

Abstract
HIV-1 entry inhibition remains an urgent need for AIDS drug discovery and development. We previously reported the discovery of cyclic peptide triazoles (cPTs) that retain the HIV-1 irreversible inactivation functions of the parent linear peptides (PTs) and have massively increased proteolytic resistance. Here, in an initial structure-activity relationship investigation, we evaluated the effects of variations in key structural and functional components of the cPT scaffold in order to produce a platform for developing next-generation cPTs. Some structural elements, including stereochemistry around the cyclization residues and Ile and Trp side chains in the gp120-binding pharmacophore, exhibited relatively low tolerance for change, reflecting the importance of these components for function. In contrast, in the pharmacophore-central triazole position, the ferrocene moiety could be successfully replaced with smaller aromatic rings, where a p-methyl-phenyl methylene moiety gave cPT 24 with an IC50 value of 180 nM. Based on the observed activity of the biphenyl moiety when installed on the triazole ring (cPT 23, IC50 ∼ 269 nM), we further developed a new on-resin synthetic method to easily access the bi-aryl system during cPT synthesis, in good yields. A thiophene-containing cPT AAR029N2 (36) showed enhanced entropically favored binding to Env gp120 and improved antiviral activity (IC50 ∼ 100 nM) compared to the ferrocene-containing analogue. This study thus provides a crucial expansion of chemical space in the pharmacophore to use as a starting point, along with other allowable structural changes, to guide future optimization and minimization for this important class of HIV-1 killing agents.
Graphical abstract
Introduction
Despite advancements in drug discovery and structure-based design of HIV-1 inhibitors, Acquired Immune Deficiency Syndrome (AIDS) is still a major global health problem with no curative agents nor protective vaccines available.1, 2 Highly Active Antiretroviral Therapy (HAART) has significantly improved survival rates3 and has also decreased the incidence of AIDS associated complications among the HIV-infected population, improving the patients' quality of life.4, 5 However, HAART does not cure HIV infection, because the virus hides by integrating proviral DNA into cells that can form long living reservoirs, and already formed viruses can persist in lymphatic tissues attached to follicular dendritic cells where they cannot be killed by current therapy.6, 7 The ability of HAART to bring the acute HIV infection into a chronic status has been associated with other chronic health problems caused by the side effects of the HAART drug cocktail. Moreover, recent global reports indicate a significant increase in HAART resistant population, increasing the demand to find novel drugs with new modes of action.
HIV-1 entry into the host cell remains an under-utilized target of the current therapy.8 This process is entirely mediated by the Env protein machinery, which functions through protein-protein interactions (PPI) with host cell receptors.9 The two components of HIV-1 Env, namely gp120 and gp41, represent attractive targets for the discovery of novel entry inhibitors. Enfuvirtide (T20), a 36 amino acid peptide, is the only FDA approved fusion inhibitor targeting the gp41 NHR region.10, 11 However, T20 is only used as a second line of treatment and has suffered from short duration of action, fast development of viral resistance and inconvenient route of administration. There are no approved gp120 inhibitors up to date, though the prodrug of BMS-626529 (1, Figure 1) is currently in phase III clinical trials as an attachment inhibitor.12 Studies have shown that the mode of action of BMS-626529 is to reversibly block the CD4 induced conformational changes in Env and consequently inhibit entry.13 Small molecule CD4 mimetics (2, Figure 1) are another class of entry inhibitors.14 They act by directly competing with CD4 for binding to the Phe43 cavity on the gp120 units. They have been shown to be useful in increasing the sensitivity of Env to antibody-dependent cell-mediated cytotoxicity (ADCC) response.15
Fig.1. Chemical structures of small molecule and small macrocyclic gp120 targeting inhibitors.

The conformational blocker attachment inhibitor BMS-626529 (1), small molecule CD4 mimetic (+)-BNM-IV-147 (2) and the conformational entrapping HIV-1 inactivator cPT AAR029b (3).
Generally, macrocyclic compounds have been found to be very effective inhibitors for poorly druggable targets such as protein-protein interactions (PPIs).16 Among all human proteins, only a relatively few can be targeted by small molecules,17 highlighting the need for novel PPI inhibitors. In fact, macrocyclic inhibitors are able to effectively inhibit PPIs by covering large hot spots between the two interacting proteins that traditional small molecules cannot reach, highlighting their importance in developing inhibitors for PPIs.18, 19 The example of HIV-1 Env and host cell receptors is an opportune PPI to target with macrocyclic inhibitors.
In this context, we have previously reported a class of small cyclic peptide triazoles (cPTs, represented by the first lead AAR029b20, 3, Figure 1) that is able to irreversibly inactivate HIV-1 virions, even before host cell encounter.20 They exert this unique mode of inactivation by hijacking the metastability of the Env protein complex,21 converting the gp120 into an inactive conformation that leads to gp120 shedding off the Env from the virion surface.20 The resultant gp41 coated virions become non-infectious. Interestingly, the binding site of cPTs seems to overlap both binding sites of BMS-626529 and the CD4 small molecule mimetics.22 Given the effectiveness of the initial cPTs to inactivate HIV-1 Env, we undertook a series of chemical explorations around the scaffold of cPT in order to better understand the structural requirements for function and to identify new methodologies to derive cPT compounds with an increased capacity for structural variation. We found in this SAR study that variations in many cPT structural elements were tolerated, though only within a narrow range of possibilities. In contrast, significant variations were tolerated in the substituted triazole component, leading to chemical space expansion, improved potency and molecular-structural properties, and establishment of a platform to guide future cPT development.
Results
I. Changing stereocenters at Lys and Asp centers
It has been previously shown that the stereocenters of the active core (Ile-triazoloPro-Trp) in the linear peptide triazoles have to be in the S configuration for optimal activity, whereas the R configuration can only be tolerated at the most N and C terminal residues.23 We examined here the effect of modifying the stereocenters at both the Asp and Lys residues, used for the cyclization (side-chain to side-chain), to investigate whether changes of these positions could tune the activity. As shown in (Figure 2), the newly introduced R configuration can only be tolerated at the Asp chiral center (center 1, cPT 5), with 7-fold decrease in activity compared to the lead cPT 3. In contrast, changing the configuration of the Lys chiral center (center 2, cPT 4) from S to R had a more devastating effect (> 50 fold decrease) on the activity. Therefore, the S configuration at both the Asp and Lys centers was judged to be optimal for cPT functions, though there is a degree of allowable tolerance at the Asp chiral center that can be an optimization point.
Fig.2. Structure of cPTs with altered stereochemistry at the Asp and Lys chiral centers.
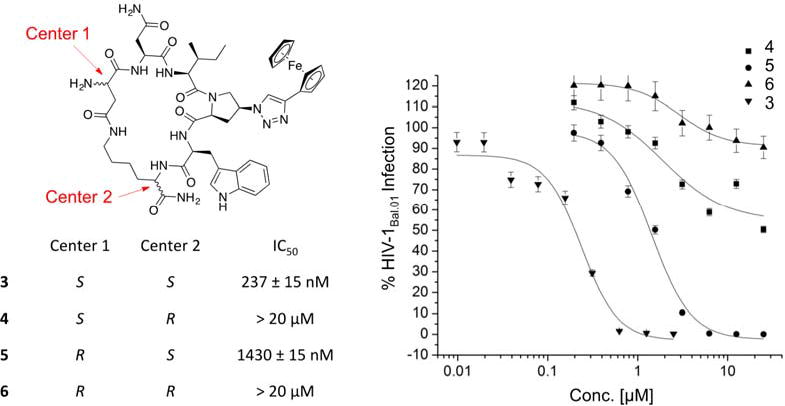
Each cPT was synthesized starting from pure L or D amino acids. The cutoff used in the infection inhibition assay was 20 μM, after which the compound was considered weak and no longer pursued.
