

Abstract
Satellite cells (SC) are muscle stem cells located between the plasma membrane of muscle fibers and the surrounding basal lamina. They are essential for muscle regeneration. Upon injury, which occurs frequently in skeletal muscles, SCs are activated. They proliferate as myoblasts and differentiate to repair muscle lesions. Among many events that take place during muscle differentiation, cytosolic Ca2+ signals are of great importance. These Ca2+ signals arise from Ca2+ release from internal Ca2+ stores, as well as from Ca2+ entry from the extracellular space, particularly the store-operated Ca2+ entry (SOCE). This paper describes a methodology used to obtain a pure population of human myoblasts from muscle samples collected after orthopedic surgery. The tissue is mechanically and enzymatically digested, and the cells are amplified and then sorted by flow cytometry according to the presence of specific membrane markers. Once obtained, human myoblasts are expanded and committed to differentiate by removing growth factors from the culture medium. The expression levels of specific transcription factors and in vitro immunofluorescence are used to assess the myogenic differentiation process in control conditions and after silencing proteins involved in Ca2+ signaling. Finally, we detail the use of Fura-2 as a ratiometric Ca2+ probe that provides reliable and reproducible measurements of SOCE.
Keywords: Developmental Biology, Issue 125, myogenic cells, myoblast differentiation, Ca2+ signaling, store-operated Ca2+ entry, markers of differentiation, fluorescence-activated cell sorting
Introduction
Human skeletal muscles are composed of groups of contractile, multinucleated muscle fibers resulting from the fusion of myogenic precursor cells. Skeletal muscles have the capacity to regenerate after injury thanks to the presence of SCs, the skeletal muscle stem cells located between the plasma membrane of myofibers (sarcolemma) and the basal lamina. In uninjured muscle, SCs are mostly present in a quiescent state. In response to mechanical stress or injury, SCs become activated (myoblasts), proliferate, and undergo either differentiation to form new myofibers or self-renewal to replenish the SC pool1,2. Over the years, several techniques were developed to isolate SCs and their progeny, the myoblasts, from skeletal muscle biopsies. With the greater understanding of the cell surface markers expressed on these cells and the methodology of fluorescence-activated cell sorting (FACS)3,4,5, it is now possible to isolate pure populations of human myoblasts from muscle samples.
Calcium signaling regulates skeletal muscle development, homeostasis, and regeneration. In particular, the Ca2+ entry that is activated following intracellular store depletion, called SOCE, is of great importance6 for these processes. Upon cell stimulation, the Ca2+ level in the endo/sarcoplasmic reticulum (ER/SR) decreases, which in turn activates plasma membrane Ca2+ channels that permit Ca2+ entry to refill the ER/SR7. The two main proteins involved in SOCE are the ER/SR Ca2+-sensing stromal interaction molecule 1 (STIM1) protein and the plasma membrane Ca2+ channel Orai1. Skeletal muscle abundantly expresses these two proteins, as well as other proteins of the same families (STIM2 and Orai2-Orai3)8,9 and several Ca2+-permeable channels of the transient receptor potential canonical (TRPC) family10,11,12,13. Ca2+ entry through the SOCE pathway is of great importance for muscle formation/regeneration14. Mutations of STIM1 or Orai1 proteins have deleterious effects on muscle function, leading mainly to muscle hypotonia15. Animal models with SOCE impairment also display reduced muscle mass and force, together with enhanced fatigability6,16,17. As mentioned, other STIM and Orai proteins, as well as many TRPC channels, are expressed in skeletal muscle, and their respective roles have not been determined so far. By knocking down their expression level, it is thus possible to investigate their implications in SOCE and their roles during human skeletal muscle differentiation.
SOCE measurement can be performed using two different approaches: electrophysiological current recordings and Ca2+ measurements. Electrophysiology is certainly a more direct method, as it measures the current of interest and its associated electrophysiological signature. However, this technique is very difficult to apply to muscle cells, mainly because of the large size of the muscle cells and the small size of the endogenous SOCE current. Cytosolic Ca2+ imaging is technically very accessible, but the measure is more indirect, as the Ca2+ level measured in the cytosol reflects both the entry of Ca2+ and the re-pumping out of the cell or in the internal stores.
A methodology that includes isolating myoblasts from small pieces of human skeletal muscle after enzymatic digestion, amplification, and FACS is explained in this paper. The process of muscle differentiation and the immunofluorescence protocol to follow the expression of differentiation markers over time are described18. Finally, the measurement of SOCE and the role of different ion channels in Ca2+ signaling and skeletal muscle differentiation are explained.
Protocol
Human muscle samples (tissues obtained from semitendinous muscles) were collected during orthopedic surgery on healthy patients as surgical waste. All methods relating to the human study were performed in accordance with the guidelines and regulations of the Swiss Regulatory Health Authorities and approved by the Commission Cantonale d'Ethique de la Recherche from the Geneva Cantonal Authorities, Switzerland (protocol CER n° 12-259). Informed and written consents were obtained from all adult subjects involved in the study.
