

Abstract
The mechanisms regulating mRNA translation are involved in various biological processes, such as germ line development, cell differentiation, and organogenesis, as well as in multiple diseases. Numerous publications have convincingly shown that specific mechanisms tightly regulate mRNA translation. Increased interest in the translation-induced regulation of protein expression has led to the development of novel methods to study and follow de novo protein synthesis in cellulo. However, most of these methods are complex, making them costly and often limiting the number of mRNA targets that can be studied. This manuscript proposes a method that requires only basic reagents and a confocal fluorescence imaging system to measure and visualize the changes in mRNA translation that occur in any cell line under various conditions. This method was recently used to show localized translation in the subcellular structures of adherent cells over a short period of time, thus offering the possibility of visualizing de novo translation for a short period during a variety of biological processes or of validating changes in translational activity in response to specific stimuli.
Keywords: Biochemistry, Issue 126, Puromycilation, localized translation, quantitative fluorescence, spreading initiation centers, translation regulation, microscopy
Introduction
The regulation of translation by different cellular functions has prompted many research teams to develop new tools and methods to determine the subcellular localization of mRNA translation and regulated protein synthesis1,2,3,4. These recent technological advances allow for an improved understanding of the mechanisms involving translation upregulation or the repression of specific mRNAs during biological processes, such as neuronal development, drug response, and metastasis5,6,7,8. However, most of these methods require expensive or hazardous reagents and specific equipment that might not be available to most laboratories. As such, a cost-effective method to allow for the rapid assessment of translation events was developed to specifically circumvent these potential issues. This method detects acute translational modulations that occur during specific cellular processes and also allows for the localization of translation using confocal microscopy.
The methods described here were used to monitor localized translation within subcellular compartments called spreading initiation centers (SIC)5. SICs are transient structures found in seeded cells that are localized on top of nascent adhesion complexes. Although SICs and adhesion complexes are distinct, their fates are closely linked. Indeed, SICs are known to gradually disappear upon focal adhesion complex maturation into an adhesion site during the initial phase of adhesion. We found that RNA-binding proteins known to specifically control mRNA translation (e.g., Sam68, FMRP, and G3BP1) and polyadenylated RNAs were enriched within these structures5. Using the methods described here, we showed that the regulation of SIC-associated mRNA translation acts as a checkpoint allowing seeded cells to consolidate cell adhesion. This method, based on puromycin incorporation, could be considered an adapted version of the surface sensing of translation assay (SunSET). Originally developed to measure global protein synthesis rates using a non-radioactively labeled amino acid, protein puromycilation offers an efficient way to visualize de novo protein synthesis9. This method relies on the intrinsic behavior of puromycin, an antibiotic that blocks translation through premature chain termination in the ribosome10. Indeed, puromycin is structurally analogous to tyrosyl-tRNA, which allows for incorporation into elongating peptide chains via the formation of a peptide bond. However, puromycin binding to a growing peptide chain prevents a new peptide bond from being formed with the next aminoacyl-tRNA, since puromycin has a non-hydrolysable amide bond instead of the hydrolysable ester bond found in tRNAs. Thus, the incorporation of puromycin into elongating polypeptides results in the premature release of numerous truncated puromycilated polypeptides corresponding to actively translated mRNA9,11,12.
Using this method, it was possible to assess active translation within a short time widow (e.g., 5 min) during cellular adhesion using a specific antibody directed against puromycin on cells that were supplemented with the antibiotic for 5-min periods at different time points during the cell adhesion process5. The precision of this assay relies on highly specific antibodies directed against the puromycilated moiety. Immunofluorescent detection of the puromycilated polypeptide provides a general subcellular repartition of the newly translated mRNA, which can also be quantified with great accuracy using confocal imaging systems.
Hence, this method offers a relevant option for a large number of laboratories studying translational regulatory mechanisms involved in processes such as neuronal granulation6,13,14,15, morphogen mRNA localization, and translation during development16,17. It is also well suited to studying localized or compartmentalized translation during rapid biological events, such as cell migration, adhesion, or invasion, or to simply assess drug treatments that might induce translational changes5,7,18. Overall, this method allows for the visualization of localized or controlled translation events in a rapid, precise, and cost-effective way.
Protocol
1. Determination of Puromycilation Conditions
NOTE: This technique describes the method used to assess localized translation during the MRC-5 cell adhesion process5. As puromycilation can be done in any cell, it is important to optimize the puromycilation conditions for the specific cell lines to be used, because the treatment conditions are not identical for each cell line in terms of the puromycin concentration and the desired incubation time. To show how these conditions are defined, three example cell lines (i.e., HeLa, MRC-5, and Huh-7) were treated with increasing concentrations of puromycin for different incubation times (Figure 1).