II. Side chain variations (Ile sec-butyl and Trp indole) in the pharmacophore region
We had previously shown the importance of the pharmacophore (IXW, where X = aryl-triazole-Pro) for the functions of the parent linear PTs.24 Here, we sought to identify non-natural replacements for this pharmacophore to allow: a) exploration of the chemical space around this pharmacophore and b) non-natural replacements which will allow functionalization and derivatization at each pharmacophore site. Figure 3 shows the variations we made for both the indole and the sec-butyl side chains of the pharmacophore. As expected, the un-substituted indole ring could be nicely replaced with the isostere25, 26 benzo[b]thiophene (14, IC50 ∼ 170 nM, Figure 3). This observation was also previously seen in a linear hexapeptide analogue.22 Additions on the indole ring with a methoxy group resulted in very weak activity (13, Figure 3), arguing that this added group might prevent docking of the indole ring onto its target site on gp120 protein. The same weakening effect was also observed when benzonitrile, as an aromatic ring carrying a H-bond acceptor, was evaluated as a side chain (cPT 15). Along with the previous findings, it was evident that un-substituted indole and benzo[b]thiophene are both optimal for the activity at that pharmacophore site.
Fig.3. Structural changes in the indole (red) and sec-butyl (bue) moieties of cPT.
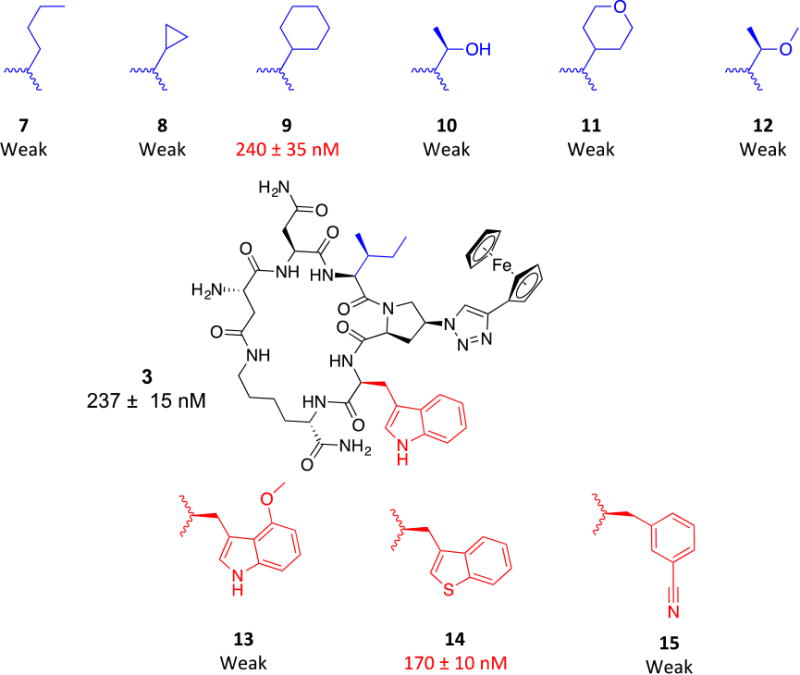
Activities shown are from HIV-1Bal.01 infection inhibition assay. The designation weak means the compound showed weak inhibition (less than 50%) at 10 μM, which was used as a cutoff point where the compound was no longer pursued.
We then moved to the Ile sec-butyl side chain, as it has not been previously explored. Interestingly, the branched nature of the sec-butyl group was found important where a methyl shuffle to the terminal carbon (7, Figure 3) resulted in massive reduction in activity. A similar effect was observed when the two terminal methyl groups were tethered in a cyclopropyl form (8, Figure 3). These weaker activities argued that increasing the bulkiness/branching of the hydrocarbon at this position might improve the activity. We therefore tried the bulky cyclohexyl group, which actually showed retained activity (9, IC50 ∼ 240 nM, Figure 3) compared to 3 (IC50 ∼ 237 nM). We next asked whether adding a polar group on this hydrophobic hydrocarbon would affect the activity. The hypothesis was that the hydrocarbon side chain might occupy a hydrophobic protein pocket and adding an electronegative atom may be able to abstract a H-bond from the protein backbone amide NHs, if they were close enough to the protein surface. In addition, adding a polar atom on these hydrocarbon groups might improve aqueous solubility. We found that adding an oxygen atom to the original sec-butyl side chain (as in 12, Figure 3) and to the cyclohexyl (as in 11, Figure 3) resulted in very weak activities. The same weakening effect also was observed when we changed the sec-butyl group with a smaller group containing unmasked OH group (10, Figure 3). The overall conclusion from this polar atom addition is that increasing the total polar surface area (tPSA) of these hydrocarbon moieties at the sec-butyl position leads to a massive reduction in functions.
III. Changes in the aryl group on the triazole moiety
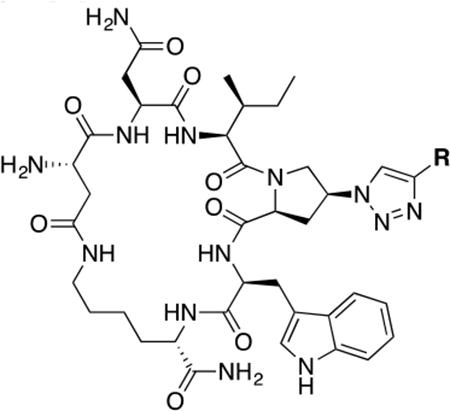
The main challenge in this work was to find alternatives for the ferrocene moiety, which has been shown to give optimal activities with both linear PTs27 as well as cPTs.20 The chemical nature of ferrocene does not allow exploration of the chemical space of triazole ring substituents. Hence, from a medicinal chemistry perspective, it was necessary to find a more acceptable moiety to replace the ferrocene ring and ultimately enhance drug development. As shown in Table 1, we used both commercially available alkynes and also synthesized additional key alkynes (Scheme 1), to identify triazole substituents that would enhance exploration of the chemical space on the triazole ring. The new triazole derivatives retained the dual host-cell receptor antagonism signature of the ferrocene-containing lead cPT 3 (Supplementary Table S1).
Table 1. Structure activity relationship around the triazole moiety.
A general structure is shown with the R group specified in the table, each with the HIV-1Bal.01 infection inhibition IC50 value. Asterisk (*) indicates that the cPT was made “in house” using a synthetic alkyne that was incorporated in the click reaction during the cPT synthesis.
| |||
|---|---|---|---|
| Compound/R group | HIV-1 IC50 (nM) | Compound/R group | HIV-1 IC50 (nM) |
|
2018 ± 9520 |
|
Inactive |
|
1400 ± 200 |
|
438 ± 12 |
|
1200 ± 500 |
|
900 ± 45 |
|
350 ± 25 |
|
269 ± 32 |
|
180 ± 9 |
|
5100 ± 220 |
|
220 ± 42 |
|
302 ± 50 |
|
2000 ± 390 |
|
8000 ± 250 |
|
5000 ± 850 |
|
1800 ± 500 |
|
7300 ± 740 |
|
6000 ± 450 |
Scheme 1. Chemical synthesis of alkynes a9-a16.
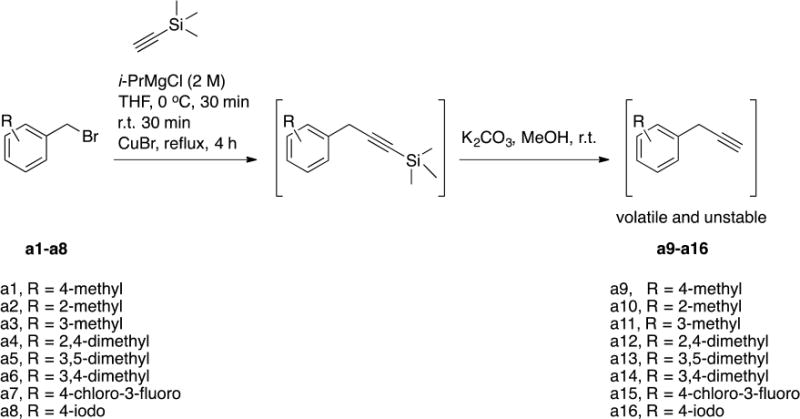
The final alkynes were volatile and unstable and hence were used immediately for the click reaction with the resin-bound azido-cPT to produce the triazole variant denoted “*” in Table 1.
We synthesized several alkynes starting from the commercially available benzyl bromides (Scheme 1). The prepared alkynes were relatively unstable and hence were utilized immediately for synthesizing cPTs (Table 1).