1. Isolation of Myoblasts from Human Skeletal Muscle
During all procedures, maintain sterile conditions by working under a level II tissue culture hood and using sterile medium and sterile buffers. Note: This procedure can be applied to many human skeletal muscles.
2. Dissociation Protocol
Transfer muscle samples (2 - 3 g) into a sterile 6 cm plate.
Wash the muscle samples with 5 - 10 mL of phosphate buffered saline (PBS). Repeat the wash.
Remove connective and adipose tissues using curved scissors and a clamp.
Weigh the muscle sample (see step 2.13) and transfer it to a new sterile 6 cm plate.
Add 5 mL of 0.05% Trypsin-EDTA.
Using scissors, cut and mince the muscle tissue into small pieces (less than 2 mm).
Using a 10 mL serological pipette, transfer the minced muscle to a 200 mL dissociation bottle containing a magnetic bar. Repeat this step until 90 mL of 0.05%Trypsin-EDTA has been transferred with all minced muscle to the dissociation box. Close the dissociation bottle and incubate at 37 °C for 60 min under mild agitation.
Remove the dissociation bottle from the heating bath and stop the enzymatic reaction by adding 10 mL of fetal calf serum (FCS; 10% final volume) to the dissociation bottle. Homogenize the mixture with a pipette.
Prepare two 50 mL tubes with a 70 µm cell strainer on each. Pass half of the cell suspension through the cell strainer on each tube by pipetting up and down on the filter until the suspension passes through.
Centrifuge the tubes at 200 x g and 20 °C for 5 min; aspirate and discard the supernatant.
Resuspend the cells with 10 mL of growth medium (GM; Table 1) and pass the cells through a 40-µm cell strainer placed on one 50 mL tube. Centrifuge the tube at 200 x g and 20 °C for 5 min; aspirate and discard the supernatant.
Resuspend the cells with 10 mL of GM and centrifuge the tube at 200 x g and 20 °C for 5 min; aspirate and discard the supernatant.
Resuspend the cells with 1 mL of GM and count the cells using a Neubauer chamber. Note: Usually, a yield of 3 x 105 cells per g of muscle should be harvested. The yield is calculated by dividing the number of cells by the weight of the muscle biopsy (step 2.4).
Put 2 x 105 cells in a sterile 6-cm plate containing 4 mL of GM and incubate at 37 °C and 7% CO2. Prepare as many plates as necessary.
Change the GM after 3 days. Note: Freshly dissociated cells need time to adhere to the culture plate. Moreover, the first myogenic cell doubling time is around 50 - 60 h. After the first division, the doubling time is around 20 h.
After 5 - 7 days, when cells reach 70 - 80% confluence (around 1.5 x 106 cells/per 6 cm plate), start antibody staining and cell sorting (step 3).
3. Antibody Staining and Cell Sorting
After rinsing once with PBS, incubate the adherent cells (in the 6-cm plate) with 1 mL of 0.05% Trypsin-EDTA at 37 °C and 7% CO2 for 3 min (or incubate at 5% CO2). Stop the reaction by adding 2 mL of GM and collect the cells in 15 mL tube. After centrifugation (200 x g for 5 min), resuspend the cells in 1 mL of GM.
Count the cells using a Neubauer chamber.
Perform the following procedures on ice. Note: The procedure for setting the correct fluorescence compensation on the cytometer is essential and should be similar on any cytometer. Tubes 1 and 2 are used as negative controls. Tubes 3 - 7 are used to set the compensation on the cytometer during the first experiment (Table 2). The final concentration used for each antibody and its corresponding isotype should be the same (around 1 µg/106 cells).
Dispatch 1 x 105 cells per tube (5-mL tube) for tubes 1 - 7 (Table 2). Place the remaining cell suspension in tube 8 for cell sorting.
Wash the cells with 1 mL of cold FACS buffer (PBS-5% FCS). Close and centrifuge the tubes at 200 x g and 4 °C for 5 min; aspirate and discard the supernatant.
Add 200 µL of FACS buffer per tube. Add antibodies to the tube according to Table 2. Incubate on ice for 30 min.
Wash the cells: add 1 mL of the FACS buffer to each tube. Close and centrifuge the tubes at 200 x g and 4 °C for 5 min. Aspirate and discard the supernatant. Repeat this step one more time.
Resuspend the cells in 500 µL of FACS buffer. Close the tubes and keep them on ice until cell sorting. Prepare 4 tubes of 1.5 mL, each containing 500 µL of GM, which is necessary to collect the cells during cell sorting. Keep them on ice.