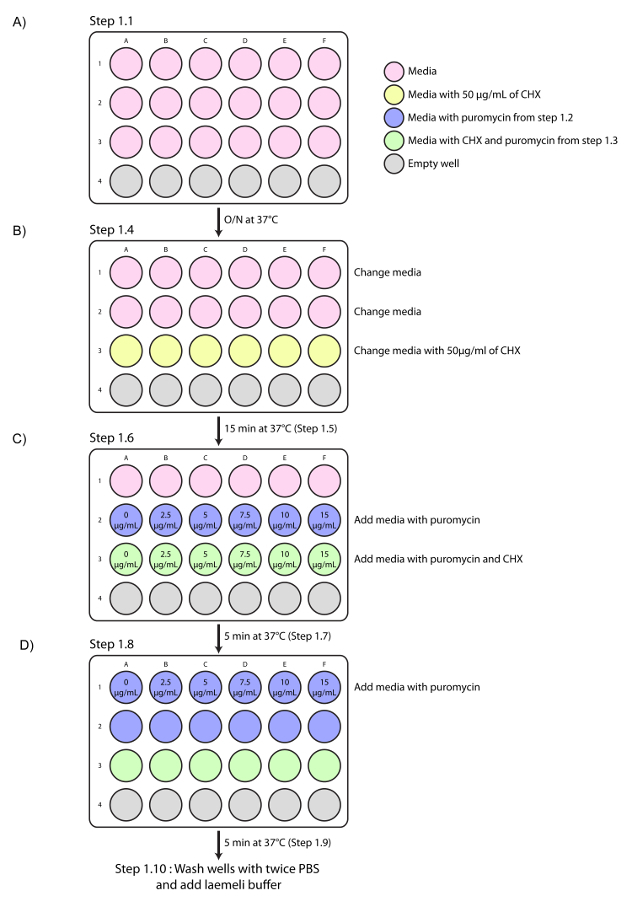
Grow identical numbers of cells in a 24-well plate in 0.5 mL of the appropriate complete culture medium 16 h before the test. Seed 30,000 MRC-5 cells, 50,000 HeLa cells, or 50,000 HuH-7 cells per well. Make sure to avoid exceeding 60-70% confluence (Figure 1A).
Prepare 6 tubes with 1.5 mL of complete cell culture medium with increasing concentrations of puromycin (0, 5, 10, 15, 20, and 30 µg/mL). Pre-warm at 37 °C.
For each concentration of puromycin medium (prepared in step 1.2), transfer 0.5 mL from each tube into 6 new tubes and add cycloheximide at a final concentration of 50 µg/mL to all 6 new tubes. Pre-warm at 37 °C.
Change the medium with 0.5 mL of complete culture medium (row 1 and 2). For the third row, add 0.5 mL of complete culture medium supplemented with 50 µg/mL of cycloheximide (Figure 1B). NOTE: Cycloheximide treatment has been established as the best negative control for protein puromycilation due to its ability to block translation elongation. Because puromycin incorporation requires translation elongation, cycloheximide treatment leads to a loss of puromycilated protein signals5,18.
Incubate for 15 min at 37 °C (5% CO2).
Add 0.5 mL of the puromycin medium prepared in step 1.2 to the second row to obtain final concentrations of 0, 2.5, 5, 7.5, 10, and 15 µg/mL, respectively, in columns A, B, C, D, E, and F. At the same time, add 0.5 mL of puromycin to the cycloheximide-supplemented medium (step 1.3) in the third row (Figure 1C).
Incubate for 5 min at 37 °C (5% CO2).
Add 0.5 mL of the puromycin medium prepared in step 1.2 to the first row to obtain final concentrations of 0.0, 2.5, 5, 7.5, 10, and 15 µg/mL (Figure 1C).
Incubate for 5 min at 37 °C (5% CO2).
Wash each well from rows 1 to 3 with 1 mL of ice-cold 1X phosphate-buffered saline (PBS) 5 min after the addition of puromycin to wells of the first row (wash twice). Add 75 µL of 1X Laemmli buffer (4% sodium dodecyl sulfate (SDS), 4% 2-mercaptoethanol, 0.120 M Tris HCl pH 6.8, 0.004% bromophenol blue, and 10% glycerol) to obtain whole-cell lysates for each condition.
- Run 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) using 1/3 of the prepared samples to assess puromycin incorporation.
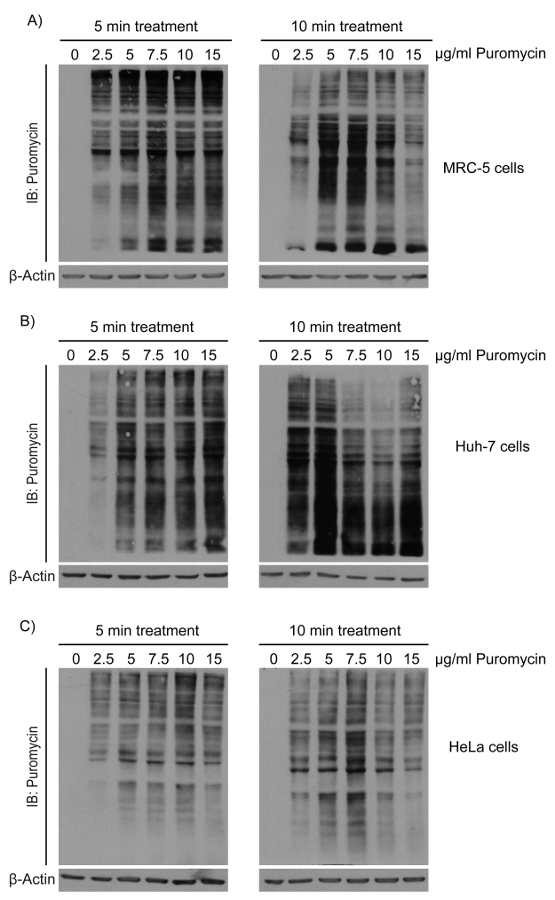
- Determine the level of puromycin incorporation by Western blot analysis using an anti-puromycin antibody (12D10) diluted at a ratio of 1:25,000 in primary antibody incubation buffer (2% bovine serum albumin (BSA)), 430 mM NaCl, 10 mM Tris pH 7.4, and 0.01% sodium azide). NOTE: Representative images corresponding to different cell line extracts made as described in step 1 are shown as examples in Figure 2.
2. In Cellulo Localized Translation Visualization
NOTE: The technique described here was used to assess localized translation within SICs in MRC-5 cells during the adhesion process5. Although MRC-5 cells are used here, other cell lines can be used with the same methodology described in step 2.1.