III.1. Structure activity relationship (SAR) of the triazole variant
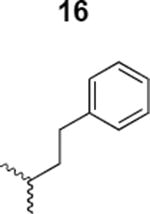
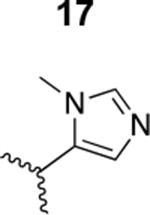
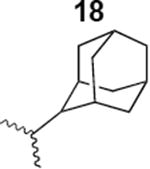
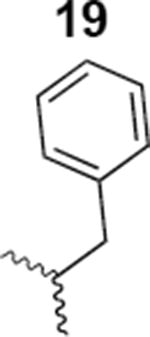
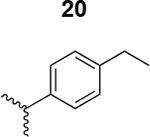
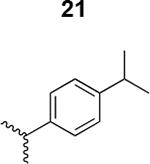
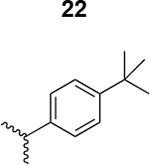
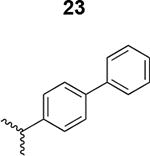
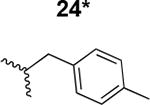
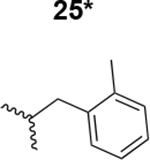
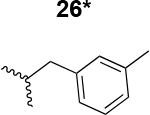
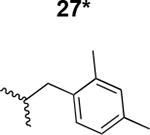
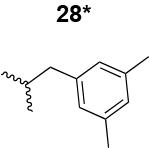
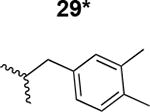
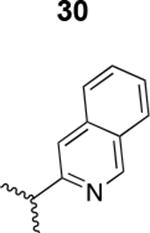
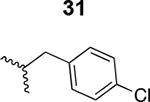
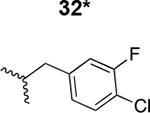
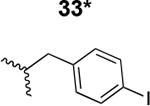
The HIV-1 cell infection inhibition activities of the new triazole derivatives were evaluated (Table 1 and Supplementary Figure S1). The hydrophobicity and aromaticity of the R group on the triazole ring were found to be important, as well as the spacing atoms between the triazole and the aryl group. Interestingly, using the basic N-methyl imidazole in cPT 17 gave the only triazole derivative was completely inactive. Comparing cPT 16 and 20, bringing the phenyl closer to the triazole ring slightly improved the activity (IC50 ∼ 1.2 μM for 20 compared to IC50 ∼ 2 μM for 16). Building up on 20 by adding extra methyl group(s) further improved the activity as shown for the isopropyl group (21, IC50 ∼ 0.9 μM) and the tert-butyl group (22, IC50 ∼ 0.35 μM). This observation indicated the importance of increasing the bulkiness at the para- position of the phenyl ring. Replacing the tert-butyl group (in 22) with a phenyl group further confirmed this hypothesis where the biphenyl containing cPT 23 exhibited an IC50 value of 269 nM. Introducing a bulky, non-aromatic moiety resulted in an active cPT (18, IC50 ∼ 1.3 μM), though less active than the aromatic biphenyl system (23). We also evaluated the isoquinoline moiety (30, IC50 ∼ 5 μM), which was not as active as the biphenyl system (23). We noticed that a single atom spacer between the triazole and the phenyl was missing in our prior PT28 and cPT20 optimization studies, and therefore evaluated the phenyl methylene moiety (in 19). Interestingly, the activity improved (IC50 ∼ 438 nM for 19) compared to the two-spacer atoms (as in 16) and the directly attached phenyl (as in 20). This enhanced potency indicated the importance of a single spacer atom in directing the phenyl ring to an optimal position. We then synthesized (Scheme 1) a series of alkynes to investigate the SAR around the phenyl of 19. Introducing an electron donating methyl group at the para- position further improved potency (24, IC50 ∼ 180 nM versus IC50 ∼ 438 nM for 19). A methyl shuffle from the para- position to the meta-position was well tolerated (26, IC50 ∼ 220 nM). However, when introduced at the ortho- position, activity was greatly decreased (25, IC50 ∼ 5.1 μM, about 28-fold lower potency compared to 24). Surprisingly, adding a para-methyl group to 25 restored the activity (27, IC50 ∼ 302 nM). This added para-methyl may have enabled a contact with a hot spot on gp120 that was not reached in the case of 25. A combination of either para-methyl and meta-methyl groups (in 29) or two meta-methyl groups (in 28) resulted in decreased anti-HIV activity (8 μM for 29 and 2 μM for 28). These results argue that increasing the bulkiness around the closer sides of the para- position may have negative effects on correctly positioning the phenyl ring on its target site on the gp120 protein, though addition of methyl far from the para- position could be tolerated (ortho- position as in 27). Introducing electron-withdrawing halogens on the phenyl ring of 19 resulted in decreased activities (31 with IC50 ∼ 1.8 μM, 32 with IC50 ∼ 7.3 μM and 33 with IC50 ∼ 6 μM). We considered the possibility that the hydrophobicity of these halogens may act similarly to the methyl groups, in addition to the possible halogen bonding with the protein carbonyl groups. However, the electron-withdrawing nature of the halogens may have reduced the electron density, on the phenyl ring, which may be required for essential π- π interactions with gp120 residues.
Based on the above-described variations (Figure 3 and Table 1), we envisioned that a combination of the pharmacophore side chain substituents would enhance the potency. We synthesized cPT 34 which has benzo[b]thiophene, cyclohexyl and ethyl-phenyl moieties, as hydrophobic groups, (Figure 4) to evaluate the activity of these combined variations. However, this cPT variant had poor solubility, and IC50 determination was challenging because of precipitation problems during the infection inhibition assay. In an attempt to increase the aqueous solubility of 34, we synthesized 35 (Figure 4) in which we replaced the amide side chain of Asn with an amine using a Dab amino acid during the cPT synthesis. As expected, 35 had better solubility because of the newly introduced amino group. However this cPT was found to be inactive. This reflects the importance of the amide functionality of the Asn residue or the unfavored positioning of the positively charged amine functionality within gp120.
Fig.4. Attempted modification in cPT scaffold to improve solubility of cPT 34.
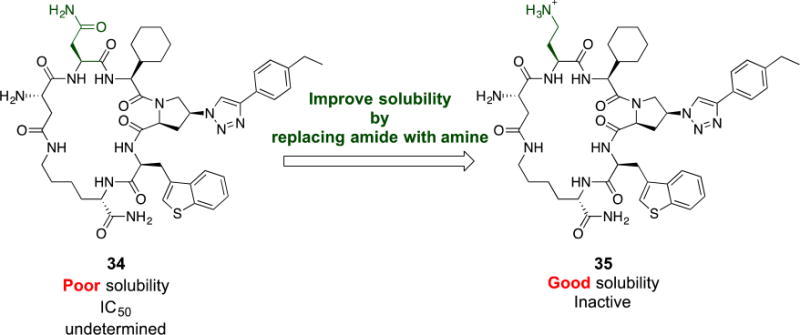
Each cPT was synthesized starting from the corresponding amino acids: Asn for 34 and Dab for 35. Designation of “poor” and “good” solubility was based on behavior of cPT in PBS buffer or PBS buffer with 5% DMSO.