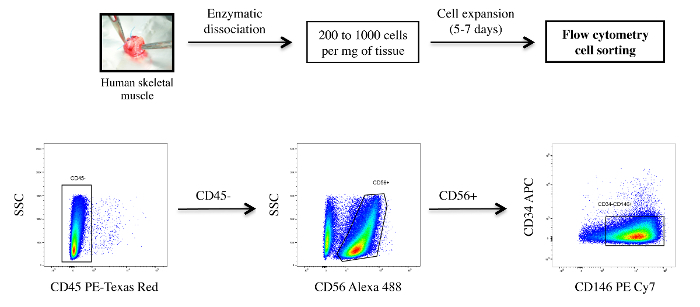
Sort human myoblasts by flow cytometry (Figure 1). After excluding CD45-positive hematopoietic cells, separate CD45-negative cells based on CD56 expression. Then, gate the CD56-positive population for CD34, CD144, and CD146 (human myoblasts are defined as CD56+/CD146+/CD45-/CD34-/CD144- cells). Reanalyze a small fraction of the sorted cell population to check the purity.
Pool the tubes containing human myoblasts in one 15-mL tube and wash one time by adding 5 mL of GM. Centrifuge the tube at 200 x g and 20 °C for 5 min; aspirate and discard the supernatant.
Resuspend the cells in 1 mL of GM and plate the cells on an uncoated 6-cm plate containing 4 mL of GM (2 x 105 cells per plate). Incubate at 37 °C and 7% CO2.
4. Cell Maintenance
Change half of the culture medium every 2 days. Remove 2 mL of old GM from the culture dish and add 2 mL of GM. Note: Myoblasts produce growth factors that are beneficial for culture conditions.
Trypsinize the cells when they reach 70 - 80% confluence to avoid pre-differentiation. After rinsing once with PBS, incubate the adherent cells (in the 6-cm plate) with 1 mL of 0.05% Trypsin-EDTA at 37 °C and 7% CO2 for 3 min. Stop the reaction by adding 2 mL of GM and collect the cells in a 15 mL tube. After centrifugation (200 x g for 5 min), resuspend the cells in 1 mL of GM.
Count the cells and prepare 6 cm plates with 2 x 105 cells per plate in 4 mL of GM.
Keep human myoblasts up to 6 passages or freeze them in GM containing 10% DMSO, 1 x 106 cells per mL. Freeze the cells in a progressive freezing box (1 °C/min); for long-term storage, place the cells in liquid nitrogen.
5. Transfection of Human Myoblasts with siRNA
Note: Myoblasts are transfected using commercial transfection reagent two days before the induction of the differentiation. The effect of specific siRNA against a gene is always compared to the effect of a control siRNA. siRNAs are double-stranded RNAs that must be kept on ice and manipulated with gloves.
Obtain a 70 - 80% confluent monolayer of human myoblasts in a 6-cm plate (around 1.5 x 106 cells). Note: 5 x 105 cells per transfection are needed to obtain a confluent monolayer of transfected myoblasts after 2 days in GM.
Prepare glass coverslips (described for the calcium imaging in step 7).
Warm GM, 0.05% Trypsin-EDTA, and PBS to 37 °C.
Perform all the following steps in a level II tissue culture hood to maintain sterility.
In a 1.5-mL tube, dispatch 500 µL of reduced serum medium, 3 µL of transfection reagent, and 1 µL of siRNA at 20 µM. Mix gently and keep at room temperature (RT) for 15 - 20 min.
During this incubation time, prepare the myoblasts for transfection. After rinsing once with PBS, incubate the adherent cells (in the 6-cm plate) with 1 mL of 0.05% Trypsin-EDTA at 37 °C and 7% CO2 for 3 min. Stop the reaction by adding 2 mL of GM and collect the cells in a 15 mL tube. After centrifugation (200 x g for 5 min), resuspend the cells in 1 mL of GM.
Count the cells using a Neubauer chamber.
Resuspend 5 x 105 cells in 200 µL of GM for each transfection (i.e., for 2 transfections, resuspend in 400 µL).
Add 200 µL of the cell suspension to each 500 µL of siRNA-transfectant mixture. Pipette up and down twice and incubate for 5 min at RT.
Add the 700 µL (cells + siRNA-transfectant) to a 3.5 cm culture dish containing a glass coverslip. Add GM to get a final volume of 2 mL.
After 24 h at 37 °C and 7% CO2, completely replace the medium with 2 mL of fresh GM. After another 24 h, a confluent monolayer of transfected cells is usually present; if so, induce myoblast differentiation by replacing the GM with 2 mL of differentiation medium (DM; Table 1).
Perform calcium imaging experiments 24 - 48 h after the induction of differentiation or on proliferating myoblasts. Note: Immunofluorescence is usually performed 48 h after the induction of differentiation.
