supplemented with 0.1% v/v F.A. Purity and exact mass of the compounds were determined using a High Resolution LC-MS system consisting of a Waters ACQUITY UPLC I-Class system coupled to a Xevo G2 quadrupole time of flight (Q-tof) system. The system comprised a Binary Solvent Manager and a Sample Manager with Fixed-Loop (SM-FL). compounds were separated (0.3 mL min–1) by the column (Polaris C18A reverse phase column 2.0 × 100 mm, Agilent) using a 15%–75% acetonitrile gradient in water supplemented with 0.1% v/v FA before analysis in positive mode in the mass spectrometer. On the basis of LC-UV data, all final compounds are ≥95% pure.
General Procedure for Suzuki Couplings Method A for the Synthesis of Compounds 13–16, 21, 22, 25, and 26
An oven-dried Schlenk tube was charged with aryl halide (1.0 equiv), boronic acid or boronic ester (1.2 equiv), KF (5.0 equiv), SPhos (0.30 equiv), and Pd2(dba)3 (0.010 equiv). The Schlenk tube was evacuated and backfilled with argon three times. Degassed dioxane/H2O (10:1 v/v, final aryl halide concentration 0.2 M) was added under positive argon flow, and the reaction was stirred at the indicated temperature for the indicated time. The reaction mixture was then allowed to cool to room temperature, passed through Celite with ethyl acetate and concentrated in vacuo. The crude product was purified by flash silica gel chromatography using the indicated eluent and concentrated in vacuo.
General Procedure for Suzuki Couplings Method B for the Synthesis of 19 and 20
An oven-dried Schlenk tube was charged with aryl halide (1.0 equiv), boronic acid or boronic ester (2.0 equiv), KOAc (3.0 equiv), and Pd(dppf)Cl2 (0.010 equiv). The Schlenk tube was evacuated and backfilled with argon three times. Degassed dioxane/H2O (5:1 v/v, final aryl halide concentration 0.4 M) was added under positive argon flow, and the reaction was stirred at the indicated temperature for the indicated time. The reaction mixture was then allowed to cool to room temperature and was separated between CH2Cl2 and H2O. The aqueous layer was extracted twice with CH2Cl2. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by flash silica gel chromatography using the indicated eluent and concentrated in vacuo.
General Procedure for Deprotection, Method A: Methyl Ethers and Methyl Esters
The compound was dissolved in dry CH2Cl2 to a final concentration of ∼0.25 M and cooled to −78 °C. A solution of BBr3 (1 M in CH2Cl2, 2.0 equiv) was added dropwise, and the reaction was stirred at −78 °C for 1 h. The temperature was raised to 0 °C, and the reaction was stirred for another hour. The reaction was then allowed to warm to room temperature and quenched with H2O. The aqueous layer was extracted with CH2Cl2 three times, and the combined organic layers washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo.
The crude product was then dissolved in dioxane/MeOH (14/5 v/v) to a concentration of ∼0.20 M. To this mixture was added NaOH (4 N in deionized water, 3.0 equiv), and the resulting mixture was stirred overnight at either room temperature or 40 °C. The solvent was then removed in vacuo, and the residue separated in H2O and CH2Cl2. The aqueous layer was extracted with CH2Cl2 three times, and the combined organic layers were washed with brine, washed over Na2SO4, filtered, and evaporated in vacuo. The products were then purified by preparative reversed-phase HPLC by UV detection.
General Procedure for Deprotection, Method B: MOM Ethers and Methyl Esters
The compound was dissolved in THF to a final concentration of ∼0.2 M. A solution of HCl (6 N in H2O, 3.0 equiv) was added, and the reaction was stirred at room temperature overnight. The reaction was then diluted with H2O and extracted with Et2O three times. The combined organic layers were washed with saturated aqueous NaHCO3 and brine, dried over Na2SO4, filtered, and concentrated in vacuo.
The crude product was then dissolved in dioxane/MeOH (14/5 v/v) to a concentration of ∼0.20 M. To this mixture was added NaOH (4 N in deionized water, 3.0 equiv), and the resulting mixture was stirred overnight at either room temperature or 40 °C. The solvent was then removed in vacuo, and the residue separated in H2O and CH2Cl2. The aqueous layer was extracted with CH2Cl2 three times, and the combined organic layers were washed with brine, washed over Na2SO4, filtered, and evaporated in vacuo. The products were then purified by preparative reversed-phase HPLC by UV detection and freeze-dried.
(E)-Methyl 3-(6-Methoxy-[1,1′:3′,1″-terphenyl]-3-yl)acrylate (13)
The described procedure for Suzuki couplings method A was used with (E)-methyl 3-(3-chloro-4-methoxyphenyl)acrylate (120 mg, 0.529 mmol), [1,1′-biphenyl]-3-ylboronic acid (130 mg, 0.656 mmol), KF (155 mg, 2.67 mmol), SPhos (67 mg, 0.16 mmol), and Pd2(dba)3 (48 mg, 0.052 mmol) at 110 °C for 18 h. The eluent used for purification was 17% v/v EtOAc in heptane to yield the title compound as a colorless oil, 180 mg, 0.52 mmol, 98% yield. Silica gel TLC Rf = 0.25 (17% v/v EtOAc in heptane); LC-MS (ESI): calcd for C23H20O3 [M + H]: 345.15 observed 345.08, LC Rt = 8.52 min; 1H NMR (400 MHz, CDCl3) δ 7.74–7.66 (m, 2H), 7.66–7.60 (m, 2H), 7.60–7.53 (m, 2H), 7.52–7.47 (m, 3H), 7.47–7.41 (m, 2H), 7.40–7.31 (m, 1H), 6.98 (d, J = 8.5 Hz, 1H), 6.36 (d, J = 16.0 Hz, 1H), 3.83 (s, 3H), 3.78 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 167.84, 158.44, 144.54, 141.26, 141.25, 138.25, 131.25, 130.64, 129.34, 128.86, 128.62, 128.50, 128.45, 127.42, 127.37, 127.33, 126.30, 115.76, 111.47, 55.87, 51.71.
(E)-3-(6-Hydroxy-[1,1′:3′,1″-terphenyl]-3-yl)acrylic Acid (2)
The described procedure for deprotection, method A was used with (E)-methyl 3-(6-methoxy-[1,1′:3′,1″-terphenyl]-3-yl) (13) (100 mg, 0.29 mmol) to afford the title compound, 2, as a white amorphous powder after preparative reverse-phase HPLC and subsequent freeze-drying (34 mg, 0.107 mmol, 37% over two steps). LC-MS (ESI): calcd for C21H16O3 [M + H]: 317.12 observed 317.17, LC Rt = 6.52 min; 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 15.9 Hz, 1H), 7.71–7.55 (m, 5H), 7.54–7.42 (m, 5H), 7.42–7.36 (m, 1H), 7.04 (d, J = 8.7 Hz, 1H), 6.36 (d, J = 15.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 171.82, 155.03, 146.63, 142.75, 140.50, 136.63, 130.84, 130.08, 129.81, 129.08, 128.83, 127.97, 127.92, 127.85, 127.35, 127.34, 127.31, 116.75, 115.09; HRMS (m/z): [M + H]+ calcd 317.1178, found 317.1179.
(E)-Methyl 3-(3′-Benzyl-6-methoxy-[1,1′-biphenyl]-3-yl)acrylate (14)
The described procedure for Suzuki couplings method A was used with (E)-methyl 3-(3-chloro-4-methoxyphenyl)acrylate (97 mg, 0.43 mmol), 2-(3-benzylphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (153 mg, 0.520 mmol), KF (125 mg, 2.15 mmol), SPhos (54 mg, 0.13 mmol), and Pd2(dba)3 (39 mg, 0.040 mmol) at 110 °C for 18 h. The eluent used for purification was 17% v/v EtOAc in heptane to yield the title compound as a colorless oil, 146 mg, 0.41 mmol, 95% yield. Silica gel TLC Rf = 0.28 (17% v/v EtOAc in heptane); LC-MS (ESI): calcd for C24H22O3 [M + H]: 359.16 observed 359.08, LC, Rt = 8.68 min; 1H NMR (400 MHz, CDCl3) δ 7.67 (d, J = 16.0 Hz, 1H), 7.48–7.42 (m, 2H), 7.37–7.25 (m, 5H), 7.24–7.14 (m, 4H), 6.92 (d, J = 8.3 Hz, 1H), 6.33 (d, J = 16.0 Hz, 1H), 4.02 (s, 2H), 3.77 (s, 3H), 3.77 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 167.80, 158.38, 144.56, 141.07, 140.98, 137.83, 131.30, 130.61, 130.15, 129.08, 128.54, 128.28, 128.02, 127.31, 127.22, 126.17, 115.64, 111.41, 55.75, 51.67, 42.05.