IV. On-resin synthesis of bi-aryl containing cPT
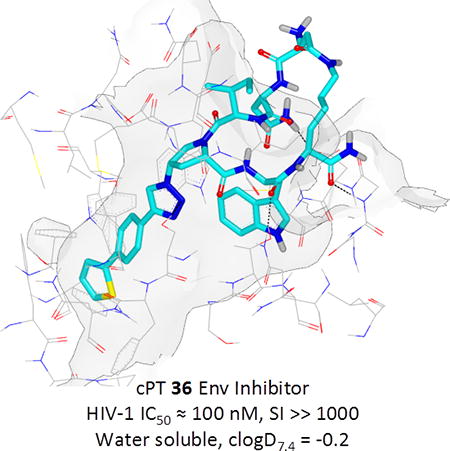
Based on the observation that the bi-phenyl containing cPT 23 (Table 1) has a comparable potency (IC50 value of 269 nM) to the parent ferrocene containing cPT 3 (IC50 value of 237 nM), we decided to explore the bi-aryl system as a potential activity-tuning moiety. We therefore designed a facile on-resin microwave-assisted29-31 synthetic pathway that will allow exploring different bi-aryl systems (Scheme 2). Briefly, after assembling the cPT sequence and on-resin cyclization, click reaction with the bromo-alkyne was achieved. The bromine handle was then used for an on-resin cross-coupling reaction using a boronic acid in presence of a palladium catalyst and a base. Mild reaction conditions were enough to achieve the complete formation of the bi-aryl system (Scheme 2). Interestingly, replacing the terminal phenyl ring of 23 (Table 1, IC50 value of 269 nM) with its isostere25, 26 thiophene32, 33 ring resulted in > 2-fold improvement in potency (36, IC50 value of 105 nM, Figure 5). This argues that the lipophilic nature of the aryl moiety in this part of the molecule is important for the activity. SPR competition assays showed that the newly derived thiophene-phenyl system retained the dual host-cell receptor antagonism signature of cPTs: 36 inhibited gp120 binding to both CD4 and 17b, the latter used as a co-receptor surrogate34 (Figure 5). However, replacing the middle phenyl ring (between the triazole and the thiophene of 36) with another thiophene ring (a bi-thiophene containing cPT 37) resulted in reduced activity (IC50 value of 405 nM for 37). This change in activity could be related to the change in the orientation of the long arm triazole-thiophene-thiophene relative to the gp120-binding site. In this view, the terminal thiophene of 37 installed in position 4 of the middle thiophene (Figure 5) could have resulted in slight bending of this long triazole arm compared to the straight arm in 36. Installing the terminal thiophene at position 5 of the middle thiophene is yet to be investigated. Cell viability was assessed for 36 and 37; no cytotoxicity was detected at the highest concentrations used (100 μM, Figure 5). Therefore, the selectivity index (SI) for 36 can be estimated to be >> 1000 based on the IC50 and the CC50 values, indicating a good cellular safety profile for this new lead compound.
Scheme 2. On-resin synthesis of the bi-aryl system on cPT.
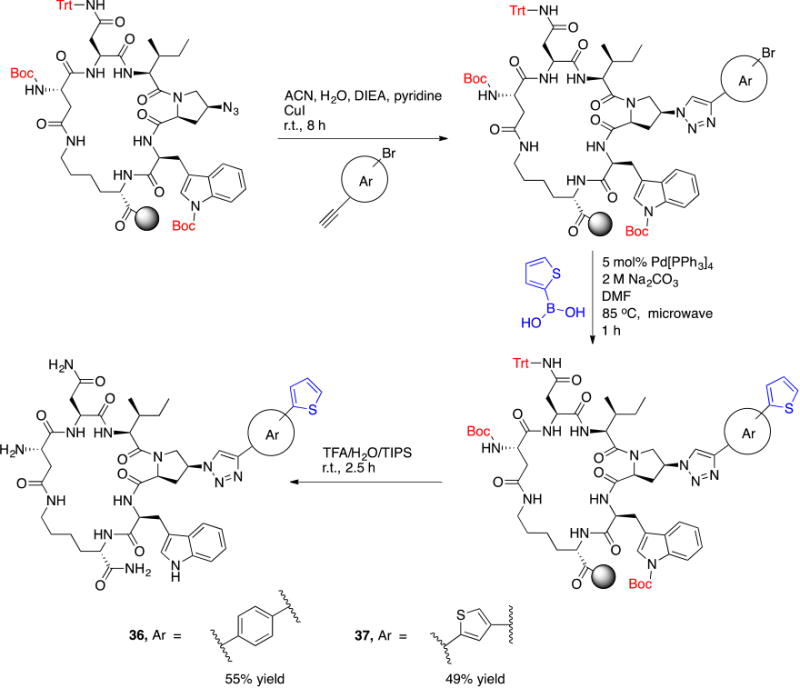
Reaction conditions and reagents are shown on the arrows.
Fig.5. Structures and functional properties of cPTs 36 & 37. (.
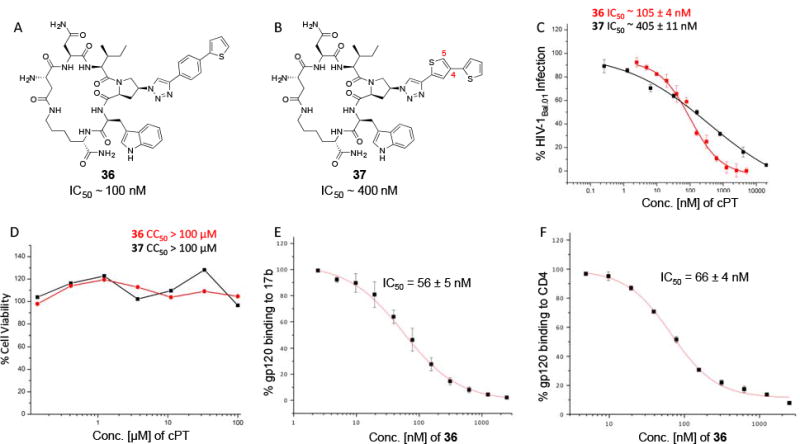
A) cPT 36, (B) cPT 37, (C) infection inhibition curves against HIV-1Bal.01 for 36 & 37, (D) cell viability assay for 36 & 37. SPR competition assay were used to determine the ability of 36 to inhibit gp120 binding to both 17b (E) and CD4 (F).
V. Isothermal titration calorimetry (ITC) of 3 versus 36
To better explain the differences in antiviral potencies between cPTs 3 and 36, we determined their binding affinities and thermodynamic profiles by isothermal titration calorimetry (ITC). Binding was measured in the monomeric gp120-YU-2 context. Glycoprotein gp120 YU-2 bound 3 with an affinity of 415 nM and a stoichiometry of one cPT per gp120 molecule (Figure 6, A). The binding mode was mainly enthalpy driven, with a ΔH value of −5.96 kcal/mol (Figure 6, C). The new cPT 36 showed improved binding affinity with dissociation constant of 350 nM and was bound by one gp120 molecule, comparable to 3 in the binding comparison (Figure 6, B). Interestingly, the thermodynamic profile of 36 indicated a slight increase towards an entropically favored (−TΔS = −4.74 kcal/mol) binding mechanism compared to 3 (−TΔS = −2.75 kcal/mol, Figure 6, C). The effect of peptide cyclization can be observed in the thermodynamic signature by comparing cPTs and PTs. In this study, both 3 and 36 showed positive ΔS values, suggesting an entropically favored binding mechanism. In contrast, the thermodynamic signature for linear PTs previously showed negative ΔS values,23 reflecting a large unfavorable entropy change.
Fig.6. cPT binding and thermodynamic profile of monomeric wild type gp120 YU-2 for cPTs 3 (A and C) and 36 (B and C).
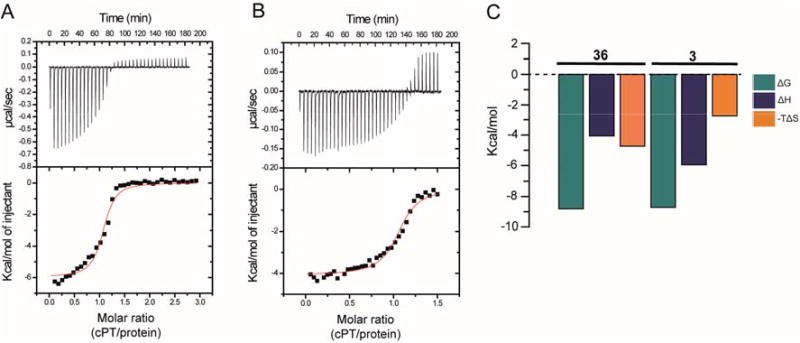
Compound solution, 390 μM (for 3) or 200 μM (for 36), was titrated in 8 μL steps into 30 μM of YU2 full-length gp120 at 25°C in the reaction chamber of a VP-ITC instrument using 1× PBS buffer at pH 7.3. The resulting heat changes were integrated, and the values obtained were fitted to a quadratic binding equation (one site binding model). The following Kd and thermodynamic values were derived: 3 (A and C): KD = 415 ± 98 nM, n = 1.06 ± 0.02, ΔH = −5.96 ± 0.13 kcal/mol, ΔS = 9.22 cal/mol; 36 (B and C): KD = 350 ± 62 nM, n = 1.06 ± 0.01, ΔH = −4.07 ± 0.06 kcal/mol, ΔS = 15.9 cal/mol.