6. Immunofluorescence Staining of Differentiation Markers
Note: Immunofluorescence staining is performed after 48 h in DM, but it can also be performed at other differentiation times. Myogenin and MEF2 are nuclear proteins only expressed in differentiated cells (before fusion into myotubes), and their staining allows for the assessment of the differentiation process. Myosin heavy chain (MyHC) is only expressed in myotubes and is used to estimate the myotube size and fusion index (number of nuclei in myotubes/total number of nuclei).
After 48 h in DM, wash the confluent monolayer of differentiated cells twice with 1 mL of PBS at RT. Do this carefully to avoid detaching the myotubes.
Fix the cells with 1 mL of 4% paraformaldehyde (PFA) for 15 min at RT. Wash the cells with PBS twice, for 5 min each.
Permeabilize the cells with 1 mL of PBS containing 0.25% Triton X-100 for 45 min and wash 3 times with 1 mL of PBS.
Block unspecific binding sites by incubating the cells in PBS supplemented with 0.2% Tween-10 and 2% goat serum (block solution) for 30 min at RT.
Prepare primary antibodies diluted in block solution (600 µL per 3.5-cm plate). Note: The following primary antibodies are used: mouse anti-myogenin (1:600), rabbit anti-MEF2 (1:300), and mouse anti-MyHC antibody (MF20, 1:1,000). It is possible to mix in the same blocking solution, mouse anti-myosin heavy chain or mouse anti-myogenin, with rabbit anti-MEF2.
Remove the block solution and add 600 µL of primary antibody solutions per 3.5-cm plate and incubate overnight at 4 °C in a humidified chamber.
Wash the cells 3 times with 1 mL of PBS. Incubate for 1 h at RT in a dark chamber with 600 µL of the secondary antibodies prepared in the block solution, as follows: goat anti-mouse Alexa 488 (1:1,000), goat anti-rabbit Alexa 546 (1:1,000), and DAPI (1:50, stock solution at 15 µM). CAUTION: DAPI is a toxic compound that should be handle with gloves.
Aspirate and wash the cells 3 times with 1 mL of PBS.
Mount glass coverslips using polyvinyl alcohol mounting medium with antifading solution. Keep them at 4 °C and protected from light until imaging the cells.
7. Calcium Imaging
Prior culture, prepare glass coverslips 25 mm in diameter. Put the coverslips in 70% ethanol for 48 h to remove any grease and dirt from the surface, which reduces cell adherence to the coverslip.
After rinsing the coverslipsseveral times with water, put them back in water and sterilize them (autoclaved).
Put one coverslip per 3.5-cm culture plate and let them dry. Store the Petri dishes for further experiments or use them immediately.
Add the transfected myoblasts (5 - 6 x 105 cells) to the glass coverslip. Note: Experiments are done 24 - 48 h after the induction of differentiation, or on proliferating myoblasts.
To perform Ca2+ imaging, wash the cells 2 times with PBS or directly with the medium used for the experiments (calcium-containing medium; Table 3). Load the cells with 2 µM Fura-2/AM plus 1 µM pluronic acid for about 30 min in the dark at RT. Note: Pluronic acid helps to solubilize Fura-2. Fura-2/AM and pluronic acid are dissolved in the calcium-containing medium.
Wash the cells twice in the same medium but without Fura-2/AM and pluronic acid and leave them in the dark for an additional 10 - 15 min to allow for the de-esterification of Fura-2/AM.
If the loading efficiency of Fura-2 is too low, add more Fura-2 (e.g., 4 µM) and/or incubate the cells for a longer time (e.g., 40 min).
Remove the coverslip from the culture plate and install it in the experimental chamber. Add ~1 mL of calcium-containing medium (depending upon the volume of the experimental chamber). Install it on the microscope stage and start the recording. Note: The fluorescent Ca2+-sensitive probe Fura-2 is excited alternately at 340 nm and 380 nm, and the emission light is collected at 510 nm. As the fluctuations of the Ca2+ concentration are relatively slow, acquire 1 ratio every 2 s.
- To activate SOCE, the following classical thapsigargin/Ca2+ re-addition protocol is used: leave the cells for 2-3 min in calcium-containing medium. Replace the medium with calcium-free medium for 1-2 min.
- Add 1 µM thapsigargin to deplete the internal calcium stores. Note: Thapsigargin is an irreversible blocker of sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) pump activity.After 8 - 10 min, rapidly replace the medium with the calcium-containing medium. Ca2+ will enter the cells through the SOCE channels. Wait around 5 min before terminating the experiments. CAUTION: Thapsigargin is a toxic compound that should be handled with gloves.
8. Ca2+ Measurement Analysis
Note: Perform the analysis with the acquisition software.