(E)-3-(3′-Benzyl-6-hydroxy-[1,1′-biphenyl]-3-yl)acrylic Acid (3)
The described procedure for deprotection, method A was used with (E)-methyl 3-(3′-benzyl-6-methoxy-[1,1′-biphenyl]-3-yl)acrylate (14) (60 mg, 0.17 mmol) to afford the title compound, 3, as a white amorphous powder after preparative reverse-phase HPLC and subsequent freeze-drying (24 mg, 0.073 mmol, 43% over two steps). LC-MS (ESI): calcd for C22H18O3 [M + H]: 331.14 observed 331.00, LC Rt = 6.65 min; 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 15.9 Hz, 1H), 7.49–7.40 (m, 3H), 7.33–7.19 (m, 8H), 6.99 (d, J = 8.4 Hz, 1H), 6.33 (d, J = 15.9 Hz, 1H), 4.05 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 172.48, 154.98, 146.74, 142.96, 140.63, 136.23, 130.81, 129.80, 129.66, 129.57, 129.15, 129.05, 128.84, 128.78, 127.17, 126.75, 126.50, 116.63, 115.08, 42.09; HRMS (m/z): [M + H]+ calcd 331.1334, found 331.1319.
(E)-Methyl 3-(3′-Isopropyl-6-methoxy-[1,1′-biphenyl]-3-yl)acrylate (15)
The described procedure for Suzuki couplings method A was used with (E)-methyl 3-(3-chloro-4-methoxyphenyl)acrylate (145 mg, 0.640 mmol), (3-isopropylphenyl)boronic acid (128 mg, 0.780 mmol), KF (187 mg, 3.22 mmol), SPhos (81 mg, 0.20 mmol), and Pd2(dba)3 (61 mg, 0.066 mmol) at 110 °C for 18 h. The eluent used for purification was 15% v/v EtOAc in heptane to yield the title compound as a colorless oil in a quantitative yield. Silica gel TLC Rf = 0.35 (15% v/v EtOAc in heptane); LC-MS (ESI): calcd for C20H22O3 [M + H]: 311.16 observed 311.08, LC, Rt = 8.52 min; 1H NMR (399 MHz, CDCl3) δ 7.70 (d, J = 16.0 Hz, 1H), 7.52–7.45 (m, 2H), 7.41–7.31 (m, 4H), 6.96 (d, J = 8.5 Hz, 1H), 6.35 (d, J = 16.0 Hz, 1H), 3.83 (s, 3H), 3.79 (s, 3H), 2.96 (hept, J = 6.9 Hz, 1H), 1.29 (d, J = 6.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 167.86, 158.44, 148.73, 144.66, 137.65, 130.68, 129.06, 128.50, 128.09, 127.75, 127.24, 127.05, 125.56, 115.60, 111.42, 55.81, 51.69, 34.25, 24.14.
(E)-3-(6-Hydroxy-3′-isopropyl-[1,1′-biphenyl]-3-yl)acrylic Acid (4)
The described procedure for deprotection, method A was used with (E)-methyl 3-(3′-isopropyl-6-methoxy-[1,1′-biphenyl]-3-yl)acrylate (15) (100 mg, 0.32 mmol) to afford the title compound, 4, as a white amorphous powder after preparative reverse-phase HPLC and subsequent freeze-drying (45 mg, 0.16 mmol, 50%,over two steps). LC-MS (ESI): calcd for C18H18O3 [M + H]: 283.13 observed 283.17, LC, Rt = 6.43 min; 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 15.9 Hz, 1H), 7.51–7.42 (m, 3H), 7.34–7.27 (m, 3H), 7.02 (d, J = 8.3 Hz, 1H), 6.34 (d, J = 15.9 Hz, 1H), 2.98 (d, J = 6.9 Hz, 1H), 1.30 (d, J = 6.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 172.43, 155.05, 150.66, 146.82, 135.97, 130.79, 129.68, 129.65, 129.17, 127.26, 127.14, 126.80, 126.38, 116.57, 115.00, 34.34, 24.14; HRMS (m/z): [M + H]+ calcd 283.1334, found 283.1334.
(E)-Methyl 3-(4-Methoxy-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)acrylate (12b)35
1-Bromo-3-propylbenzene
To a solution of 3-bromopropiophenone (2.98 g, 14.0 mmol) in TFA (30 mL, 0.40 M) was added dropwise triethylsilane (11.5 mL, 72.0 mmol) at 0 °C in 5 min, and the mixture was stirred for additional 20 min. The reaction mixture was heated to 80 °C and stirred overnight. The reaction mixture was allowed to cool to room temperature and concentrated in vacuo. Toluene was added, and the mixture was again concentrated in vacuo to obtain the crude material. The product was purified via flash silica gel chromatography eluting with hexane to yield the title compound, 78 mg, 0.39 mmol, 3% yield; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.38–7.27 (m, 2H), 7.18–7.06 (m, 2H), 2.59–2.49 (m, 2H), 1.72–1.56 (m, 2H), 0.93 (td, J = 7.3, 0.9 Hz, 3H); 13C NMR (100 MHz, CDCl3): 145.16, 131.65, 129.91, 128.87, 127.28, 122.45, 37.81, 24.49, 13.87.
(E)-Methyl 3-(6-methoxy-3′-propyl-[1,1′-biphenyl]-3-yl)acrylate (16)
The described procedure for Suzuki couplings method A was used with (E)-methyl 3-(4-methoxy-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)acrylate (152 mg, 0.478 mmol), 1-bromo-3-propylbenzene (78 mg, 0.39 mmol), KF (114 mg, 1.96 mmol), SPhos (49 mg, 0.12 mmol), and Pd2(dba)3 (37 mg, 0.040 mmol) at 100 °C for 7 h. The eluent used for purification was 5%–10% v/v EtOAc in hexane to yield the title compound as a dark yellow oil, 95 mg, 0.31 mmol, 78% yield; 1H NMR (400 MHz, CDCl3): 7.69 (d, J = 16.0 Hz, 1H), 7.55–7.45 (m, 2H), 7.36–7.30 (m, 3H), 7.23–7.13 (m, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.35 (d, J = 16.0 Hz, 1H), 3.85 (s, 3H), 3.80 (s, 3H), 2.75–2.57 (m, 2H), 1.76–1.61 (m, 2H), 0.98 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3): 167.90, 158.50, 144.68, 142.66, 137.65, 130.69, 129.73, 129.12, 128.54, 128.08, 127.68, 127.30, 126.94, 115.68, 111.45, 55.89, 51.75, 38.27, 24.73, 14.07.
(E)-3-(6-Hydroxy-3′-propyl-[1,1′-biphenyl]-3-yl)acrylic Acid (5)
The described procedure for deprotection, method A was used with (E)-methyl 3-(6-methoxy-3′-propyl-[1,1′-biphenyl]-3-yl)acrylate (16), with the adjustment that the intermediate after treatment with BBr3 was purified using silica gel chromoatography eluting with 5%–10% v/v EtOAc in hexane to yield the title compound, 5, as a yellow oil, 25 mg, 0.085 mmol, 42% yield. On repeating the reaction a second time (25.1 mg, 0.084 mmol), the title compound was isolated in a 37% yield over two steps (8.8 mg, 0.03 mmol) as a white amorphous powder after preparative reverse-phase HPLC and subsequent freeze-drying. LC-MS (ESI): calcd for C18H18O3 [M + H]: 283.13, observed 283.17, LC, Rt = 6.47; 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 15.9 Hz, 1H), 7.53–7.36 (m, 3H), 7.27–7.23 (m, 3H), 7.01 (d, J = 8.2 Hz, 1H), 6.33 (d, J = 15.9 Hz, 1H), 2.72–2.62 (m, 2H), 1.75–1.66 (m, 2H), 0.98 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.22, 155.07, 146.83, 144.45, 135.93, 130.77, 129.64, 129.58, 129.19, 129.08, 128.78, 127.14, 126.26, 116.58, 114.95, 38.16, 24.69, 14.02.