VI. Binding model of 36 with HIV-1 Env gp120 unit (pdb: 5FUU35)
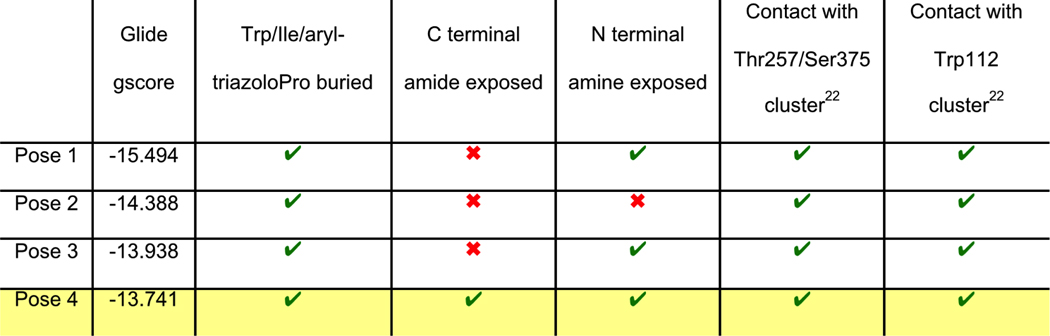
PTs and cPTs bind to an inactive state of the conformationally dynamic36 37 HIV-1 Env that lacks a formed bridging sheet, possibly by inserting a hydrophobic part of the molecule under the β20/21 loop of the bridging sheet.38, 39 To investigate the binding mode of new cPT 36 with the dynamic HIV-1 Env gp120, we used Glide InducedFit Extended Sampling to allow flexibility of the protein at the active site. Multiple conformers of 36 were generated (Schrödinger Macrocycle Conformational Sampling)40, 41 and clustered using heavy atoms. This yielded 30 clusters, for each of which one conformer was selected as a cluster representative. The 30 cluster representatives were docked (using Schrödinger InducedFit Extended Sampling), and the returning pose/protein complexes were sorted according to their Glide gscores. Examination of the top scored poses was based on the selection criteria (Table 2) that were extracted from structure activity relationships in prior work20, 22, 27, 28 as well as from the current study. The first pose that meets all the selection criteria (pose 4 from Table 2) was selected as a representative putative binding mode (Figure 7) for 36 within gp120. The overall ligand-protein interaction energy for this pose was calculated using SZYBKI (v1.8.0.1: OpenEye Scientific Software, Santa Fe, NM. http://www.eyesopen.com) and found to be −27.13 kcal/mol, indicating high stability within this binding conformation. Interestingly, the pose shows a similar orientation to what has been previously illustrated for different cPTs.20, 38 The newly introduced thiophene-phenyl on the triazole nicely penetrates through the pre-identified sub-site 222 within gp120 located under the α1-helix and β20/21 loop (yellow shading in Figure 7), and the Trp indole buries itself in sub-site 122 in proximity to residue Thr257 and the α5-helix residue Asn478 (light blue shading in Figure 7). The bi-aromatic system stabilizes itself in the gp120 pocket through two possible T-shaped and edge-to-face stacking interactions. The thiophene stacks to the important residue Trp112 indole ring,22 whereas the phenyl (between the triazole and thiophene) stacks with residue Phe382 (Figure 7). Such interactions could be the reason for activity retention by the bi-aryl systems comparable to the bulkier ferrocene moiety. The putative model also shows the sec-butyl moiety of the cPT-Ile buried between the β20/21 loop and the CD4 binding loop (Figure 7). This finding might explain the importance of this branched/bulky moiety compared to less branched or tethered moieties (Figure 3). In addition, this model is consistent with the finding that adding polar atoms to this cPT-Ile side chain moiety/substituent would cause solvation of this part of the molecule, therefore could be pulling the molecule out into the solvent and preventing the proper docking onto the active site.
Table 2. Selection criteria of a putative binding pose of 36 with gp120 (pdb code 5FUU35).
Green “correct” marks mean that the pose fits the criterion, the red “cross” marks mean that the pose doesn't fit the criterion. The first pose, which meets all criteria, is highlighted in yellow.
|
Fig.7. Putative binding pose 4 (from.
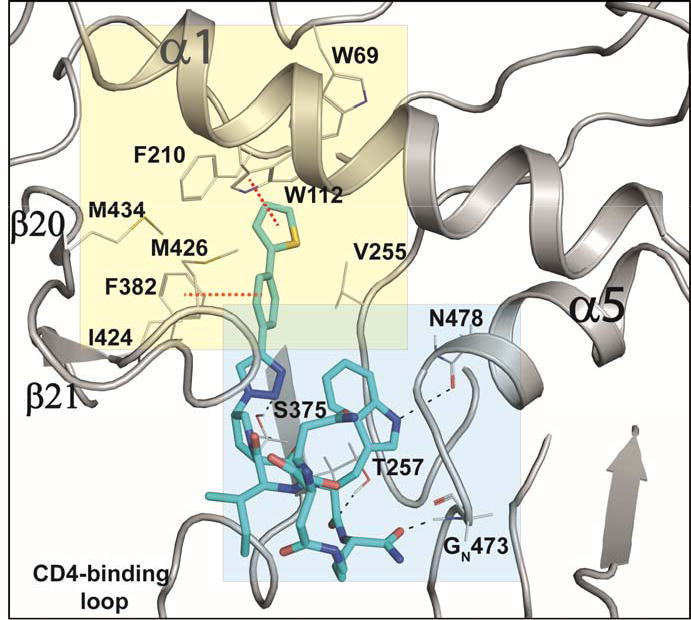
Table 2) of cPT 36 docked onto gp120 from the pdb 5FUU35. cPT 36 is shown in cyan sticks, H-bonds are shown as black dashed lines and π-π stacking interactions are shown in red dashed lines.
VII. Effect of removing of the primary amine group from 36
The above-described model (Figure 7) showed that the N terminal of 36 is solvent exposed. We investigated the potential of removing this primary NH2 from the cPT scaffold. This would allow exploring the importance of this part of the scaffold, which acts as a connecting bridge to the important cPT pharmacophore. Additionally, this NH2 removal would reduce the total polar surface area of the scaffold and increase the lipophilicity. Controlled lipophilicity42 could be a step towards optimizing the scaffold for enhanced oral bioavailability. We designed a synthetic route to achieve the cyclization using a non-chiral, NH2-free linker (Scheme 3). The cyclization intermediate 4-((9H-fluoren-9-yl)methoxy)-4-oxobutanoic acid (a19) was synthesized as previously reported starting from 9-fluorenylmethanol (FmOH, a17) and succinic anhydride (a18).43 The resultant cPT 38 showed HIV-1 IC50 value of ∼ 882 ± 44 nM (Supplementary Figure S2), with about 8-fold decrease in potency compared to the NH2-containing analogue 36 (IC50 ∼ 100 nM). Interestingly, this 8-fold decrease in potency is comparable to that observed with changing the chirality of the Asp residue in cPT 5 from the S to R configuration (Figure 2). This potency decrease could therefore be related to disturbing the cPT conformation. In this view, losing the chirality by removing the NH2 results in more flexibility, and therefore a less ordered conformation of cPTs, leading to decreased potency. As expected by the binding model, the role of NH2 in interaction with gp120 could then be ruled out. Supporting this hypothesis, we previously showed that this amino terminal could be extended through amide bond formation with additional residues without potency decrease.20 However, other possible electrostatic interactions by the amide bond cannot be precluded and should be investigated. This is an important result, which will need further confirmation to direct further chemical optimization and designs at this part of the cPT scaffold, where we envision that rigidification will be important.
Scheme 3. Synthesis of the cPT 38, with no free amine group.