Open the experiment. Draw region 1 in an area where there are no cells (background region), and draw the other regions in the cytoplasm of the cells to be analyzed (the regions can be drawn on any image: the 340-nm, the 380-nm, or the ratio image). Go to "Run experiments" > "reference images." Under the 340 and 380 channels, select "region 1" and then click on "Subtract background."
Run the entire experiment (F4: forward) to ensure that nothing interferes with the background region, such as a fluorescent dust spot that travels through the background region, and that the cells are not moving during the time of the experiments. Note: The regions of interest that are selected should not include dark areas; otherwise, the ratio value will appear noisy, as part of the background will be included in the measurement.
Click on "Log data" and play the entire experiment again. Note: This will open a spreadsheet in which the time and the ratio are logged. It is also possible to save the individual 340 nm and 380 nm wavelengths.
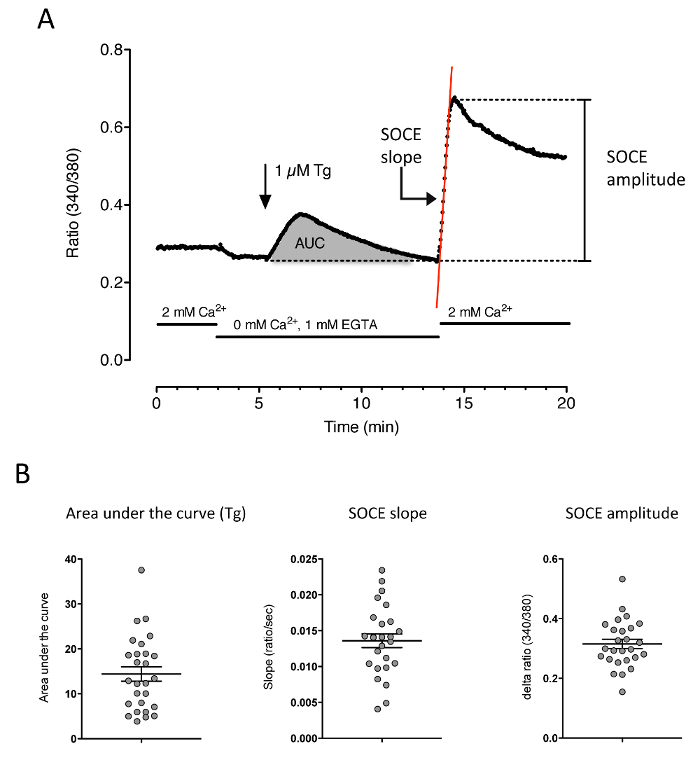
To get information about SOCE, extract the two important parameters from the experiment: the amplitude of the Ca2+ re-addition and the slope of the Ca2+ entry phase (Figure 3).
Obtain the amplitude by subtracting the ratio value just before Ca2+ re-addition from the maximal amplitude of the Ca2+ elevation. Obtain the maximal slope by fitting a linear regression of the first seconds after Ca2+ re-addition. Note: The area under the curve of the thapsigargin response is also an informative parameter to extract, as it reflects the amount of free calcium stored in the SR.
Representative Results
After the enzymatic dissociation of a human muscle sample, cells were amplified in GM. Human myoblasts, defined as CD56+/CD146+/CD45-/CD34-/CD144- cells, were obtained after FACS. Myoblasts represented more than 60% of the analyzed population (Figure 1). Primary human myoblasts were replated, grown to confluence, and cultured in DM for 48 h. After immunostaining for the expression of the transcription factor MEF2 and the muscle-specific protein myosin heavy chain (MyHC), we observed that a majority of cells formed myotubes in vitro (MyHC-positive cells), with fusion index values of 60.9 ± 1.3% (n = 5; Figure 2). We also observed that around 40% of human myoblasts, called reserve cells, escaped this terminal differentiation and remained undifferentiated mononucleated cells. For functional studies, we analyzed the Ca2+ response of myoblasts or myotubes 48 h after the initiation of differentiation. Figure 3 shows a representative protocol used to induced SOCE and the key parameters to extract from such a recording. To discern information related to the molecules involved in SOCE, cells were transfected with siRNA against the molecule of interest. Figure 4 shows the impact of silencing Ca2+-permeable channels of the TRPC family on SOCE in myoblasts.