- Cell preparation.
- Detach MRC-5 cells using 0.25% trypsin/2.21 mM ethylenediaminetetraacetic acid (EDTA) in Hank's Balanced Salt Solution (HBSS). Pellet the cells by centrifugation (5 min at 200 x g) in a 15-mL conical centrifuge tube and suspend them in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 40,000 cells/mL after counting with a counting chamber.
- Incubate the suspended cells at 37 °C under gentle rotation using a tube rotator for 20 min to maintain them in suspension. NOTE: This step is critical to allow for the complete dissociation of the focal adhesion complexes.
- Plate 2 mL of suspended MRC-5 cells (40,000 cells/mL) in two 35-mm glass-bottom dishes. Use the first 35-mm glass-bottom dish for the puromycilation assay (see step 2.1.5) and use the second as a negative control (see step 2.1.6).
- Keep the cells at 37 °C (5% CO2) for 55 min to allow for cellular adhesion and SIC formation.
- Add puromycin to the medium in the 35-mm glass-bottom dishes (assay) using the concentration previously defined in section 1. To treat MRC-5 cells, use 10 µg/mL puromycin for 5 min at 37 °C (5% CO2).
- As a negative control, add cycloheximide to the second 35-mm glass-bottom dish 40 min after seeding the cells to pre-treat them with 50 µg/mL cycloheximide (Figure 2B). Then, 15 min after cycloheximide addition, add puromycin to the medium using the same concentration and incubation time as in step 2.1.5 (e.g., use 10 µg/mL of puromycin for 5 min at 37 °C (5% CO2) for MRC-5).
- Wash each plate twice with 2 mL of ice-cold 1X PBS and fix the cells with 1 mL of 4% formaldehyde (diluted in 1X PBS) for 15 min at room temperature (RT). Caution: Paraformaldehyde must be used strictly in a chemical hood.
- Immunostaining.
- Following paraformaldehyde fixation, wash three times with 2 mL of 1X PBS and incubate in 500 µL of PBS-TritonX-100 (0.5%) for 20 min at RT to permeabilize the cells.
- To prevent nonspecific antibody binding, block the sample with 500 µL PBS – BSA (1%) for 20 min at RT.
- Wash three times with 2 mL of PBS-Tween20 (0.1%) and incubate with 300 µL of anti-puromycin antibody (12D10) diluted 1:12,500 in 1X PBS for 1 h at RT.
- Wash 3 times with 2 mL of PBS-Tween20 (0.1%) and incubate with 300 µL of anti-mouse IgG conjugated to a fluorophore (here, 488 nm) diluted in PBS to visualize puromycilated polypeptides and with phalloidin-conjugated to another fluorophore (here, 555 nm) to visualize F-actin. Incubate for 1 h at RT.
- Wash three times with 2 mL of PBS-Tween20 (0.1%) and then incubate for 5 min at RT in 300 µL of PBS-Tween20 (0.1%) supplemented with 1 µg/mL 4',6-diamidino-2-phenylindole (DAPI).
- Wash three times with 2 mL of PBS-Tween20 (0.1%) and then wash with 2 mL of 1X PBS. Prevent the cells from drying by keeping the sample in 2 mL of 1X PBS during image acquisition.
3. Immunofluorescent Image Acquisition
NOTE: The following methodology can be used with any commercially available confocal imaging system.
- Determine the appropriate settings for each fluorophore to maximize the dynamic range for quantification. NOTE: Representative quantification can only be obtained if pixel saturation is avoided and the signal threshold is adjusted properly. These settings can be attained by carefully adjusting the laser power, high voltage (HV), gain, and offset.
- Adjust the zoom factor (5X) and the number of pixels (512 x 512) to optimize the pixel size. NOTE: This should be at least 2.3-times smaller than the optical resolution, according to the Nyquist theorem.
- Decrease the scan speed (12.5-20 µs/pixel) and use averaging (2-3 times) to improve the signal-to-noise ratio according to the settings mentioned above.
Manually determine the appropriate top and bottom focal planes along the Z-axis with the optimal step size (0.4 µm/slice). NOTE: The step size plane is determined by the axial resolution divided by 2.3, according to the Nyquist theorem. Typically, 20-25 layers are necessary to cover the entire cell depth of adherent cells.
Acquire a confocal image of an entire single cell with a 60X Plan Apo oil immersion objective (1.42 numerical aperture).
4. Immunofluorescent Image Quantification
- Determination of enrichment.
- Open an image corresponding to the z-plane layer of interest from the acquired cell data file using the ImageJ software (freeware available at http://fiji.sc).
- Draw a line using the line drawing tool (Tool bar → Straight) through the regions of the cell where quantification is desired. Modify the line width by double-clicking on "straight;" the intensity values will be averaged through the width of the line (set at 8 in Figure 3). NOTE: A thinner line is preferred to avoid signal overlap from different structures.
- After the line is properly set, profile the signal density along the determined axis (menu bar → Analyze → Plot Profile). NOTE: A new window will open that shows a profile corresponding to the signal intensity for each pixel of the selected channel (specific fluorophore) along the line for the selected z-plane section.
- Repeat the process for each channel corresponding to the fluorophore used (i.e., one quantification for 488, one for 568, and one for 405). NOTE: The signal intensity value will always be ordered following the orientation of the drawn line.
- To obtain the numerical gray-scaled values for each pixel of the profile corresponding to the quantifications of the selected z-plane layer, copy the profile data (menu bar in image window → Copy) and paste it in any spreadsheet software.