3-Chloro-2-methyl-1,1′-biphenyl (19)
The described general Suzuki coupling conditions method B was used with 1-chloro-3-iodo-2-methylbenzene (275 μL, 1.97 mmol), phenylboronic acid (364 mg, 2.99 mmol), KOAc (584 mg, 5.95 mmol), and Pd(dppf)Cl2 (148 mg, 0.181 mmol) at 90 °C for 5.5 h. The eluent used for purification was heptane to yield the title compound as a colorless oil, 229 mg, 1.13 mmol, 57% yield. Silica gel TLC Rf = 0.56 (heptane); GC-MS (EI) m/z calc. for C13H11Cl: 202.05, most abundant peaks observed: 202, 167; 1H NMR (400 MHz, CDCl3) δ 7.43–7.29 (m, 4H), 7.28–7.21 (m, 2H), 7.18–7.03 (m, 2H), 2.27 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 144.11, 141.57, 135.40, 133.84, 129.27, 128.42, 128.29, 127.28, 126.49, 17.99.
(E)-Methyl 3-(6-(Methoxymethoxy)-2′-methyl-[1,1′:3′,1″-terphenyl]-3-yl)acrylate (21)
The described general Suzuki coupling conditions method A was used with (E)-methyl 3-(4-(methoxymethoxy)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)acrylate (104 mg, 0.300 mmol), 3-chloro-2-methyl-1,1′-biphenyl (52 mg, 0.26 mmol), KF (72 mg, 1.2 mmol), SPhos (32 mg, 0.078 mmol), and Pd2(dba)3 (25 mg, 0.027 mmol) at 110 °C for 21 h. The eluent used for purification was 17% v/v EtOAc in heptane to yield the title compound as a colorless oil in a quantitative yield. Silica gel TLC Rf = 0.31 (heptane/EtOAc 17% v/v); LC-MS (ESI): calcd for C25H24O4 [M + H]: 389.18 observed 389.08, LC, Rt = 8.63 min. 1H NMR (400 MHz, CDCl3): δ 7.68 (d, J = 16.0 Hz, 1H), 7.49 (dd, J = 8.6, 2.3 Hz, 1H), 7.45–7.32 (m, 6H), 7.31–7.20 (m, 3H), 7.18 (dd, J = 7.0, 2.1 Hz, 1H), 6.35 (d, J = 16.0 Hz, 1H), 5.14 (s, 2H), 3.79 (s, 3H), 3.37 (s, 3H), 2.02 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 167.77, 156.38, 144.46, 142.45, 142.43, 138.75, 134.27, 132.90, 131.03, 129.50, 129.16, 129.10, 128.26, 128.19, 126.92, 125.33, 116.25, 115.08, 94.72, 56.33, 51.74, 18.18.
(E)-3-(6-Hydroxy-2′-methyl-[1,1′:3′,1″-terphenyl]-3-yl)acrylic Acid (6)
The described procedure for deprotection, method B was used at 40 °C with (E)-methyl 3-(6-(methoxymethoxy)-2′-methyl-[1,1′:3′,1″-terphenyl]-3-yl)acrylate (21). LC-MS (ESI): calcd for C22H18O3 [M + H]: 331.13 observed 331.17, LC, Rt = 6.67 min; 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 15.9 Hz, 1H), 7.51 (dd, J = 8.5, 2.2 Hz, 1H), 7.47–7.32 (m, 8H), 7.24 (dd, J = 6.7, 2.1 Hz, 1H), 7.04 (d, J = 8.4 Hz, 1H), 6.34 (d, J = 15.9 Hz, 1H), 2.04 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 172.46, 155.20, 146.81, 143.84, 141.79, 135.45, 135.00, 130.90, 130.83, 129.85, 129.67, 129.37, 129.05, 128.37, 127.26, 127.03, 126.53, 116.25, 115.05, 17.94. HRMS (m/z): [M + H]+ calcd for C22H18O3, 331.1334, found 331.1331.
3-Chloro-4-methyl-1,1′-biphenyl (20)
The described general Suzuki coupling conditions method B was used with 2-chloro-4-iodo-1-methylbenzene (321 mg, 1.27 mmol), phenylboronic acid (226 mg, 1.85 mmol), KOAc (354 mg, 3.61 mmol), and Pd(dppf)Cl2 (95 mg, 0.12 mmol) at 90 °C for 3.5 h. The eluent used for purification was heptane to yield the title compound as a colorless oil, 124 mg, 0.61 mmol, 51% yield. Silica gel TLC Rf = 0.49 (heptane); GC-MS (ESI) m/z calc. for C13H11Cl: 202.05, most abundant peaks observed: 202, 167; 1H NMR (400 MHz, CDCl3) δ 7.62–7.49 (m, 3H), 7.47–7.29 (m, 4H), 7.29–7.22 (m, 1H), 2.39 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 140.56, 139.88, 134.98, 134.87, 131.35, 128.96, 127.70, 127.67, 127.04, 125.37, 19.85.
(E)-Methyl 3-(6-(Methoxymethoxy)-6′-methyl-[1,1′:3′,1″-terphenyl]-3-yl)acrylate (22)
The described general Suzuki coupling conditions method A was used with (E)-methyl 3-(4-(methoxymethoxy)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)acrylate (103 mg, 0.300 mmol), 3-chloro-4-methyl-1,1′-biphenyl (48 mg, 0.24 mmol), KF (72 mg, 1.2 mmol), SPhos (32 mg, 0.078 mmol), and Pd2(dba)3 (23 mg, 0.025 mmol) at 110 °C for 21 h. The eluent used for purification was 17% v/v EtOAc in heptane to yield the title compound 65 mg, 0.17 mmol, 70% yield. Silica gel TLC Rf = 0.29 (heptane/EtOAc 17% v/v); LC-MS (ESI): calcd for C25H24O4 [M + H]: 389.18 observed 389.08, LC, Rt = 8.65 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.69 (d, J = 16.0 Hz, 1H), 7.63–7.59 (m, 2H), 7.57–7.32 (m, 9H), 6.36 (d, J = 16.0 Hz, 1H), 5.13 (s, 2H), 3.79 (s, 3H), 3.36 (s, 3H), 2.21 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 167.76, 156.42, 144.42, 140.90, 138.60, 138.39, 135.96, 132.41, 130.98, 130.30, 129.20, 128.87, 128.73, 128.30, 127.22, 127.08, 126.37, 116.30, 115.18, 94.80, 56.35, 51.75, 19.79.
(E)-3-(6-Hydroxy-6′-methyl-[1,1′:3′,1″-terphenyl]-3-yl)acrylic Acid (7)
The described procedure for deprotection, method B was used at 40 °C with (E)-methyl 3-(6-(methoxymethoxy)-6′-methyl-[1,1′:3′,1″-terphenyl]-3-yl)acrylate (22) (32 mg, 0.082 mmol LC-MS (ESI): calcd for C22H18O3 [M + H]: 331.13 observed 331.17, LC, Rt = 6.67 min; 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 15.9 Hz, 1H), 7.64–7.56 (m, 3H), 7.52 (dd, J = 8.5, 2.2 Hz, 1H), 7.48 (d, J = 2.0 Hz, 1H), 7.47–7.38 (m, 4H), 7.38–7.31 (m, 1H), 7.04 (d, J = 8.5 Hz, 1H), 6.33 (d, J = 15.9 Hz, 1H), 2.21 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 172.26, 155.19, 146.75, 140.26, 139.90, 136.44, 135.16, 131.56, 130.85, 129.92, 129.15, 129.02, 128.52, 127.73, 127.63, 127.10, 127.06, 116.30, 115.05, 19.54. HRMS (m/z): [M + H]+ calcd for C22H18O3, 331.1334, found 331.1328.