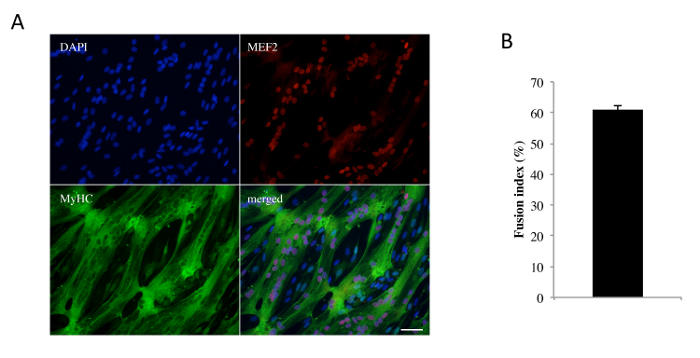
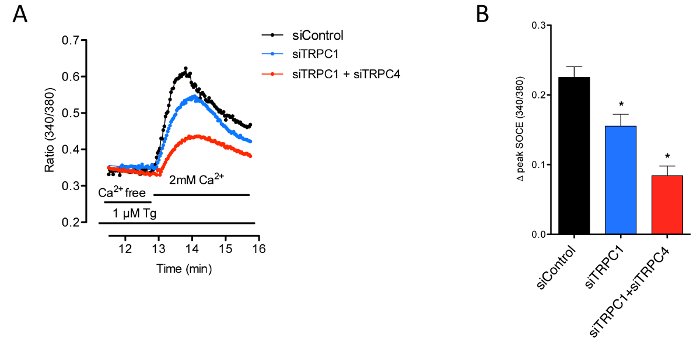
Figure 1: FACS of Human Myoblasts. Dissociated human cells were immunostained with antibodies for the following cell surface markers: CD45, CD56, CD34, CD144, and CD146. After excluding CD45-positive hematopoietic cells, CD45-negative cells were separated based on CD56 expression. The CD56-positive population was then gated for CD34, CD144, and CD146. Human myoblasts were defined as CD56+/CD146+/CD45-/CD34-/CD144- cells, as previously described19,4. Please click here to view a larger version of this figure.
Figure 2: Human Myoblast Differentiation. (A) A confluent monolayer of human myoblasts was exposed to DM for 48 h; fixed; and stained with antibodies against MEF2 (red), MyHC (green), and DAPI (blue) to identify nuclei. Scale bar = 50 µm. (B) The fusion index represents the number of nuclei incorporated in myotubes divided by the total number of nuclei on a certain field. Data are mean ± SEM of 5 independent experiments. Please click here to view a larger version of this figure.
Figure 3: Representative Calcium Measurement of Thapsigargin-induced SOCE. Myotubes were loaded with Fura-2 and the cytosolic Ca2+ concentration was measured. (A) The cells sit for a few minutes in a calcium-containing medium, followed by 2 min in a calcium-free medium. 1 µM thapsigargin is then used for 8 min, followed by the calcium re-addition. The 3 important parameters to extract from such recording are: the area under the curve of the thapsigargin response, the slope of the SOCE, and the amplitude of the SOCE. (B) Quantification of the 3 parameters . The area under the curve provides information about the amount of Ca2+ stored in the ER/SR, and the two other parameters allow for the quantification of the SOCE. Please click here to view a larger version of this figure.
Figure 4: Involvement of TRPC1 and TRPC4 in SOCE. (A) Cytosolic Ca2+ concentration was assessed using Fura-2 in proliferating myoblasts transfected with siTRPC1 or siTRPC1 and siTRPC4. SOCE was triggered by internal store depletion with 1 µM thapsigargin in the absence of external Ca2+ (Ca2+ free) and measured during the re-addition of 2 mM external Ca2+. Each trace represents the mean of a representative coverslip. (B) Quantification of the effects of siRNA on SOCE amplitude. This figure has been modified from reference12, with permission. Bars are mean ± SEM, *p <0.05. Please click here to view a larger version of this figure.
| Growth medium (GM): | Concentration | Differentiation medium (DM): | Concentration | |
| Ham's F10 | DMEM | |||
| FCS | 15% | bovine serum albumin | 0.5 mg/ml | |
| bovine serum albumin | 0.5 mg/ml | epidermal growth factor | 10 ng/ml | |
| fetuin | 0.5 mg/ml | insulin | 0.01 mg/ml | |
| epidermal growth factor | 10 ng/ml | creatine | 1 mM | |
| dexamethasone | 0.39 µg/ml | pyruvate | 100 µg/ml | |
| insulin | 0.04 mg/ml | uridine | 50 µg/ml | |
| creatine | 1 mM | gentamycin | 10 µg/ml | |
| pyruvate | 100 µg/ml | |||
| uridine | 50 µg/ml | |||
| gentamycin | 5 µg/ml |
Table 1: Medium Composition for Myoblast Culture.
| Tubes | Antibodies |
| 1 | None |
| 2 | Isotype AlexaFluor 488-, Isotype PE-CF594, Isotype PE, Isotype PECy7Isotype APC |
| 3 | Mouse anti-human CD56-AlexaFluor 488 |
| 4 | Mouse anti-human CD45 PE-CF594 |
| 5 | Mouse anti-human CD144-PE |
| 6 | Mouse anti-human CD146-PECy7 |
| 7 | Mouse anti-human CD34-APC |
| 8 | Mouse anti-human CD56-AlexaFluor 488-, Mouse anti-human CD45 PE-CF594, Mouse anti-human CD144-PE, Mouse anti-human CD34-APC, Mouse anti-human CD146-PECy7 |
Table 2: Antibodies used for the Cell Sorting.