- Repeat steps 4.1.1-4.1.4 for each z-plane section of the acquired confocal image and each channel where quantification is desired. NOTE: The quantification of different z-plane layers can be pooled to obtain a general assessment of the special enrichment of the signal by calculating the sum value of each pixel along the line for each z-plane layer. This type of pooling can only be achieved if the line drawn is identical for each z-plane layer included in the mean values.
- As an alternative method for the whole-cell quantification of different channels with a large number of layers, use the macro called StackprofileData (see the Supplemental File). NOTE: This macro is accessible freely at https://imagej.nih.gov/ij/macros/StackProfileData.txt and can be used as follows:
- Paste the macro into a text file and save it.
- Open the image file that includes all of the confocal layers of the cell.
- Draw a line, as described in step 4.1.2, open a new window to import the macro (menu bar → Plugins → Macros→ Run), choose the previously saved text file corresponding to the StackprofileData macro, and click "Open." NOTE: Following macro importation, a new window named "Results" will open and all of the quantification along the determined axis will be listed for each z-plane layer and channel present in the image files.
- Copy the data for each channel (menu bar → Edit → Copy) to obtain the graphical profile representation corresponding to the signal quantifications. Paste it into any spreadsheet software. Calculate the sum value of each channel for each pixel along the line for each z-plane layer. Use these values to create a graphical representation similar to the one presented in Figure 3.
- Quantification of signal in a designated cellular area or volume.
- Open the image file with ImageJ software.
- Draw an area to denote the area of interest using the geometrical form function (tool bar →"rectangle," "oval," or "polygon"). NOTE: The quantification value will be determined for the area corresponding to the drawn form.
- Adjust the threshold level to avoid any background signal (menu bar → Image Adjust → Threshold); a new window will appear to represent the pixel intensities in histogram form. Change the values to include/exclude pixels in the quantification using the slider. Select a threshold that eliminates red pixels outside the cell, as pixels highlighted in red will be included in the quantification.
- Define the measurement parameters to quantify the pixel signal within the selected area, without background signal (menu bar → Analyze → Set Measurements), and make sure to check the "Limit to Threshold" and the "Integrated Density" boxes.
- Measure the quantitative values for the selected area (menu bar → Analyze → Measure). NOTE: Signal intensity quantification will be presented in a new window under the "IntDen" identification, which represents the product of the mean gray values and the number of pixels within the selected area.
- Draw an area that includes the entire cell using a geometrical form (tool bar →"rectangle," "oval," or "polygon") and proceed with steps 4.2.3-4.2.5 to obtain a value corresponding to the total signal. Calculate the ratio of the signal within the selected area over the whole signal to obtain the percent of the signal within the area of interest.
- Repeat steps 4.2.2-4.2.6 to obtain the quantification of cell volume for each z-plane. Adapt the geometrical form as needed.
- Copy/paste the data obtained from the "Results" windows for each z-plane layer into a spreadsheet for graphical depiction and to find a mean value.
Representative Results
To accurately observe translation events using puromycin incorporation, it is critical to determine the optimal conditions for each cell line because each shows different puromycin incorporation kinetics (Figure 1)9,11,12,18. Hence, to validate puromycin incorporation, it is necessary to treat the desired cell line with a standardized spectrum of increasing concentrations of puromycin (1.0, 2.5, 5.0, 7.5, 10.0, and 15.0 µg/mL) for different periods of time (5 and 10 min). To assess localized translation or an early response to drug treatment, a shorter incubation time is preferred (e.g., less than 5 min), as it allows for the direct evaluation of the translational events directly following a specific treatment. Ideally, the puromycin signal should be easily detectable but should also avoid the saturation point (Figure 2). As depicted in Figure 2 (left panels), an acceptable puromycin concentration is 7.5-10.0 µg/mL for MRC-5 and HeLa, whereas 5.0-7.5 µg/mL of puromycin is suggested for Huh-7. Optimization of the experimental parameters is crucial because the goal of this method is not to completely inhibit all translational elongation at the moment of initiation but to tag newly synthetized polypeptide chains during translation to detect acute variations in translation. This initial phase in the protocol should not be neglected, as excessive puromycin availability and extended treatment time will cause major undesired consequences. As observed in Figure 2 (right panels), cells treated for 10 min with a high puromycin concentration will generate mostly smaller puromycilated polypeptides (e.g., Huh-7, 7.5 µg/mL or more for 10 min). These smaller polypeptides will diffuse more rapidly in the cell, which might interfere with accurately assessing subcellular localization. Another problem is that increased proteolysis occurs upon excessive puromycin treatment19. This result was observed in MRC-5 and HeLa cells treated for 10 min, which showed decreased signal intensity in the presence of 10 or 15 µg/mL of puromycin. Both of these phenomena are misleading if puromycin incorporation is assessed by immunofluorescence because increased diffusion prevents the accurate visualization of translation localization, whereas puromycin-induced degradation greatly decreases detection sensitivity. These unwanted consequences are easily avoided by properly defining optimal experimental conditions, as described in section 1 of the Protocol.