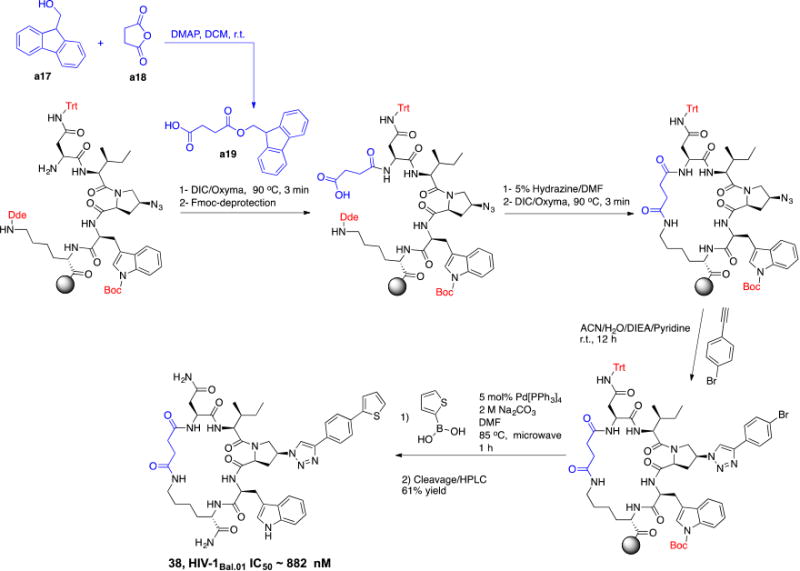
The cyclization linker a19 (shown in blue) was synthesized using FmOH (a17) and succinic anhydride (a18)43 and was incorporated in the cPT synthesis.
Discussion
We have previously discovered small cPTs that are metabolically stable and have great potential to be developed as HIV-1 therapeutics.20 Moreover, we have recently shown that gold nanoparticles coated with linear PTs are not only able to efficiently kill HIV-1 virions but also to kill HIV-1 infected cells via irreversible cell lysis.44 As cPTs have all the phenotypes of the parent linear PTs, the important infected-cell-killing ability can also be recapitulated with the metabolically stable cPTs, highlighting the potential of formulating these agents as a possible HIV cure.
The primary goal in this work was to carry out a first SAR investigation of the chemical space around the cPT scaffold in order to identify paths for chemical and potency optimization. This was achieved by examining the effects of chemical variations in different parts of the cPT scaffold, including the pharmacophore Ile-X-Trp (where X is the aryl-triazolo-Pro) and the other half of the molecule (the cyclization bridge). We found that, within the pharmacophore Ile-X-Trp, the aryl moiety on the triazole can act as an activity tuner, whereas the SAR around the Ile and Trp side chains was more limited and changes were less permissible.
The major finding in this work was the successful replacement of the less drug-like ferrocene moiety (on the triazole ring) with other moieties that retained similar activities. Previously, the bulky ferrocene moiety on the triazole ring was found, after a series of optimizations,27, 28 to be optimal for activity. However, since varying the chemical substituents around the ferrocene is limited, this hindered exploration of different chemical environments in that part of the pharmacophore. Here, we evaluated new triazole substituent moieties that had not been previously explored and found hits that can allow further chemical optimization.
We found that installing a long planer arm on the triazole ring that contains aromatic electron system(s) not only can preserve the activity previously observed with the bulky ferrocene moiety, but also can tune the activity to improve potency and obtain more drug-like cPTs. A first promising moiety was the p-methyl-phenyl methylene moiety, where we observed slight improvement in activity (cPT 24, IC50 ∼ 180 nM) compared to the lead cPT 3. The second promising system found in this work was the bi-aryl system (cPTs 23 and 36), wherein we first observed the activity retention with the bi-phenyl moiety. Looking at cPTs 24 and 36, an obvious future effort would be to hybridize these two cPTs by introducing a methylene atom between the thiophene-phenyl moiety of 36 and the triazole ring; this could potentially improve the activities of 36. One advantage of cPTs as an emerging class of HIV-1 inhibitors is the facile short solid phase mediated synthesis, an important feature in inhibitor development. We therefore developed an on-resin method to install different bi-aryl systems on the triazole ring, keeping the ease of synthesis unchanged.
Since newly discovered triazole substituent components come from synthetic building blocks, rather than the amino acids, it can also be helpful to tune the physiochemical properties of the molecule. One example was replacing the terminal phenyl ring in cPT 23 with a thiophene isostere, in cPT 36, which resulted in improved anti-HIV potency. Similar approaches therefore could be utilized to improve aqueous solubility, by introducing heteroatoms on selected positions of the scaffold, and to increase metabolic stability, by introducing metabolic blocking groups or even locking the conformation of the bi-aryl system that may have favorable effects on activity and/or physiochemical properties.
Overall, findings from optimizing the pharmacophore could suggest simultaneous multiple changes in the pharmacophore Ile-X-Trp as a future strategy to improve potency. The challenge here will be selection of moieties that can be combined together to maintain a balance between compound hydrophobicity and solubility. Utilizing the “activity tuner” as mentioned above could be a strategy to improve solubility while allowing combinations of structural changes to be made.
In the current work, we also found that the other half of the cPT scaffold, the cyclization bridge, greatly affects the anti-HIV activity. This could occur not through the direct effect of losing or gaining contacts with the target gp120 protein by this part of the molecule, but rather through correctly/incorrectly directing the pharmacophore Ile-X-Trp within the active binding site on gp120. This part of the scaffold, therefore, can be referred to as a “conformational tuner”. We previously found that varying the length of this cyclization bridge has powerful impact on activities.20 Here, we further confirmed this fact by observing the changes in activities in response to changes in the cyclization bridge. Changing the chirality of cPTAsp residue from the S to R configuration was more tolerated (7-fold change) than the same change in the cPTLys residue. One explanation for that could be the proximity of the cPTLys residue to the pharmacophore-Trp, whereas the cPTAsp residue is one-residue apart from the pharmacophore-Ile. Therefore, conformational changes at the cPTAsp residue could be more easily overcome than those caused by cPTLys residue disruption. Interestingly, complete removal of the chiral center at the cPTAsp residue resulted in comparable 8-fold decrease in activity observed with the S to R conversion. And since an N terminal extension is tolerated in cPTs, the direct role of the missing NH3+ as a binding group can be ruled out. The results argue instead that removing the chirality by removing the NH3+ group had the same conformational disturbance as did the S to R conversion. For future optimization, it must be taken into consideration where optimizing this cyclization bridge will benefit from modifying rigidification, yet retaining functional conformation. The scheme in Figure 8 summarizes an overall SAR perspective that can be extracted from this study to guide future synthetic modifications.
Fig.8. Overall structure activity relationship of cPTs depicted with cPT 36.
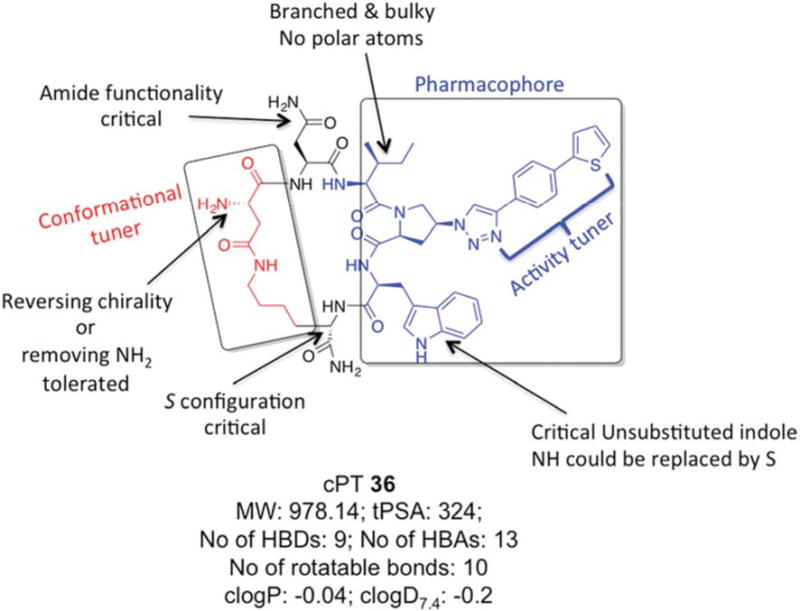
Molecular properties of 36 were calculated using MarvinSketch software (version 15.11.9.0, 2015, ChemAxon, http://www.chemaxon.com).