(E)-methyl 3-(5′-allyl-6-(methoxymethoxy)-2′-propoxy-[1,1′-biphenyl]-3-yl)acrylate (25)
The described general Suzuki coupling conditions method A was used with (E)-methyl 3-(4-(methoxymethoxy)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)acrylate (72 mg, 0.21 mmol), 4-allyl-2-chloro-1-propoxybenzene (40 mg, 0.19 mmol), KF (52 mg, 0.90 mmol), SPhos (22.2 mg, 0.054 mmol), and Pd2(dba)3 (17 mg, 0.020 mmol) at 110 °C for 21 h. The eluent used for purification was 17% v/v EtOAc in heptane to yield the title compound, 67 mg, 0.17 mmol, 94% yield. Silica gel TLC Rf = 0.29 (heptane/EtOAc 17% v/v); LC-MS (ESI): calcd for C24H28O5 [M + H]: 397.20, observed 397.00, LC, Rt = 8.80; 1H NMR (400 MHz, Chloroform-d) δ 7.67 (d, J = 16.0 Hz, 1H), 7.49–7.38 (m, 2H), 7.19 (d, J = 9.1 Hz, 1H), 7.13 (dd, J = 8.3, 2.3 Hz, 1H), 7.06 (d, J = 2.4 Hz, 1H), 6.89 (d, J = 8.4 Hz, 1H), 6.33 (d, J = 16.0 Hz, 1H), 5.98 (ddt, J = 16.8, 10.0, 6.7 Hz, 1H), 5.17–4.95 (m, 4H), 3.85 (t, J = 6.5 Hz, 2H), 3.78 (s, 3H), 3.44–3.28 (m, 5H), 1.68–1.56 (m, 2H), 0.83 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 167.73, 156.79, 154.86, 144.64, 137.71, 131.64, 131.45, 131.44, 129.55, 128.77, 128.75, 127.73, 127.29, 115.67, 115.55, 115.02, 112.43, 94.78, 70.18, 56.00, 51.54, 39.36, 22.56, 10.40.
(E)-3-(5′-Allyl-6-hydroxy-2′-propoxy-[1,1′-biphenyl]-3-yl)acrylic Acid (9)
The described procedure for deprotection, method B was used at room temperature with methyl (E)-3-(5′-allyl-6-(methoxymethoxy)-2′-propoxy-[1,1′-biphenyl]-3-yl)acrylate (25) (31 mg, 0.078 mmol) LC-MS (ESI): calcd for C21H22O4 [M + H]: 339.16 observed 339.0, LC, Rt = 6.82 min. 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 15.9 Hz, 1H), 7.52 (dd, J = 8.4, 2.2 Hz, 1H), 7.47 (d, J = 2.2 Hz, 1H), 7.22 (dd, J = 8.4, 2.2 Hz, 1H), 7.16 (d, J = 2.2 Hz, 1H), 7.04 (d, J = 8.4 Hz, 1H), 7.00 (d, J = 8.4 Hz, 1H), 6.35 (d, J = 15.9 Hz, 1H), 5.98 (ddt, J = 16.8, 10.1, 6.7 Hz, 1H), 5.17–5.04 (m, 2H), 4.02 (t, J = 6.7 Hz, 2H), 3.44–3.37 (m, 2H), 1.76 (dtd, J = 13.8, 7.4, 6.5 Hz, 2H), 0.94 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.78, 156.66, 153.39, 147.04, 137.37, 134.45, 132.68, 132.27, 129.84, 129.43, 127.28, 127.21, 126.82, 118.47, 116.25, 114.65, 113.69, 71.77, 39.50, 31.09, 22.61, 10.51; HRMS (m/z): [M + H]+ calcd for C21H22O4, 339.1596, found 339.1592.
(E)-Methyl 3-(5′-Allyl-2′-(hexyloxy)-6-(methoxymethoxy)-[1,1′-biphenyl]-3-yl)acrylate (26)
The described general Suzuki coupling conditions method A was used with (E)-methyl 3-(4-(methoxymethoxy)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)acrylate (66 mg, 0.19 mmol), 4-allyl-2-chloro-1-(hexyloxy)benzene (40 mg, 0.16 mmol), KF (49 mg, 0.84 mmol), SPhos (20 mg, 0.049 mmol), and Pd2(dba)3 (16 mg, 0.017 mmol) at 110 °C for 21 h. The eluent used for purification was 17% v/v EtOAc in heptane to yield the title compound, 68 mg, 0.15 mmol, 98% yield. Silica gel TLC Rf = 0.39 (heptane/EtOAc 17% v/v); LC-MS (ESI): calcd for C27H34O5 [M + H]: 439.25, observed 439.08, LC, Rt = 9.63 min; 1H NMR (399 MHz, Chloroform-d) δ 7.67 (d, J = 16.0 Hz, 1H), 7.48–7.39 (m, 3H), 7.19 (d, J = 9.1 Hz, 1H), 7.16–7.04 (m, 1H), 6.90 (d, J = 8.3 Hz, 1H), 6.33 (d, J = 16.0 Hz, 1H), 5.98 (ddt, J = 16.8, 10.0, 6.7 Hz, 1H), 5.17–4.97 (m, 4H), 3.88 (t, J = 6.5 Hz, 2H), 3.78 (s, 3H), 3.37 (s, 5H), 1.65–1.48 (m, 2H), 1.37–1.08 (m, 6H), 0.85–0.76 (m, 3H); 13C NMR (100 MHz, cdcl3) δ 167.88, 156.92, 155.02, 144.80, 137.86, 131.77, 131.61, 131.58, 129.10, 128.92, 128.53, 127.86, 127.41, 115.80, 115.72, 115.15, 112.61, 94.89, 68.87, 56.16, 51.70, 39.52, 31.58, 29.32, 25.77, 22.68, 14.11.
(E)-3-(5′-Allyl-2′-(hexyloxy)-6-hydroxy-[1,1′-biphenyl]-3-yl)acrylic Acid (10)
The described procedure for deprotection, method B was used at room temperature with methyl (E)-3-(5′-allyl-2′-(hexyloxy)-6-(methoxymethoxy)-[1,1′-biphenyl]-3-yl)acrylate (26) (17 mg, 0.039 mmol) LC-MS (ESI): calcd for C24H28O4 [M + H]: 381.21 observed 381.00, LC, Rt = 7.83 min; 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 15.9 Hz, 1H), 7.51 (dd, J = 8.4, 2.2 Hz, 1H), 7.47 (d, J = 2.2 Hz, 1H), 7.22 (dd, J = 8.3, 2.2 Hz, 1H), 7.16 (d, J = 2.2 Hz, 1H), 7.04 (d, J = 8.3 Hz, 1H), 7.00 (d, J = 8.3 Hz, 1H), 6.35 (d, J = 15.9 Hz, 1H), 5.98 (ddt, J = 16.8, 10.1, 6.7 Hz, 1H), 5.20–5.01 (m, 2H), 4.04 (t, J = 6.7 Hz, 2H), 3.41 (d, J = 6.7 Hz, 2H), 1.72 (p, J = 6.7 Hz, 2H), 1.44–1.12 (m, 6H), 0.83 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.27, 156.68, 153.45, 147.06, 137.38, 134.46, 132.64, 132.26, 129.84, 129.44, 127.30, 127.22, 126.88, 118.49, 116.24, 114.76, 113.78, 70.35, 39.51, 31.48, 29.13, 25.57, 22.59, 14.05; HRMS (m/z): [M + H]+ calcd for C24H28O4, 381.2066, found 381.2063.
4-Bromo-2-chloro-1-(methoxymethoxy)benzene
To a solution of 4-bromo-2-chlorophenol (7.35 g, 35.4 mmol, 1.0 equiv) in CH2Cl2 (50 mL, 0.70 M) was added N,N-diisopropylethylamine (18.5 mL, 106 mmol, 3.0 equiv) and MOMCl (5.38 mL, 70.8 mmol, 2.0 equiv). The reaction was stirred at room temperature for 17 h. The reaction mixture was separated between CH2Cl2 and H2O and the aqueous layer was extracted twice with CH2Cl2. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The product was purified via flash silica gel chromatography eluting with 50% v/v CH2Cl2 in heptane to yield the title compound as a colorless oil, 8.9 g, 35 mmol, 99% yield. Silica gel TLC Rf = 0.51 (CH2Cl2/heptane 1/1 v/v); 1H NMR (400 MHz, CDCl3) δ 7.51 (d, J = 2.4 Hz, 1H), 7.30 (dd, J = 8.8, 2.4 Hz, 1H), 7.05 (d, J = 8.8 Hz, 1H), 5.22 (s, 2H), 3.50 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 152.30, 132.84, 130.72, 124.88, 117.74, 114.19, 95.36, 56.54.