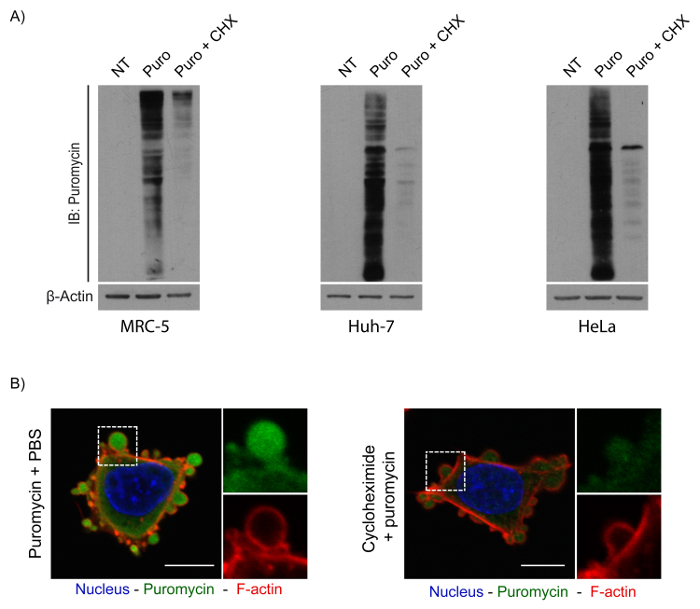
While visualizing puromycin incorporation, it is also important to perform proper controls. As shown in Figure 3A, puromycin incorporation can be efficiently blocked by the addition of 50 µg/mL cycloheximide, a compound known to inhibit translation elongation20 and thus puromycin incorporation. This behavior is shown in Figure 3B, which shows that cycloheximide treatment efficiently prevented most of the signal corresponding to puromycin incorporation in adherent MRC-5 cells. Indeed, while puromycilated peptides can be visualized in cells treated with puromycin only, a weak signal is detected in cells that were also treated with cycloheximide under identical staining conditions. Alternatively, another antibiotic (anisomycin) can also be used as a negative control, as it has the ability to block peptidyl transferase activity5 and has been shown to be as efficient as cycloheximide at blocking puromycin incorporation5,18
Following immunofluorescent image acquisition, it is possible to assess the general localization of puromycilated polypeptides corresponding to newly synthetized proteins (Figure 4). Image layers are selected at different depths within the adherent cells, allowing the signal intensity in subcellular compartments to be assessed in different cell planes. As such, it is possible to compare the localized puromycilation reaction at the bottom of the cell (Figure 4A), the middle of the cell (Figure 4B), or the top of the cell (Figure 4C). Situated slightly above nascent and maturating adhesion structures, SICs are mostly observed in the middle of the cell. As expected, intense puromycilation is detected in SICs, suggesting that mRNA translation is isolated to these subcellular structures. As mentioned previously, it is possible to compare signal intensity along a desired axis. This comparison is only possible if the images acquired do not include saturated pixels, which appear red in the grayscale image (second panels from the top) obtained using the microscope operating software. These images can be imported into the ImageJ software for quantification and analysis of the pixel intensity along a designated axis. The pixel quantification can be represented graphically (middle panel) to illustrate the relative pixel intensity at designated positions along the axis. A closer look at the insert in the upper panels shows SIC-actin boundaries (in red) and a green signal within the structure that corresponds to highly active translation events, which are enriched within these structures (bottom section). Although this quantification method is not the best way to determine the exact percentage of total translation occurring in these structures, it is visually informative by allowing for the rapid assessment of signal intensity and is a good approximation of the signal distribution throughout the cell.
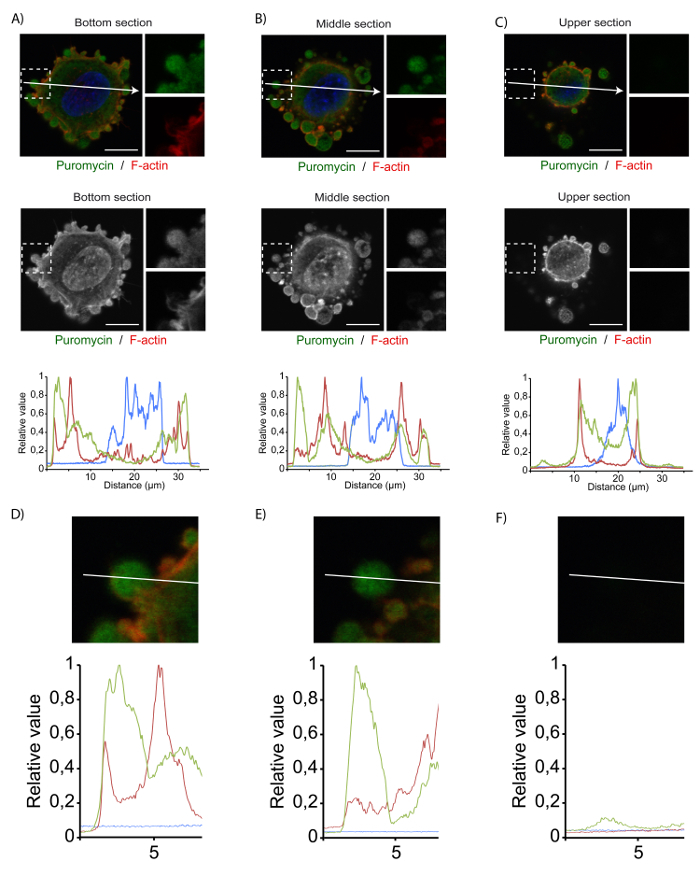
To obtain a quantitative distribution of the signal throughout the whole cell, another method should be used. As shown in Figure 5, one of the key steps is to denote the areas of interest that need to be quantified for every z-plane composing the confocal image files. This assignment can be achieved using a different drawing tool found in ImageJ. Using this method, it is possible to quantify either a single area or multiple areas, which can then be compared, subtracted, or even compiled. The main difficulties are to: (i) find the optimal imaging conditions that avoid pixel saturation and background signal, which might misrepresent the quantitative value of the obtained signal, and (ii) establish optimal calibration of the threshold in ImageJ. Assuming that both of these conditions are optimal, this quantification method is highly reproducible while still remaining easy to perform. Prior to image acquisition, it is also important to carefully determine the lower and upper limits of the z-planes of the cell selected for quantification. In the case presented in Figure 4, the bottom section corresponds to the resolution limit of the objective, which often includes contaminating evanescent fluorescence, which can decrease the SIC/cell body signal ratio. As previously mentioned, SICs are involved in adhesion site formation and maturation; therefore, SIC structures are situated slightly above newly forming adhesion complexes, which excludes evanescent fluorescence from the bottom-most layers. Thus, the SIC-bound puromycilation signal is barely visible in the bottom sections, whereas we can clearly observe it in the middle section, explaining the increased ratio of SIC/cell body signal in the middle region compared to the lower or upper planes.