| Calcium-containing medium | concentration | Calcium-free medium | concentration | |
| NaCl | 135 mM | NaCl | 135 mM | |
| KCl | 5 mM | KCl | 5 mM | |
| MgCl2 | 1 mM | MgCl2 | 1 mM | |
| CaCl2 | 2 mM | EGTA | 1 mM | |
| Hepes | 10 mM | Hepes | 10 mM | |
| Glucose | 10 mM | Glucose | 10 mM | |
| pH 7.45 adjusted with NaOH | pH 7.45 adjusted with NaOH |
Table 3: Medium Composition for Calcium Imaging Experiments.
Discussion
The isolation and culture of human myoblasts from adult skeletal muscle offers an in vitro model to study muscle differentiation and muscle regeneration. In this paper, we provide a protocol that allows for the purification of high yields of human myoblasts in a simple and cost-limited way. In addition, this technique provides reliable and reproducible results in terms of percentage of myoblasts isolated and their myogenic differentiation efficiency. Indeed, we obtained around 60% of human myoblasts after cell sorting. These cells can be maintained in culture for up to 6 passages (around 30 doublings) and keep their capacities to differentiate. Furthermore, myogenic differentiation occurs rapidly after changing the medium from GM to DM, as after 2 to 3 days in DM, myoblast fusion is maximal. In this model, around 60% of the myoblasts fuse into myotubes after 2 days in DM. Another advantage is that these primary human cell cultures are easily transfected with siRNA. The GM and DM media used in our laboratory are homemade. Alternatively, commercial media may be used, but we never tried their efficiency in human primary cultures.
One drawback of the method mentioned is the use of trypsin during tissue enzymatic digestion. This procedure removes a large fraction of the cell surface markers and implies that we leave the cells in culture for some days before performing FACS . Collagenase could be used instead of trypsin to better preserve the membrane proteins and allow for the sorting of human myoblasts immediately after tissue dissociation. However, the cost of collagenase is significantly higher than trypsin, and an amplification step would still be required after cell sorting to obtain sufficient cell number for the subsequent experiments. Another limitation of the technique is the "quality" of the muscle samples obtained from orthopedic surgery. Indeed, the pieces of tissue obtained could be more or less damaged due to surgery, and the time between the surgery and the cell dissociation in the laboratory can vary from day to day. However, for myoblast isolation, purification, and differentiation into myotubes, we routinely obtain satisfactory results. Finally, the age of the patient can be an issue, as the amount and regenerative capacity of satellite cells decline with age20. Thus, we restrict our experiments to young patients (less than 40 years old).
As mentioned, myoblasts differentiate rapidly and efficiently, which is a great advantage of studying the initial events of skeletal muscle differentiation. However, as myotubes are formed rapidly, they also have a tendency to spontaneously contract and detach from the culture plates. This precludes the analysis of the later fusion events. To study these later events, one would need to coat the culture plate with extracellular matrix to improve the cell adherence and to decrease myotube detachment21.
Calcium imaging using the fluorescent probe Fura-2 is a widely used, reproducible technique that is easy to perform. The loading of Fura-2 is efficient, and the use of a ratiometric probe such as Fura-2 has several advantages over a single-wavelength dye. It normalizes changes of focus or differences in the loading between cells and within a cell (e.g., nucleus versus cytosol). The protocol we described to elicit and measure SOCE is regularly used in many different cellular systems. The silencing of different channels or calcium regulators permits to elucidation of the molecular composition of SOCE and the relative importance of the different players, both in myoblasts and in myotubes. An alternative approach using Fura-2 and Ca2+ imaging is the application of the Mn2+ quench technique. Mn2+ enters the cells through Ca2+-permeable channels and quenches the fura-2 fluorescence. The measure of the quench rate gives a more direct measure of the SOCE22,23. This, however, requires the utilization of slightly different equipment. Ca2+ signaling could also be performed using genetically encoded Ca2+ indicators, which have the great advantage of being precisely localized in different cell compartments. The drawbacks are the requirement for cell transfection and the dynamics of the signal, which is usually less than with chemical dyes24. To measure Ca2+ in organelles like mitochondria or the SR, the genetic probes are the gold standard. However, for cytosolic Ca2+ measurements, the benefit of using Fura-2 over genetic probes remains.
Disclosures
The authors have no competing financial interests.
Acknowledgments
This work was supported by the Swiss National Science Foundation (grant number 310030-166313), The "Fondation Suisse pour la recherche sur les maladies musculaires," the "Foundation Marcel Levaillant," and the "Fondation pour la recherche ostéo-articulaire."