Generally, macrocyclic drugs, both naturally derived and synthetic, have received increasing attention in the past years owing to their excellent ability to target poor druggable targets such as PPIs. Macrocycles possess good pharmaceutical properties despite their relatively larger molecular weights compared to the conventional small molecule drugs.19 The evolving cPT inhibitors of HIV-1 Env PPI interactions with the host cellular receptor proteins are a potentially important class of inhibitors that could lead to novel anti-HIV therapeutics, especially with their unique modes of action, namely inhibition of Env-cellular protein interactions and triggering gp120 shedding from the virions. The pioneering work of Villar et al.18 helped derive rules, “Villar's rule of macrocycles”, for designing orally bioavailable macrocyclic drugs by controlling macrocyclic physiochemical properties. According to Villar's rules,18 a macrocycle should be orally bioavailable if it has 1) molecular weight of 600-1200 daltons, 2) clogP value of −2 to 6, 3) polar surface area of 180-320 Å2, 4) number of H-bond donors < 12, 5) number of H-bond acceptors in the range of 12-16 and 6) number of rotatable bonds < 15. Interestingly, analysis of our lead compound 36 discovered in this study (Figure 8) shows that it actually fits these rules, leading to the question whether these molecules might be orally bioavailable. If true, the potential for oral bioavailability, proteolytic stability and good aqueous solubility (reflected by clogD7.4) make this class of cPTs viable candidates for development as HIV therapeutics.
Conclusions
In summary, we have explored the effect of chemical changes around the cPT scaffold including the pharmacophore region as well as the cyclization bridge. A major task going forward will be to further utilize the SAR data derived from this work at both the “activity tuner” and “conformational tuner” sites for optimizing the scaffold to increase the potency and further improve the molecular properties of cPTs. This will help identify lead cPTs suitable for advanced ADME (absorption, distribution, metabolism and excretion) as well as pharmacokinetics studies.
Experimental
I. Chemistry
I.1. Chemical synthesis of cPTs (3-38)
A CEM microwave synthesizer (Liberty Blue) was used for solid phase peptide synthesis of cPTs as previously described.20 Fmoc-4-azido-Proline was synthesized as previously reported.20 All other Fmoc-, Boc- protected amino acids, N,N'-diisopropylcarbodiimide (DIC), ethyl (hydroxyimino) cyanoacetate (OxymaPure) and rink amide resin (100-200 mesh size, 0.53 meq/g substitution) were purchased from Chem-Impex International, INC. Alkynes and CuI catalyst for the click reaction, and hydrazine, were purchased from Sigma Aldrich. Benzyl bromides were purchased form Alfa Aesar. HPLC purifications were performed using a Waters® HPLC system with reverse phase (RP) C18 prep columns. All synthesized cPTs were >95 % pure as judged by purity checks using an analytical C18 RP-HPLC column and BeckmanCoulter® HPLC system. HPLC grade ACN, Millipore-MilliQ water and 0.1% TFA were used as solvents for the HPLC purification. Alkynes a9-a16 were synthesized from the corresponding benzyl bromides a1-a8 following the procedures previously described45 (see below). Alkynes a9-a16 were used in click reactions without purification because they are volatile and unstable upon storage. Mass validation of all cPTs was performed using Thermo Scientific LTQ XL Ion Trap LC/MS (Supplementary Table S2).
I.1.a General synthesis of alkynes a9-a16
To a solution of ethynyltrimethylsilane (40 mmol) in 20 mL THF at 0 °C, was added i-PrMgCl (2 M solution in THF) dropwise and the mixture was stirred at 0 °C for 30 min and allowed to warm up to r.t. for 30 min. CuBr (6 mmol) was added and the mixture was stirred at r.t. for 1 h. The corresponding benzyl bromide a1-a8 was added and the mixture was heated under reflux for 4 h. The reaction mixture was then cooled to r.t. before pouring onto saturated NH4Cl solution (500 mL). The mixture was extracted twice with diethyl ether, the combined organic layers were washed with water and brine and then dried over MgSO4. Diethyl ether was evaporated under vacuum and the crude residues were used for the next step without purification. The residues were dissolved in MeOH and the cooled to 0 °C before adding K2CO3 (40 mmol) and the mixture was stirred at 0 °C for 2 h before adding 20 mL water. After layer separation, the aqueous layer was extensively extracted with diethyl ether and the combined ethereal layer was washed with brine and dried over MgSO4. Ether was removed under vacuum and the remaining liquid alkyne was immediately used for the click reaction with the resin-bound cyclic peptide.
I.1.b On-resin synthesis of the bi-aryl system on cPT
After the on-resin click reaction with the bromo-alkyne (Scheme 2), the resin-bound protected cPT was washed sequentially with 5% HCl, DMF and DCM before transferring to a 25 mL round bottom flask for Suzuki reaction. The flask was charged with 5 mol% tetrakis(triphenylphosphine)palladium(0) (Pd[PPh3]4), aqueous 2 M Na2CO3 (5 equivalents) and 5 equivalents of thiophen-2-ylboronic acid dissolved in 5 mL DMF and the reaction mixture was flushed with N2 for 10 min. The flask was then capped and microwave irradiated at 85 °C for 30 min to 1 h. Few resin beads were collected and subjected to cleavage conditions to check the coupling which indicated complete reaction, without traces of the starting material (∼ 87% yield for the coupling step based on cleavage from 0.1 g resin). The vessel was then cooled and the resin was extensively washed with 5% HCl, water, DMF and DCM. Cleavage from resin, deprotection and HPLC purification were performed as previously described.20 The overall yield of 36 and 37 was 55% and 49%, respectively. The same coupling procedures were applied during the synthesis of the amine-free cPT 38 with overall yield of 61%
II. Pseudovirus production, antiviral assay and cytotoxicity assay
Pseudoviruses were produced as previously described.24,46 Briefly, HEK 293T cells (3 × 106) were co-transfected with 4 μg of BaL.01 gp160 plasmid and 8 μg of NL4-3 R- E- Luc+ core DNA (obtained from the NIH AIDS Reagent Program as a kind gift from Dr. John Mascola and Dr. Nathaniel Landau, respectively), using Polyethyleneimine (PEI) as a transfection vehicle. After 72 hours, the supernatant containing virus was collected and filtered using a 0.45 μm syringe filter (Corning). The supernatant was then loaded on a 10 ml Iodixanol gradient; 6%-20% (Optiprep, Sigma Aldrich) and centrifuged on SW41Ti rotor (Beckman Coulter) at . 30,000 RPM for 2 hours at 4°C. Virus samples were pooled from fractions 6 through 9 in 1 ml aliquots and diluted in serum free media before storage in −80°C. All batches of virus were titrated for infectivity and p24 content immediately after production.
Pseudoviral infection assays were carried out as previously described.39 Briefly, 7500 HOS.T4.R5 cells were seeded in 96 well plates on day one. 24 hours later, virus stocks were diluted in growth media such that the final dilution gave 1 × 106 luminescence counts. Inhibitors to be tested were solubilized in 1× PBS containing 2% DMSO and serially diluted in 1.5 mL tubes. Virus was then added to these inhibitors-containing tubes, 1:1 (v/v), and the tubes were mixed by repeated inversions. Positive control (100%) contained virus treated with PBS while the negative control (0%) contained no virus. The samples were incubated at 37 °C for 45 minutes before addition to the cells. The plates were incubated for 24 hours at 37 °C before the medium was changed. 24 hours post media change, the cells were lysed using Passive Lysis Buffer(Promega). Lysed cells were then transferred to a 96 well white plate (Greiner) and mixed with Luciferin salt (Anaspec) in 0.1 M potassium phosphate buffer containing 0.1 M magnesium sulfate and the luminescence measured using a Wallace 1450 Microbeta Luminescence reader. Luminiscence reading for each inhibitor concentration was normalized to the controls and plotted against the inhibitor concentration. Inhibition potencies were then determined by calculating the inhibitor concentration required for 50% inhibition of maximal binding (IC50) after fitting the plot using the four parameter Logistic sigmoidal fit in Origin 8.0.
Cytotoxicity assay: cPTs 36 and 37 were tested for cytotoxicity in vitro with HOS.T4.R5 cells. The latter were seeded at 75,00 cells per well and incubated with peptides as described above for the infection inhibition assays. Cell viability was determined using the tetrazolium salt premix reagent WST-1 from TaKaRa Bio, Inc., by following the manufacturer's protocol. The formazan product was measured 24 h post-exposure using a microplate reader (Molecular Devices) at an absorbance wavelength of 450 nm.