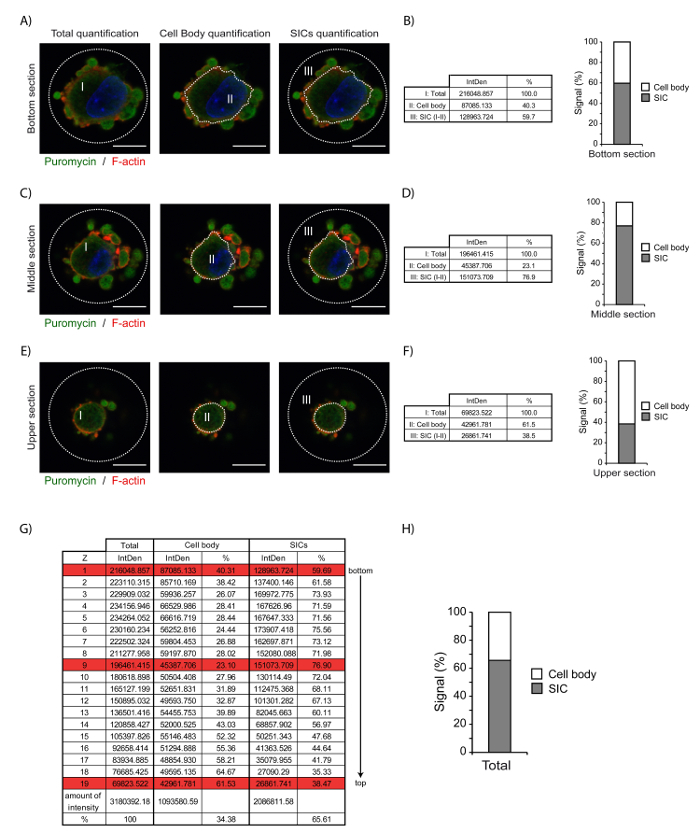
Figure 1. Experimental representation of the puromycin treatment optimization described in section 1. (A) A 24-well representation of cells being seeded for the experiment (step 1.1). (B) Schematic representation of step 1.4 showing the medium change in rows 1 and 2 of the 24-well plate and the addition of cycloheximide (CHX) to each well of the third row for a final concentration of 50 µg/mL. (C) Schematic representation of the puromycin treatment in the second and third rows (step 1.6) 15 min after step 1.4. (D) Schematic representation of the puromycin treatment in the first row (step 1.8) 5 min after step 1.6. Please click here to view a larger version of this figure.
Figure 2. Determination of the optimal conditions for puromycin incorporation. Western blot analysis of the whole-cell extract from (A)MRC-5, (B) Huh-7, and (C) HeLa cells, incubated with increasing concentrations of puromycin (0, 2.5, 5, 7.5, 10, and 15 µg/mL) for a period of 5 min (left panels) or 10 min (right panels). Puromycilated peptides were detected using an antibody against puromycin. Please click here to view a larger version of this figure.
Figure 3. Inhibition of puromycilation using cycloheximide. (A) Western blot showing the effect of cycloheximide on a 5-min puromycilation assay. Incorporation efficiency in MRC-5, Huh-7, and HeLa cells following mock (PBS 1X) or cycloheximide treatment. (B) Confocal image showing the effect of cycloheximide on puromycin incorporation. MRC-5 cells were pre-treated with either PBS (mock) or cycloheximide for 15 min and then supplemented with 10 µg/mL of puromycin + PBS 1X (left panel) or 10 µg/mL of puromycin + 50 µg/mL of cycloheximide (right panel) for 5 min. Puromycilated proteins were detected using an antibody against puromycin (green). F-actin was detected using phalloidin (red), and the nucleus was stained with DAPI (blue). Inserts on the right correspond to a 2.5x magnification of the white box. Bars = 10 μm. Please click here to view a larger version of this figure.
Figure 4. Quantification of the translational activity in SICs in adherent MRC-5 cells. (A-C) Representative confocal image of adherent MRC-5 cells treated with 10 µg/mL puromycin for 5 min. The images presented correspond to the bottom (A), middle (B), and upper (C) portions of the cell. The upper panels show the puromycilated proteins detected using an antibody against puromycin (green). F-actin was detected using phalloidin (red), and the nucleus was stained with DAPI (blue). Inserts on the right correspond to a 2x magnification of the white box. Bars = 10 μm. The middle panels show the absence of saturated pixels, which would have been highlighted in red. The lower panels show a graphical representation of puromycilated protein enrichment in an adherent MRC-5 cell by comparing the signal intensity for puromycin (green), F-actin (red), and DAPI (blue) along the cell axis indicated by the white arrow. (D-F) The images presented correspond to the4.4x magnification insert of the SICs presented in (A-C). The quantification shows the SIC border (red peaks: actin), as well as localized translation within the SICs (green peaks: puromycin) in the lowest (D), the middle (E),and the upper (F) regions of the cell. Please click here to view a larger version of this figure.