References
- Dumont NA, Bentzinger CF, Sincennes MC, Rudnicki MA. Satellite Cells and Skeletal Muscle Regeneration. Compr Physiol. 2015;5(3):1027–1059. doi: 10.1002/cphy.c140068. [DOI] [PubMed] [Google Scholar]
- Sambasivan R, Tajbakhsh S. Adult skeletal muscle stem cells. Results Probl Cell Differ. 2015;56:191–213. doi: 10.1007/978-3-662-44608-9_9. [DOI] [PubMed] [Google Scholar]
- Chen WC, et al. Isolation of blood-vessel-derived multipotent precursors from human skeletal muscle. J Vis Exp. 2014. p. e51195. [DOI] [PMC free article] [PubMed]
- Zheng B, et al. Isolation of myogenic stem cells from cultures of cryopreserved human skeletal muscle. Cell Transplant. 2012;21(6):1087–1093. doi: 10.3727/096368912X636876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charville GW, et al. Ex Vivo Expansion and In Vivo Self-Renewal of Human Muscle Stem Cells. Stem Cell Reports. 2015;5(4):621–632. doi: 10.1016/j.stemcr.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiber J, et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat Cell Biol. 2008;10(6):688–697. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Lewis RS. Store-Operated Calcium Channels. Physiol Rev. 2015;95(4):1383–1436. doi: 10.1152/physrev.00020.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbellay B, et al. STIM1- and Orai1-dependent store-operated calcium entry regulates human myoblast differentiation. J Biol Chem. 2009;284(8):5370–5380. doi: 10.1074/jbc.M806726200. [DOI] [PubMed] [Google Scholar]
- Darbellay B, et al. Human muscle economy myoblast differentiation and excitation-contraction coupling use the same molecular partners, STIM1 and STIM2. J Biol Chem. 2010;285(29):22437–22447. doi: 10.1074/jbc.M110.118984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87(1):165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- Vandebrouck C, Martin D, Colson-Van Schoor M, Debaix H, Gailly P. Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J Cell Bio. 2002;158(6):1089–1096. doi: 10.1083/jcb.200203091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antigny F, Koenig S, Bernheim L, Frieden M. During post-natal human myogenesis, normal myotube size requires TRPC1- and TRPC4-mediated Ca(2)(+) entry. J Cell Sci. 2013;126(11):2525–2533. doi: 10.1242/jcs.122911. [DOI] [PubMed] [Google Scholar]
- Brinkmeier H. TRP channels in skeletal muscle: gene expression, function and implications for disease. Adv Exp Med Biol. 2011;704:749–758. doi: 10.1007/978-94-007-0265-3_39. [DOI] [PubMed] [Google Scholar]
- Pan Z, Brotto M, Ma J. Store-operated Ca2+ entry in muscle physiology and diseases. BMB Rep. 2014;47(2):69–79. doi: 10.5483/BMBRep.2014.47.2.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacruz RS, Feske S. Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci. 2015;1356:45–79. doi: 10.1111/nyas.12938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, et al. STIM1-Ca(2+) signaling is required for the hypertrophic growth of skeletal muscle in mice. Mol Cell Biol. 2012;32(15):3009–3017. doi: 10.1128/MCB.06599-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei-Lapierre L, Carrell EM, Boncompagni S, Protasi F, Dirksen RT. Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat Commun. 2013;4:2805. doi: 10.1038/ncomms3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig S, et al. Membrane hyperpolarization triggers myogenin and myocyte enhancer factor-2 expression during human myoblast differentiation. J Biol Chem. 2004;279(27):28187–28196. doi: 10.1074/jbc.M313932200. [DOI] [PubMed] [Google Scholar]
- Crisan M, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3(3):301–313. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Bigot A, et al. Age-Associated Methylation Suppresses SPRY1, Leading to a Failure of Re-quiescence and Loss of the Reserve Stem Cell Pool in Elderly Muscle. Cell Rep. 2015;13(6):1172–1182. doi: 10.1016/j.celrep.2015.09.067. [DOI] [PubMed] [Google Scholar]
- Bettadapur A, et al. Prolonged Culture of Aligned Skeletal Myotubes on Micromolded Gelatin Hydrogels. Sci Rep. 2016;6:28855. doi: 10.1038/srep28855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbellay B, Arnaudeau S, Bader CR, Konig S, Bernheim L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J Cell Biol. 2011;194(2):335–346. doi: 10.1083/jcb.201012157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauc S, et al. STIM1L traps and gates Orai1 channels without remodeling the cortical ER. J Cell Sci. 2015;128(8):1568–1579. doi: 10.1242/jcs.164228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaurex N. Calcium measurements in organelles with Ca2+-sensitive fluorescent proteins. Cell Calcium. 2005;38(3-4):213–222. doi: 10.1016/j.ceca.2005.06.026. [DOI] [PubMed] [Google Scholar]