(E)-Methyl 3-(3-Chloro-4-(methoxymethoxy)phenyl)acrylate (17)
An oven-dried Schlenk tube was charged with 4-bromo-2-chloro-1-(methoxymethoxy)benzene (2.0 g, 8.0 mmol, 1.0 equiv), methyl acrylate (3.0 mL, 33.1 mmol, 4.1 equiv), Pd(dppf)Cl2 (0.94 g, 1.2 mmol, 0.15 equiv), NEt3 (35 mL, 251 mmol, 31 equiv), and DMF (20 mL, 0.40 M), and the reaction was stirred at 110 °C. After 20 h, the reaction mixture was poured into H2O and extracted with CH2Cl2 three times. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The product was purified via flash silica gel chromatography, eluting with 20% v/v EtOAc in heptane to yield the title compound as a white solid, 1.6 g, 6.2 mmol, 78% yield. Silica gel TLC Rf = 0.34 (heptane/EtOAc 20% v/v); GC-MS (ESI) m/z calcd for C12H13ClO4: 256.05, most abundant peaks observed: 256, 226, 195; 1H NMR (400 MHz, CDCl3) δ 7.61–7.55 (m, 2H), 7.36 (dd, J = 8.6, 2.0 Hz, 1H), 7.18 (d, J = 8.6 Hz, 1H), 6.33 (d, J = 15.9 Hz, 1H), 5.28 (s, 2H), 3.80 (s, 3H), 3.52 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 167.41, 154.45, 143.15, 129.75, 129.27, 128.04, 124.24, 117.37, 116.14, 95.08, 56.63, 51.87.
(E)-Methyl 3-(4-(Methoxymethoxy)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)acrylate (18)
An oven-dried Schlenk tube was charged with (E)-methyl 3-(3-chloro-4-(methoxymethoxy)phenyl)acrylate (489 mg, 1.91 mmol, 1.0 equiv), KOAc (573 mg, 5.84 mmol, 3.1 equiv), bis(pinacolato)diboron (1.34 g, 5.28 mmol, 2.8 equiv), Xphos (78 mg, 0.16 mmol, 0.080 equiv), and Pd(OAc)2 (22 mg, 0.098 mmol, 0.051 equiv). The Schlenk tube was evacuated and backfilled with argon three times. Dioxane (6.5 mL, aryl halide concentration 0.30 M) was added under a positive argon flow, and the reaction was stirred at 110 °C for 5 h. The reaction mixture was then allowed to cool to room temperature and passed through Celite, eluting with EtOAc. The product was purified via flash silica gel chromatography eluting with 25% v/v EtOAc in heptane to yield the title compound, 524 mg, 1.51 mmol, 80% yield. Silica gel TLC Rf = 0.25 (heptane/EtOAc 25% v/v); LC-MS (ESI): calcd for C18H25BO6 [M + H]: 349.18, observed 348.92, LC, Rt = 7.42; 1H NMR (400 MHz, CDCl3) δ 7.87 (d, J = 2.3 Hz, 1H), 7.65 (d, J = 16.0 Hz, 1H), 7.53 (dd, J = 8.6, 2.3 Hz, 1H), 7.03 (d, J = 8.6 Hz, 1H), 6.36 (d, J = 16.0 Hz, 1H), 5.22 (s, 2H), 3.78 (s, 3H), 3.49 (s, 3H), 1.35 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 167.82, 163.36, 144.49, 136.92, 132.38, 128.00, 116.05, 115.18, 94.78, 83.89, 56.35, 51.71, 24.97.
4-Bromo-2-chloro-1-propoxybenzene
4-Bromo-2-chlorophenol (4.0 g, 19 mmol, 1.0 equiv) was dissolved in dry DMF (100 mL, concentration 0.20 M) in an oven-dried round-bottom flask. To this solution, K2CO3 (8.0 g, 58 mmol, 3.1 equiv) and 1-bromopropane (8.8 mL, 96 mmol, 5.1 equiv) were added, and the reaction was stirred at 70 °C for 22 h. The reaction was then quenched with H2O and extracted with CH2Cl2 three times. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The product was purified via flash silica gel chromatography, eluting with 5% v/v EtOAc in heptane to yield fourthe title compound as a colorless oil, 4.7 g, 19 mmol, 98% yield. Silica gel TLC Rf = 0.46 (heptane/EtOAc 5% v/v); GC-MS (ESI) m/z calcd for C9H10BrClO: 249.53, most abundant peaks observed: 250, 210, 208, Rt = 5.04; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.45 (d, J = 2.4 Hz, 1H), 7.25 (dd, J = 8.7, 2.4 Hz, 1H), 6.72 (d, J = 8.7 Hz, 1H), 3.94 (t, J = 6.6 Hz, 2H), 1.53–1.40 (m, 2H), 0.95–0.82 (m, 3H).); 13C NMR (50 MHz, CDCl3) δ 153.99, 132.62, 130.48, 124.07, 114.49, 112.26, 70.84, 22.52, 10.54.
4-Allyl-2-chloro-1-propoxybenzene (23)
An oven dried Schlenk flask was charged with 4-bromo-2-chloro-1-propoxybenzene (1.02 g, 4.09 mmol, 1.0 equiv), CsF (1.31 g, 8.62 mmol, 2.1 equiv), and Pd(PPh3)4 (467 mg, 0.404 mmol, 0.099 equiv). The flask was then evacuated and backfilled with argon three times, and THF (34 mL, aryl halide concentration 0.12 M) was added. The mixture was stirred for 30 min at room temperature before 2-allyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.35 mL, 7.21 mmol, 1.8 equiv) was added, and the reaction was stirred at 78 °C for 22 h. Another portion of CsF (1.26 g, 8.29 mmol, 2.0 equiv), Pd(PPh3)4 (467 mg, 0.404 mmol, 0.099 equiv), and THF (30 mL) was added, and the reaction was stirred at 78 °C for another 24 h. The reaction was then allowed to cool to room temperature and was separated between pentane and H2O. The aqueous layer was washed with pentane twice, and the combined organic layers were washed with brine, dried over Na2SO4, filtered twice, and concentrated in vacuo. The product was purified via flash silica gel chromatography eluting with 5% v/v EtOAc in heptane to yield the title compound as colorless oil, 690 mg, 3.27 mmol, 82% yield. Silica gel TLC Rf = 0.58 (heptane/EtOAc 5% v/v); GC-MS (ESI) m/z calcd for C12H15ClO: 210.70, most abundant peaks observed: 210, 168, 133, Rt = 5.02; 1H NMR (400 MHz, CDCl3) δ 7.18 (d, J = 2.2 Hz, 1H), 6.98 (dd, J = 8.4, 2.2 Hz, 1H), 6.82 (d, J = 8.4 Hz, 1H), 6.04–5.70 (m, 1H), 5.10–5.06 (m, 1H), 5.06–5.01 (m, 1H), 3.95 (t, J = 6.5 Hz, 2H), 3.54–2.99 (m, 2H), 1.91–1.76 (m, 2H), 1.05 (t, J = 7.4 Hz, 3H); 13C NMR (50 MHz, CDCl3) δ 153.04, 137.21, 133.11, 130.36, 127.70, 122.87, 116.11, 113.56, 70.82, 39.14, 22.66, 10.61.
4-Bromo-2-chloro-1-(hexyloxy)benzene
4-Bromo-2-chlorophenol (4.0 g, 19 mmol) was dissolved in dry DMF (100 mL, final concentration 0.19 M) in an oven-dried round-bottom flask. To this solution was added K2CO3 (8.0 g, 58 mmol, 3.1 equiv) and 1-bromohexane (14 mL, 1.0 × 102 mmol, 5.3 equiv), and the reaction was stirred at 70 °C for 18 h. The reaction was separated between H2O and CH2Cl2 and the aqueous layer was washed with CH2Cl2 twice. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The product was purified via flash silica gel chromatography eluting with 5% v/v EtOAc in hexane to yield the title compound as a colorless oil, 5.5 g, 19 mmol, 98% yield. Silica gel TLC Rf = 0.70 (hexane/EtOAc 5% v/v). GC-MS (ESI) m/z calcd for C12H16BrClO: 291.6, most abundant peaks observed: 292, 210, 208, Rt = 6.25; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.45 (d, J = 2.5 Hz, 1H), 7.25 (dd, J = 8.7, 2.4 Hz, 1H), 6.72 (d, J = 8.8 Hz, 1H), 3.94 (t, J = 6.6 Hz, 2H), 1.87–1.71 (m, 2H), 1.53–1.40 (m, 2H), 1.40–1.24 (m, 4H), 0.95–0.82 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 154.04, 132.68, 130.53, 124.10, 114.47, 112.26, 69.44, 31.60, 29.08, 25.69, 22.68, 14.11.