Figure 5. Whole-cell signal quantification of puromycilated proteins in adherent MRC-5 cells. (A) Determination of the signal within different areas of the bottom z-plane layer of an adherent MRC-5 cell. (Left panel) Selection of area (I) covering the totality of the cell to obtain a quantitative value corresponding to the total cellular signal for this z-plane layer. (Middle panel) Selection of the cellular area corresponding to the nucleus and the cytoplasmic portion that does not includes SIC (II). (Right panel) Visualization of the area corresponding to the SICs (III) by subtracting the value obtained from area II from the value obtained for the whole cell (area I). Bars = 10 μm. (B) Quantitative pixel values for each area are listed in the table for the image depicting the area selection in (A), followed by a graph showing the proportion (%) of signal found in SICs. (C) Determination of the signal within different areas of the mid z-plane layer of an adherent MRC-5 cell. (Left panel) Selection of the area (I) covering the totality of the cell to obtain a quantitative value corresponding to the total cellular signal for this z-plane layer. (Middle panel) Selection of the cell area corresponding to the nucleus and the cytoplasmic portion that does not includes SIC (II). (Right panel) Visualization of the area corresponding to the SICs (III) by subtracting the value obtained for area II from the value obtained for the whole cell (area I). (D) Quantitative pixel values for each area are listed in the table for the image depicting the area selection in (C), followed by a graph showing the proportion (%) of the signal found in SICs. (E) Determination of the signal within different areas of the upper z-plane layer of an adherent MRC-5 cell. (Left panel) Selection of the area (I) covering the totality of the cell to obtain a quantitative value corresponding to the total cell signal for this z-plane layer. (Middle panel) Selection of the cell area corresponding to the nucleus and the cytoplasmic portion that does not includes SICs (II). (Right panel) Visualization of the area corresponding to the SICs (III) by subtracting the value obtained for area II from the value obtained for the whole cell (area I). (F) Quantitative pixel values for each area are listed in the table for the image depicting the area selection in (E), followed by a graph showing the proportion (%) of the signal found in SICs. (G) Quantification values for all of the z-plane layers from the cell presented above, including one calculated for z-planes presented in A (z = 1), C (z = 9), and E (z = 19), which are highlighted in red on the table. (H) Graphical representation of the percentage of signal found in SICs throughout the entire cell volume. Please click here to view a larger version of this figure.
Discussion
Recent technological advances have allowed for a better understanding of the mechanisms involved in translational upregulation or the repression of specific mRNAs in biological processes, such as neuronal development, drug response, and metastasis. The cost-effective methodology described here allows translation events to be visualized in cells to study how RNA-binding proteins regulate metastatic processes, such as cellular adhesion, migration, and invasion.
Although numerous methods to assess de novo protein translation have been developed, they all share the same strategy, in which the elongating polypeptide incorporates modified amino acids tagged with a detectable moiety. The first of these methods relies on 35S-radiolabeled arginine incorporation into the newly translated protein. Although it is efficient for comparing general rates of translation under specific conditions, the use of radiolabeled amino acids makes this method unappealing to most research teams. Hence, the development of novel methods using non-radioactive labels, such as SunSET, Click-it, or the TRICK approach, offered new opportunities to assess and observe in vivo translation events. Although ingenious, most of these methods only allow for the assessment of translation events following drug treatment or specific cellular events using a specific exogenous cDNA construct of specific targets, limiting experiments to already identified targets. This is true for the TRICK approach4, which allows for the translational regulation of a single exogenously expressed mRNA target to be assessed. Although it allows for the validation of the translational activation of endogenous mRNAs, the "Click-IT" method minimal incubation time for AHA incorporation is at least 1 h13, which is considered too long for most acute translation mechanisms.
While highly versatile and easy to set up, the method described here requires that the critical steps be followed in the initial phase of the protocol. As shown in the representative results, different cell lines will have different puromycin incorporation kinetics, which might simply be due to the metabolic rate of each cell line, as seems to be the case for MRC-5 cells5,21. Indeed, non-transformed cells are not as proliferative as HeLa, and therefore, protein mass renewal is less robust than in transformed cells. This should be taken into account when using the protocol. Accordingly, the first portion of the protocol is critical for the rest of the method. Carefully determining the concentration of puromycin and the length of incubation will allow for a better assessment of the translation occurring during the experiment. As mentioned above, treatment with an excessive amount of puromycin increases the ratio of small puromycilated polypeptides and seems to stimulate proteolysis, which could easily lead to a misinterpretation of normally occurring translational events19. As a rule of thumb, the longer the incubation time, the less puromycin is needed, whereas a shorter incubation time requires a higher puromycin concentration. The concentration/time can be adapted to particular experimental limitations.
The methods presented here are highly adaptable and can easily be modified according to the treatment applied or the biological process studied. Puromycin incorporation allows for the rapid assessment of changes in translation kinetics over a short period of time, while the quantification method provides highly informative images of cellular events. Although powerful, these methods have limitations that must be considered. Puromycin will be incorporated into proteins newly synthesized from all mRNAs that are actively translated. For this reason, the puromycilation method described here might not be suited to study specific targets (i.e., single mRNAs), but it is ideal to obtain a general overview of translational activity within a single cell or a cell population. As mentioned above, excessive puromycin has major drawbacks, and puromycin dosages and treatment times should be carefully defined, as proposed in the protocol. Indeed, as shown in Figure 2, excessive puromycin availability will result in the formation of smaller puromycilated polypeptides that diffuse more rapidly in the cell, leading to inaccurate subcellular localization data. Additionally, increased proteolysis is known to occur after excessive puromycin treatment19, an effect that might result in a complete loss of signal.
