
Figure 1.
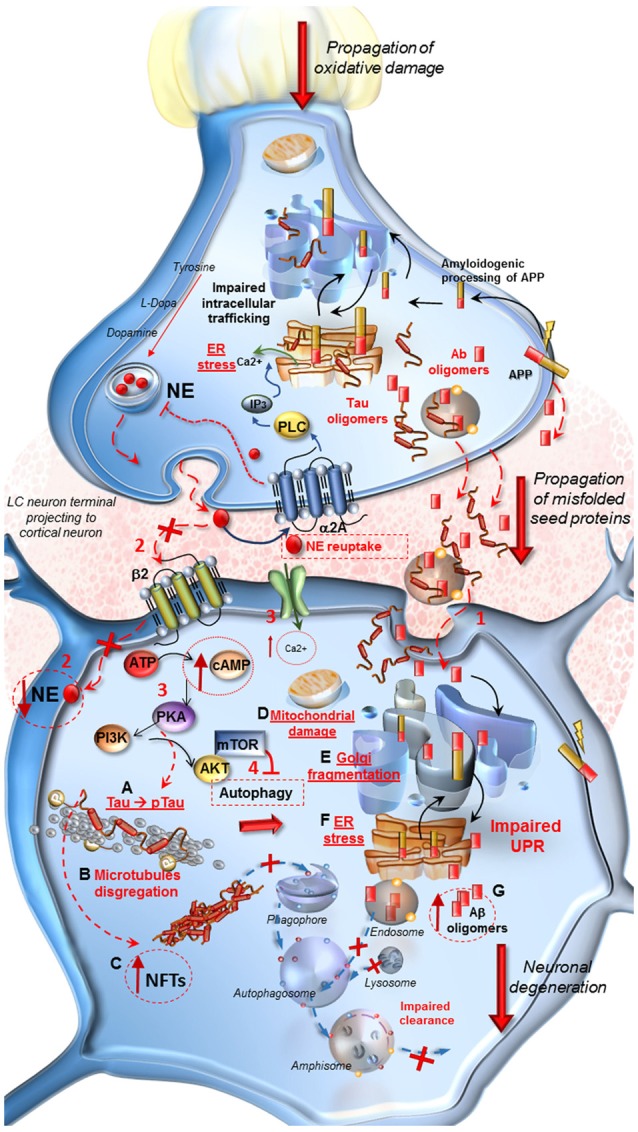
Molecular events occurring in Alzheimer’s Disease (AD) following locus coeruleus (LC) norepinephrine (NE) loss and autophagy impairment. At LC pre-synaptic terminal. Decreased expression of β2-adrenergic receptors in the pre-synaptic terminal leads to autophagy suppression. Oxidative stress, NE decrease and IP3 all contribute to endoplasmic reticulum stress and impairment of proteins processing, folding and trafficking in the trans-Golgi network. Aβ oligomers seeds, deriving from an amyloidogenic cleavage of APP, as well as Tau protein, are engulfed into vesicles and released into the axo-somatic synaptic cleft. At post-synaptic cortical neuron. (1) Aβ and Tau seeds are spread into the synaptic cleft and internalized into the cell body of the cortical neuron. (2) The loss of β2-adrenergic receptors (β2R) stimulation decreases autophagy. Thus the lack of NE, cannot exert its protective effect in the cell, it drives a cascade of detrimental intracellular effects instead. (3,4) Ca2+ entry into the cell, increase of cAMP levels, activation of PKA and PI3K/AKT/mTOR pathway lead to a further inhibition of autophagy. (A–G) PKA hyper-phosphorylates Tau protein (A) which leads to microtubules disgregation (B) and to the formation of neurofibrillary tangles (NFTs). (C) These effects translate into mitochondrial damage (D), Golgi fragmentation due to the inactivation of associated binding proteins (E) and ER stress (F). These events disrupt the Unfolded Protein Response (UPR) which does not provide for a proper intracellular trafficking, processing and sorting of misfolded Aβ, leading to further increase of harmful oligomers into the cell (G). Misfolded proteins are internalized into the endosomal compartment but autophagy impairment does not allow their removal and fosters trans-synaptic propagations.