4-Allyl-2-chloro-1-(hexyloxy)benzene (24)
An oven-dried Schlenk flask was charged with 4-bromo-2-chloro-1-(hexyloxy)benzene (1.01 g, 3.47 mmol, 1.0 equiv), CsF (1.12 g, 7.37 mmol, 2.1 equiv), and Pd(PPh3)4 (397 mg, 0.344 mmol, 0.099 equiv). The flask was evacuated and backfilled with argon three times, and THF (22 mL, aryl halide concentration 0.16 M) was added. The mixture was stirred for 30 min at room temperature, and then 2-allyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1.16 mL, 6.17 mmol, 1.8 equiv) and THF (7.5 mL) were added. The reaction was stirred at 78 °C for 21 h. Another portion of CsF (1.12 g, 7.37 mmol, 2.1 equiv), Pd(PPh3)4 (401 mg, 0.347 mmol, 0.010 equiv), and THF (30 mL) was added, and the mixture was stirred at 78 °C for another 24 h. The mixture was allowed to cool to room temperature and was separated between pentane and H2O. The aqueous layer was washed with pentane twice and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The product was purified via flash silica gel chromatography eluting with 3% v/v EtOAc in heptane to yield the title compound as a colorless oil, 789 mg, 3.12 mmol, 91% yield. Silica gel TLC Rf = 0.38 (heptane/EtOAc 3% v/v); GC-MS (ESI) m/z calcd for C15H21ClO: 252.78, most abundant peaks observed: 252, 168, 133, Rt = 6.21; 1H NMR (400 MHz, CDCl3) δ 7.18 (d, J = 2.2 Hz, 1H), 7.00 (dd, J = 8.4, 2.2 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 5.92 (ddt, J = 17.6, 9.5, 6.7 Hz, 1H), 5.12–5.05 (m, 1H), 5.08–5.01 (m, 1H), 4.00 (t, J = 6.7 Hz, 2H), 3.30 (dt, J = 6.7, 1.5 Hz, 2H), 1.88–1.72 (m, 2H), 1.57–1.42 (m, 2H), 1.41–1.28 (m, 4H), 0.90 (td, J = 5.9, 4.7, 3.5 Hz, 3H)); 13C NMR (100 MHz, CDCl3) δ 153.11, 137.26, 133.16, 130.41, 127.71, 122.94, 116.15, 113.64, 69.47, 39.18, 31.69, 29.27, 25.79, 22.74, 14.17.
Mammalian Two-Hybrid (M2H) Assays
Mammalian two-hybrid (M2H) assays were performed as previously described.35
General Considerations for Protein Expression and Purification
All solutions and equipment used in the handling of microbial cultures were autoclaved or sterile filtered. Media, plastic, and glassware were autoclaved at 121 °C for 20 min prior to use. Bacterial cultures were incubated in a New Brunswick Series 25 shaker. Centrifugation was performed in a Beckman Coulter Avanti J-25 centrifuge. Microcentrifugation was performed in an Eppendorf centrifuge 5415R or a Beckman Coulter microfuge 18. All biological laboratory buffers and media were bought from common suppliers and used as purchased. BL21(DE3) and NovaBlue Escherichia coli competent cells were purchased from Novagen, XL-10. DNA and protein concentration was determined using a NanoDrop 1000 spectrometer from Thermo Scientific using 260 and 280 nm wavelength, respectively. Gel electrophoresis for proteins was performed using 12% SDS-PAGE gels in running buffer and visualized using InstantBlue stain. Protein concentration was determined using a NanoDrop 1000 spectrometer with a wavelength ratio of 280–260 nm. The fluorescent D22 coactivator peptide was purchased from Invitrogen life technologies. RXRα-NURR1 heterodimerization and RXRα homodimerization BRET2 assays were performed as described.2,33 Briefly, RXRα and NURR1 receptors were tagged with GFP and renilla luciferase. pEC50 is the negative logarithm of the EC50 in molar, and efficacies were compared to that of LG100268 (100%). Values represent mean (SD) of three or more independent experiments.
Fluorescence Polarization Assay
His6-RXRα-LBD (1 μM), fluorescein-labeled D22 coactivator peptide (0.1 μM), and the ligand at the indicated concentration in buffer containing 100 mM HEPES (pH 7.5), 100 mM NaCl, 5 mM DTT, and 0.1% bovine serum albumin were incubated for 60 min at 4 °C and protected from light. Conditions for the competition assay: His6-RXRα-LBD (500 nM), fluorescein-labeled D22 coactivator peptide (50 nM), LG100268 (50 nM). Fluorescent polarization signals (mP) were measured with a Tecan Infinite F500 plate reader. Experiments were performed in triplicate, and the data were analyzed using Origin software.
Expression, Purification, and Crystallization of the RXRα LBD
The histidine-tagged LBD of human RXRα (in a pET15b vector) was expressed in E. coli BL21(DE3). Cells were grown at 37 °C in LB medium supplemented with 100 mg mL–1 ampicillin until OD600 reached about 0.7. Expression of T7 polymerase was induced by addition of isopropyl-b-d-thiogalactoside (IPTG) to a final concentration of 0.1 mM. After an additional incubation for 15 h at 15 °C, and cell cultures were harvested by centrifugation at 8000g for 20 min. The cell pellet from 2 L of RXRα LBD was resuspended in 50 mL buffer A (20 mM Tris-HCl pH 8.0, 500 mM NaCl, 5 mM imidazole) supplemented with a protease inhibitor (PMSF) and DNase I. The suspension was then lysed by sonication and centrifuged at 35 000g and 4 °C for 45 min. The supernatant was loaded onto a 5 mL Ni2+-affinity column, preequilibrated with buffer A. The column was washed with 10 volumes of buffer A and 10 volumes of buffer A supplemented with 50 mM imidazole. Bound proteins were eluted with buffer A containing 200 mM imidazole. The fractions containing RXR LBD were pooled, concentrated, and desalted to buffer B (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM DTT). To remove the histidine-tag, the protein was incubated for 16 h at 4 °C with thrombin (1 unit/mg RXR). The protein was passed through a Ni2+ column and a superdex gel filtration column. The protein was concentrated and stored at −80 °C until further use.
Before crystallization, the protein was mixed with a 1.5-fold molar excess of ligand and a 3-fold excess of TIF2 NR2 cofactor peptide (686-KHKILHRLLQDSS-698). The complex was incubated for 1h at 4 °C. Drops with a size of 2–3 μL using different reservoir to protein ratio were manually mixed and equilibrated against reservoirs with a volume of 500 mL. Optimal crystals were grown in a week in 3 μL drops with protein solution to reservoir ratio of 2:1 with 0.1 M PIPES, pH 7.0, 0.1 M NaCl, 22% PEG 2K MME. The crystals were cryo-cooled in liquid nitrogen using sucrose as cryo-protectant for X-ray data collection. Diffraction data for RXR were collected at the DESY beamline (Deutsches Elektronen-Synchrotron, Hamburg, Germany). The data set was indexed and integrated using iMosflm and scaled using SCALA. The structure was phased by molecular replacement using PDB ID 5EC9 as search model in Phaser. Coot and phenix.refine were used in alternating cycles of model building and refinement. All data collection, refinement, and validation statistics are shown in Table S1.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.7b00216.
Supporting table and figures; 1H NMR, 13C NMR, and LC-MS analytical data of all new compounds; protein crystallography data (PDF)
Author Contributions
The manuscript was written through contributions of all authors. M.S. and F.A.M. performed synthesis; M.S., F.A.M., and J.-N.M. performed biochemical and cellular studies; M.S., S.A.A., R.M.J.M.d.V., and C.O. performed structural characterization and analysis; E.S.B., R.O., L.-G.M., and L.B. designed the studies; M.S., L.-G.M., and L.B. wrote the manuscript. All authors have given approval to the final version of the manuscript.
The authors thank the Michael J. Fox Foundation for Parkinson’s Research, The Netherlands Organization for Scientific Research via Gravity program 024.001.035, ECHO Grants 711011017 and 717014001, and VICI Grant 016150366.