Although not as specific as the Click-it or TRICK methods, the approach proposed here is highly accessible and allows for the rapid assessment of general changes in translation kinetics. Even if other methods are better suited for specific applications, puromycin incorporation assays can be performed as a supplementary control. Indeed, if the experiment needs to show that a specific mRNA target is the only one affected in a particular condition, it is necessary to show that this is not caused by a general increase in translation. As such, negative puromycin incorporation could provide evidence for this case. Moreover, this method is well suited to observing general changes in translation rates. Hence, it can be used to show a translation repression mechanism, as in the case of stress granule formation9, or to validate if cycloheximide treatment used in the initial phase of translational complex fractionation is efficiently blocking elongation22. It is also important to mention that, although the quantification methods described in sections 2.3 and 2.4 relate to experiments focusing on puromycin staining, they could be applied to any type of compartmentalized staining using various types of fluorophores. Indeed, these quantification methods could be used to quantify the cellular distribution of a molecule or protein and can even validate the localization of a target in a specific subcellular compartment.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Dr. Rachid Mazroui (Université Laval, Québec, Canada) for the critical reading of the manuscript. We thank the Cell Imaging Unit of the Research Center for their technical assistance. M.-É. Huot is a Junior 1 Research Scholar of the Fonds de Recherche du Québec-Santé (FRQ-S). This work was supported by the Canadian Institutes of Health Research (grant number CIHR, MOP-286437 to M.-É. Huot).
References
- Yan X, Hoek TA, Vale RD, Tanenbaum ME. Dynamics of Translation of Single mRNA Molecules In Vivo. Cell. 2016;165:976–989. doi: 10.1016/j.cell.2016.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisaki T, et al. Real-time quantification of single RNA translation dynamics in living cells. Science. 2016;352:1425–1429. doi: 10.1126/science.aaf0899. [DOI] [PubMed] [Google Scholar]
- Wu B, Eliscovich C, Yoon YJ, Singer RH. Translation dynamics of single mRNAs in live cells and neurons. Science. 2016;352:1430–1435. doi: 10.1126/science.aaf1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstead JM, et al. TRICK: A Single-Molecule Method for Imaging the First Round of Translation in Living Cells and Animals. Methods Enzymol. 2016;572:123–157. doi: 10.1016/bs.mie.2016.02.027. [DOI] [PubMed] [Google Scholar]
- Bergeman J, Caillier A, Houle F, Gagne LM, Huot ME. Localized translation regulates cell adhesion and transendothelial migration. J Cell Sci. 2016;129:4105–4117. doi: 10.1242/jcs.191320. [DOI] [PubMed] [Google Scholar]
- El Fatimy R, et al. Tracking the Fragile X Mental Retardation Protein in a Highly Ordered Neuronal RiboNucleoParticles Population: A Link between Stalled Polyribosomes and RNA Granules. PLoS Genet. 2016;12:e1006192. doi: 10.1371/journal.pgen.1006192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adjibade P, et al. Sorafenib, a multikinase inhibitor, induces formation of stress granules in hepatocarcinoma cells. Oncotarget. 2015;6:43927–43943. doi: 10.18632/oncotarget.5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paronetto MP, et al. Sam68 regulates translation of target mRNAs in male germ cells, necessary for mouse spermatogenesis. J Cell Biol. 2009;185:235–249. doi: 10.1083/jcb.200811138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, Pierre PSUnSET. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- Azzam ME, Algranati ID. Mechanism of puromycin action: fate of ribosomes after release of nascent protein chains from polysomes. Proc Natl Acad Sci U S A. 1973;70:3866–3869. doi: 10.1073/pnas.70.12.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman CA, Hornberger TA. Measuring protein synthesis with SUnSET: a valid alternative to traditional techniques. Exerc Sport Sci Rev. 2013;41:107–115. doi: 10.1097/JES.0b013e3182798a95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David A, et al. Nuclear translation visualized by ribosome-bound nascent chain puromycylation. J Cell Biol. 2012;197:45–57. doi: 10.1083/jcb.201112145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C, Bassell GJ, Rossoll W. Spinal muscular atrophy: the role of SMN in axonal mRNA regulation. Brain Res. 2012;1462:81–92. doi: 10.1016/j.brainres.2012.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, et al. Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature. 2005;436:1166–1173. doi: 10.1038/nature03897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du TG, Schmid M, Jansen RP. Why cells move messages: the biological functions of mRNA localization. Semin Cell Dev Biol. 2007;18:171–177. doi: 10.1016/j.semcdb.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Ephrussi A, St Johnston D. Seeing is believing: the bicoid morphogen gradient matures. Cell. 2004;116:143–152. doi: 10.1016/s0092-8674(04)00037-6. [DOI] [PubMed] [Google Scholar]
- Mardakheh FK, et al. Global Analysis of mRNA, Translation, and Protein Localization: Local Translation Is a Key Regulator of Cell Protrusions. Dev Cell. 2015;35:344–357. doi: 10.1016/j.devcel.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacsina JR, et al. Premature translational termination products are rapidly degraded substrates for MHC class I presentation. PLoS One. 2012;7:e51968. doi: 10.1371/journal.pone.0051968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Poetsch T, et al. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol. 2010;6:209–217. doi: 10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JP, Jones CM, Baille JP. Characteristics of a human diploid cell designated MRC-5. Nature. 1970;227:168–170. doi: 10.1038/227168a0. [DOI] [PubMed] [Google Scholar]
- Gandin V, et al. Polysome fractionation and analysis of mammalian translatomes on a genome-wide scale. J Vis Exp. 2014. [DOI] [PMC free article] [PubMed]