The authors declare no competing financial interest.
Supplementary Material
References
- Skerrett R.; Malm T.; Landreth G. (2014) Nuclear receptors in neurodegenerative diseases. Neurobiol. Dis. 72, 104–116. 10.1016/j.nbd.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K.; Spalding T. A.; Hubbard D.; Ma J.-N.; Olsson R.; Burstein E. S. (2013) Low dose bexarotene treatment rescues dopamine neurons and restores behavioral function in models of Parkinson’s disease. ACS Chem. Neurosci. 4, 1430–1438. 10.1021/cn400100f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S.; Yadav A.; Chaturvedi R. K. (2017) Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem. Biophys. Res. Commun. 483, 1166–1177. 10.1016/j.bbrc.2016.08.043. [DOI] [PubMed] [Google Scholar]
- Spathis A. D.; Asvos X.; Ziavra D.; Karampelas T.; Topouzis S.; Cournia Z.; Qing X.; Alexakos P.; Smits L. M.; Dalla C.; Rideout H. J.; Schwamborn J. C.; Tamvakopoulos C.; Fokas D.; Vassilatis D. K. (2017) NURR1:RXRα heterodimer activation as monotherapy for Parkinson’s disease. Proc. Natl. Acad. Sci. U. S. A. 114, 3999–4004. 10.1073/pnas.1616874114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lera Á. R.; Krezel W.; Rühl R. (2016) An Endogenous Mammalian Retinoid X Receptor Ligand, At Last!. ChemMedChem 11, 1027–1037. 10.1002/cmdc.201600105. [DOI] [PubMed] [Google Scholar]
- Bourguet W.; Ruff M.; Chambon P.; Gronemeyer H.; Moras D. (1995) Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-alpha. Nature 375, 377–382. 10.1038/375377a0. [DOI] [PubMed] [Google Scholar]
- Egea P. F.; Mitschler A.; Rochel N.; Ruff M.; Chambon P.; Moras D. (2000) Crystal structure of the human RXRalpha ligand-binding domain bound to its natural ligand: 9-cis retinoic acid. EMBO J. 19, 2592–2601. 10.1093/emboj/19.11.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard J. H.; Collings J. C.; Whiting A.; Przyborski S. A.; Marder T. B. (2009) Synthetic Retinoids: Structure–Activity Relationships. Chem. - Eur. J. 15, 11430–11442. 10.1002/chem.200901952. [DOI] [PubMed] [Google Scholar]
- Dawson M. I.; Xia Z. (2012) The retinoid X receptors and their ligands. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1821, 21–56. 10.1016/j.bbalip.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz B.; de Lera Á. R. (2012) Advances in drug design with RXR modulators. Expert Opin. Drug Discovery 7, 1003–1016. 10.1517/17460441.2012.722992. [DOI] [PubMed] [Google Scholar]
- Pérez E.; Bourguet W.; Gronemeyer H.; de Lera A. R. (2012) Modulation of RXR function through ligand design. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1821, 57–69. 10.1016/j.bbalip.2011.04.003. [DOI] [PubMed] [Google Scholar]
- Huang P.; Chandra V.; Rastinejad F. (2014) Retinoic Acid Actions through Mammalian Nuclear Receptors. Chem. Rev. 114, 233–254. 10.1021/cr400161b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez R.; Vaz B.; Gronemeyer H.; de Lera Á. R. (2014) Functions, Therapeutic Applications, and Synthesis of Retinoids and Carotenoids. Chem. Rev. 114, 1–125. 10.1021/cr400126u. [DOI] [PubMed] [Google Scholar]
- Burris T. P.; Solt L. A.; Wang Y.; Crumbley C.; Banerjee S.; Griffett K.; Lundasen T.; Hughes T.; Kojetin D. J. (2013) Nuclear Receptors and Their Selective Pharmacologic Modulators. Pharmacol. Rev. 65, 710–778. 10.1124/pr.112.006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre P.; Benomar Y.; Staels B. (2010) Retinoid X receptors: common heterodimerization partners with distinct functions. Trends Endocrinol. Metab. 21, 676–683. 10.1016/j.tem.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Cramer P. E.; Cirrito J. R.; Wesson D. W.; Lee C. Y. D.; Karlo J. C.; Zinn A. E.; Casali B. T.; Restivo J. L.; Goebel W. D.; James M. J.; Brunden K. R.; Wilson D. A.; Landreth G. E. (2012) ApoE-Directed Therapeutics Rapidly Clear β-Amyloid and Reverse Deficits in AD Mouse Models. Science 335, 1503–1506. 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster K. P.; Smith C.; Valencia-Olvera A. C.; Thatcher G. R. J.; Tai L. M.; LaDu M. J. (2017) Rexinoids as therapeutics for Alzheimer' disease: Role of APOE. Curr. Top. Med. Chem. 17, 708–720. 10.2174/1568026616666160617090227. [DOI] [PubMed] [Google Scholar]
- Michellys P.-Y.; Ardecky R. J.; Chen J.-H.; D’Arrigo J.; Grese T. A.; Karanewsky D. S.; Leibowitz M. D.; Liu S.; Mais D. A.; Mapes C. M.; Montrose-Rafizadeh C.; Ogilvie K. M.; Reifel-Miller A.; Rungta D.; Thompson A. W.; Tyhonas J. S.; Boehm M. F. (2003) Design, Synthesis, and Structure–Activity Relationship Studies of Novel 6,7-Locked-[7-(2-alkoxy-3,5-dialkylbenzene)-3-methylocta]-2,4,6-trienoic Acids. J. Med. Chem. 46, 4087–4103. 10.1021/jm020401k. [DOI] [PubMed] [Google Scholar]
- Leibowitz M. D.; Ardecky R. J.; Boehm M. F.; Broderick C. L.; Carfagna M. A.; Crombie D. L.; D’Arrigo J.; Etgen G. J.; Faul M. M.; Grese T. A.; Havel H.; Hein N. I.; Heyman R. A.; Jolley D.; Klausing K.; Liu S.; Mais D. E.; Mapes C. M.; Marschke K. B.; Michellys P.-Y.; Montrose-Rafizadeh C.; Ogilvie K. M.; Pascual B.; Rungta D.; Tyhonas J. S.; Urcan M. S.; Wardlow M.; Yumibe N.; Reifel-Miller A. (2006) Biological characterization of a heterodimer-selective retinoid X receptor modulator: potential benefits for the treatment of type 2 diabetes. Endocrinology 147, 1044–1053. 10.1210/en.2005-0690. [DOI] [PubMed] [Google Scholar]
- Wallén A.; Zetterström R. H.; Solomin L.; Arvidsson M.; Olson L.; Perlmann T. (1999) Fate of mesencephalic AHD2-expressing dopamine progenitor cells in NURR1 mutant mice. Exp. Cell Res. 253, 737–746. 10.1006/excr.1999.4691. [DOI] [PubMed] [Google Scholar]
- Jankovic J.; Chen S.; Le W. D. (2005) The role of NURR1 in the development of dopaminergic neurons and Parkinson’s disease. Prog. Neurobiol. 77, 128–138. 10.1016/j.pneurobio.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Decressac M.; Volakakis N.; Björklund A.; Perlmann T. (2013) NURR1 in Parkinson disease—from pathogenesis to therapeutic potential. Nat. Rev. Neurol. 9, 629–636. 10.1038/nrneurol.2013.209. [DOI] [PubMed] [Google Scholar]
- Zetterström R. H. (1997) Dopamine Neuron Agenesis in NURR1-Deficient Mice. Science 276, 248–250. 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- Saijo K.; Winner B.; Carson C. T.; Collier J. G.; Boyer L.; Rosenfeld M. G.; Gage F. H.; Glass C. K. (2009) A NURR1/CoREST Pathway in Microglia and Astrocytes Protects Dopaminergic Neurons from Inflammation-Induced Death. Cell 137, 47–59. 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le W.-D.; Xu P.; Jankovic J.; Jiang H.; Appel S. H.; Smith R. G.; Vassilatis D. K. (2003) Mutations in NR4A2 associated with familial Parkinson disease. Nat. Genet. 33, 85–89. 10.1038/ng1066. [DOI] [PubMed] [Google Scholar]
- Sleiman P. M. A.; Healy D. G.; Muqit M. M. K.; Yang Y. X.; Van Der Brug M.; Holton J. L.; Revesz T.; Quinn N. P.; Bhatia K.; Diss J. K. J.; Lees A. J.; Cookson M. R.; Latchman D. S.; Wood N. W. (2009) Characterisation of a novel NR4A2 mutation in Parkinson’s disease brain. Neurosci. Lett. 457, 75–79. 10.1016/j.neulet.2009.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Benoit G.; Liu J.; Prasad S.; Aarnisalo P.; Liu X.; Xu H.; Walker N. P. C.; Perlmann T. (2003) Structure and function of NURR1 identifies a class of ligand-independent nuclear receptors. Nature 423, 555–560. 10.1038/nature01645. [DOI] [PubMed] [Google Scholar]
- Tice C. M.; Zheng Y.-J. (2016) Non-canonical modulators of nuclear receptors. Bioorg. Med. Chem. Lett. 26, 4157–4164. 10.1016/j.bmcl.2016.07.067. [DOI] [PubMed] [Google Scholar]
- Friling S.; Bergsland M.; Kjellander S. (2009) Activation of Retinoid X Receptor increases dopamine cell survival in models for Parkinson’s disease. BMC Neurosci. 10, 146. 10.1186/1471-2202-10-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Bi W.; Zhao W.; Varghese M.; Koch R. J.; Walker R. H.; Chandraratna R. A.; Sanders M. E.; Janesick A.; Blumberg B.; Ward L.; Ho L.; Pasinetti G. M. (2016) Selective brain penetrable NURR1 transactivator for treating Parkinson’s disease. Oncotarget 7, 7469–7479. 10.18632/oncotarget.7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giner X. C.; Cotnoir-White D.; Mader S.; Levesque D. (2015) Selective ligand activity at Nur/retinoid X receptor complexes revealed by dimer-specific bioluminescence resonance energy transfer-based sensors. FASEB J. 29, 4256–4267. 10.1096/fj.14-259804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K.; Kawana K.; Sodeyama M.; Shimomura I.; Kagechika H.; Makishima M. (2005) Selective allosteric ligand activation of the retinoid X receptor heterodimers of NGFI-B and NURR1. Biochem. Pharmacol. 71, 98–107. 10.1016/j.bcp.2005.10.017. [DOI] [PubMed] [Google Scholar]
- Sundén H.; Schäfer A.; Scheepstra M.; Leysen S.; Malo M.; Ma J.-N.; Burstein E. S.; Ottmann C.; Brunsveld L.; Olsson R. (2016) Chiral Dihydrobenzofuran Acids Show Potent Retinoid X Receptor-Nuclear Receptor Related 1 Protein Dimer Activation. J. Med. Chem. 59, 1232–1238. 10.1021/acs.jmedchem.5b01702. [DOI] [PubMed] [Google Scholar]
- Rodrigues T.; Reker D.; Schneider P.; Schneider G. (2016) Counting on natural products for drug design. Nat. Chem. 8, 531–541. 10.1038/nchem.2479. [DOI] [PubMed] [Google Scholar]
- Scheepstra M.; Nieto L.; Hirsch A. K. H.; Fuchs S.; Leysen S.; Lam C. V.; in het Panhuis L.; van Boeckel C. A. A.; Wienk H.; Boelens R.; Ottmann C.; Milroy L.-G.; Brunsveld L. (2014) A Natural-Product Switch for a Dynamic Protein Interface. Angew. Chem., Int. Ed. 53, 6443–6448. 10.1002/anie.201403773. [DOI] [PubMed] [Google Scholar]
- Nahoum V.; Perez E.; Germain P.; Rodriguez-Barrios F.; Manzo F.; Kammerer S.; Lemaire G.; Hirsch O.; Royer C. A.; Gronemeyer H.; de Lera A. R.; Bourguet W. (2007) Modulators of the structural dynamics of the retinoid X receptor to reveal receptor function. Proc. Natl. Acad. Sci. U. S. A. 104, 17323–17328. 10.1073/pnas.0705356104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingsley K. L.; Barder T. E.; Buchwald S. L. (2007) Palladium-catalyzed borylation of aryl chlorides: scope, applications, and computational studies. Angew. Chem., Int. Ed. 46, 5359–5363. 10.1002/anie.200701551. [DOI] [PubMed] [Google Scholar]
- Takahashi B.; Ohta K.; Kawachi E.; Fukasawa H.; Hashimoto Y.; Kagechika H. (2002) Novel Retinoid X Receptor Antagonists: Specific Inhibition of Retinoid Synergism in RXR–RAR Heterodimer Actions. J. Med. Chem. 45, 3327–3330. 10.1021/jm0255320. [DOI] [PubMed] [Google Scholar]
- Gunther J. R.; Parent A. A.; Katzenellenbogen J. A. (2009) Alternative inhibition of androgen receptor signaling: peptidomimetic pyrimidines as direct androgen receptor/coactivator disruptors. ACS Chem. Biol. 4, 435–440. 10.1021/cb900043e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFrate A. L.; Gunther J. R.; Carlson K. E.; Katzenellenbogen J. A. (2008) Synthesis and biological evaluation of guanylhydrazone coactivator binding inhibitors for the estrogen receptor. Bioorg. Med. Chem. 16, 10075–10084. 10.1016/j.bmc.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan P. K.; Wang J.; Littler P.-L. H.; Wong K. K.; Sweetnam T. A.; Keefe W.; Nash N. R.; Reding E. C.; Piu F.; Brann M. R.; Schiffer H. H. (2007) Monitoring interactions between receptor tyrosine kinases and their downstream effector proteins in living cells using bioluminescence resonance energy transfer. Mol. Pharmacol. 72, 1440–1446. 10.1124/mol.107.039636. [DOI] [PubMed] [Google Scholar]
- Ohsawa F.; Morishita K.-I.; Yamada S.; Makishima M.; Kakuta H. (2010) Modification at the Lipophilic Domain of RXR Agonists Differentially Influences Activation of RXR Heterodimers. ACS Med. Chem. Lett. 1, 521–525. 10.1021/ml100184k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawata K.; Morishita K.; Nakayama M.; Yamada S.; Kobayashi T.; Furusawa Y.; Arimoto-Kobayashi S.; Oohashi T.; Makishima M.; Naitou H.; Ishitsubo E.; Tokiwa H.; Tai A.; Kakuta H. (2015) RXR partial agonist produced by side chain repositioning of alkoxy RXR full agonist retains antitype 2 diabetes activity without the adverse effects. J. Med. Chem. 58, 912–926. 10.1021/jm501863r. [DOI] [PubMed] [Google Scholar]
- Kojetin D. J.; Matta-Camacho E.; Hughes T. S.; Srinivasan S.; Nwachukwu J. C.; Cavett V.; Nowak J.; Chalmers M. J.; Marciano D. P.; Kamenecka T. M.; Shulman A. I.; Rance M.; Griffin P. R.; Bruning J. B.; Nettles K. W. (2015) Structural mechanism for signal transduction in RXR nuclear receptor heterodimers. Nat. Commun. 6, 8013. 10.1038/ncomms9013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gampe R. T.; Montana V. G.; Lambert M. H.; Miller A. B.; Bledsoe R. K.; Milburn M. V.; Kliewer S. A.; Willson T. M.; Xu H. E. (2000) Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol. Cell 5, 545–555. 10.1016/S1097-2765(00)80448-7. [DOI] [PubMed] [Google Scholar]
- Flaig R.; Greschik H.; Peluso-Iltis C.; Moras D. (2005) Structural Basis for the Cell-specific Activities of the NGFI-B and the NURR1 Ligand-binding Domain. J. Biol. Chem. 280, 19250–19258. 10.1074/jbc.M413175200. [DOI] [PubMed] [Google Scholar]
- Chandra V.; Huang P.; Hamuro Y.; Raghuram S.; Wang Y.; Burris T. P.; Rastinejad F. (2008) Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature 456, 350–356. 10.1038/nature07413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M.; Fukuzawa K.; Mochizuki Y.; Nakano T.; Tanaka S. (2007) Ab initio fragment molecular orbital study of molecular interactions between liganded retinoid X receptor and its coactivator: roles of helix 12 in the coactivator binding mechanism. J. Phys. Chem. B 111, 3525–3533. 10.1021/jp070054w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.