III. Production of HIV-1 gp120 and soluble CD4 proteins
III.1. Reagents
Escherichia coli strain Stbl2 cells were products of Novagen Inc. (Madison, WI). DNA plasmids encoding BaL.01 gp160 and NL4-3 R- E- Luc+ were obtained from the NIH AIDS Reagent Program, Division of AIDS, NIAID, and were kind gifts of Dr. J Mascola and Dr. N Landau, respectively. pcDNA3.1 vector carrying CD4 was a gift from Navid Madani. 17b IgG was purchased from Strategic Diagnostics Inc (Newark, DE). All other reagents used were of the highest analytical grade available.
III.2. Expression and purification of wild-type gp120YU-2
The DNA for gp120YU-2 in pcDNA3.1 vector for transient transfection was purified using a Qiagen MaxiPrep kit (Qiagen) after transforming into Stbl2 competent cells. The purified DNA encoding gp120 YU-2 was transfected into HEK 293F cells according to manufacturer's protocol (Invitrogen). Five days after transfection was initiated, cells were harvested and spun down (3000 RPM), and the supernatant was filtered through 0.2 μm filters. Purification was performed over a 17b antibody-coupled column prepared using an NHS-activated Sepharose, HiTrap HP column (GE Healthcare). gp120 was eluted from the column using 0.1M Glycine buffer pH 2.4. The pH of the eluted protein was rapidly neutralized by addition of 1M Tris pH 8.0. Identity of the eluted fractions was confirmed by SDS-PAGE and Western blotting using antibody D7324 (Aalto Bioreagents). Eluted protein was immediately buffer exchanged into PBS using spin-columns (Amicon Ultra Ultracell-30K, Millipore). Protein was filtered through 0.45 μm syringe filters (Millex-LH, Millipore) and separated by size exclusion on a HiLoad 26/60 Superdex 200 HR prepacked gel filtration column (GE). Purity of eluted fractions and monomeric state of gp120 were identified by SDS-PAGE. Monomeric fractions were pooled, concentrated, frozen and stored at −80°C.
III.3. Four domain soluble CD4 production
Hexa histidine-tagged 4-domain soluble CD4 (CD4) was produced by transient transfection into 293F cells using standard protocols (Gibco). CD4 was separated from the expression medium by Nickel affinity purification on HiTrap columns (GE) using an Akta FPLC System (GE). CD4 was further purified by size-exclusion on a Superdex 200 column (GE). Protein size and functionality were verified by SDS PAGE and anti-gp120 ELISA, respectively.
IV. Surface plasmon resonance (SPR) assays
SPR experiments were performed on a Biacore 3000 optical biosensor (GE Healthcare). All experiments were carried out at 25°C using standard PBS bufferpH 7.4 with 0.005% surfactant Tween and 2% DMSO. Three flow cells in the CM5 chip were used for amine coupling of different ligands through standard 1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide (EDC)/N-hydroxysuccinamide (NHS) chemistry. Flow cell 1 containing 2000 RUs of immobilized antibody 2B6R (α-human IL5R) served as a negative control for flow cells 2 and 3 each of which contained 2000 RUs of immobilized CD4 and 17b respectively.
Analysis of cPT-mediated inhibition of gp120 binding to sCD4 and mAb 17b was achieved by injecting a fixed concentration of HIV-1 YU-2 gp120 (250 nM), with increasing peptide concentrations, over flow cells containing sCD4 and mAb 17b surfaces for 5 minute association and 5 minute dissociation at a flow rate of 100 μl min-1 in PBS. Regeneration of the surface was achieved by injecting 10 ul of 10mM HCl at flowrate of 75ul/min.. All analyses were performed in triplicate.
Data analysis of SPR competition data was performed using BIAevaluation v4.1.1 software (GE). To correct for nonspecific binding, response signals from buffer injection and from control flow cell were subtracted from all sensorgrams. Inhibition potencies were determined by calculating the inhibitor concentration required for 50% inhibition of maximal binding (IC50). The inhibition curve was plotted and then fitted using the four-parameter equation as shown below using OriginPro 8 graphing software.
where, Rhigh is the response at the highest inhibitor concentration and Rlow at low inhibitor concentration; concn is the concentration of inhibitor and A1 and A2 are fitting parameters.
V. Binding studies of cPTs 3 & 36 to gp120 YU-2 using ITC
cPT dissociation constants were determined at 25 °C on a VP-Isothermal Titration Calorimeter (VP-ITC) system (MicroCalTM, GE Healthcare, Freiburg). 200 μM (for 36) or 390 μM (for 3) compound solutions were in 8 μL steps titrated into 30 μM of wild type gp120 YU-2 at 25 °C using 1× PBS buffer at pH 7.3. The resulting heat change upon injection was integrated over a time range of 300 sec, and the obtained values were fitted to a standard single-site binding model using Origin®.
Supplementary Material
Acknowledgments
This work was funded by the National Institute of Health through the NIH Program Project GM 56550-17, Structure Based Antagonism of HIV-1 Envelope Function in Cell Entry. We thank Prof. Amos B. Smith III (University of Pennsylvania) for helpful discussions during this project. We thank Openeye Scientific Software (Santa Fe, NM. http://www.eyesopen.com) for providing a complimentary academic license of their software package.
Abbreviations
- ACN
acetonitrile
- Boc
Tertbutyloxycarbonyl
- cPT
cyclic peptide triazole
- DIC
N,N′-Diisopropylcarbodiimide
- DMF
Dimethylformamide
- Env
HIV envelope gp160
- Fmoc
9-Fluorenylmethoxycarbonyl
- HPLC
high performance liquid chromatography
- IXW
Ile-ferrocenyltriazolePro-Trp
- PT
Peptide triazole
- SAR
Structure Activity Relationship
- SI
selectivity index
- SPR
Surface Plasmon Resonance
- tBu
Tert-butyl
- TIPS
Triisoproylsilane
- Trt
Triphenylmethyl
- TFA
Trifluoroacetic acid
- wt
wild type
Footnotes
Competing Interests: The authors declare no competing financial interest.
Supplementary information. Additional tables and figures available.
Notes and References
- 1.Sanders RW, Moore JP. Immunol Rev. 2017;275:161–182. doi: 10.1111/imr.12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanders RW, Sanders IA, Moore JP. Sci Am. 2016;315:50–55. doi: 10.1038/scientificamerican1216-50. [DOI] [PubMed] [Google Scholar]
- 3.van Sighem AI, Gras LA, Reiss P, Brinkman K, de Wolf F A. n. o. c. study. AIDS. 2010;24:1527–1535. doi: 10.1097/QAD.0b013e32833a3946. [DOI] [PubMed] [Google Scholar]
- 4.Arts EJ, Hazuda DJ. Csh Perspect Med. 2012;2 doi: 10.1101/cshperspect.a007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Volberding PA, Deeks SG. Lancet. 2010;376:49–62. doi: 10.1016/S0140-6736(10)60676-9. [DOI] [PubMed] [Google Scholar]
- 6.Guaraldi G, Orlando G, Zona S, Menozzi M, Carli F, Garlassi E, Berti A, Rossi E, Roverato A, Palella F. Clin Infect Dis. 2011;53:1120–1126. doi: 10.1093/cid/cir627. [DOI] [PubMed] [Google Scholar]
- 7.Mzingwane ML, Tiemessen CT. Rev Med Virol. 2017 doi: 10.1002/rmv.1924. [DOI] [PubMed] [Google Scholar]
- 8.Acharya P, Lusvarghi S, Bewley CA, Kwong PD. Expert Opin Ther Tar. 2015;19:765–783. doi: 10.1517/14728222.2015.1010513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ward AB, Wilson IA. Immunol Rev. 2017;275:21–32. doi: 10.1111/imr.12507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burton A. Lancet Infect Dis. 2003;3:260. doi: 10.1016/s1473-3099(03)00620-0. [DOI] [PubMed] [Google Scholar]
- 11.Kitchen CM, Nuno M, Kitchen SG, Krogstad P. Ther Clin Risk Manag. 2008;4:433–439. doi: 10.2147/tcrm.s1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Landry I, Zhu L, Abu Tarif M, Hruska M, Sadler BM, Pitsiu M, Joshi S, Hanna GJ, Lataillade M, Boulton DW, Bertz RJ. Antimicrob Agents Chemother. 2016;60:2782–2789. doi: 10.1128/AAC.02503-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Si Z, Madani N, Cox JM, Chruma JJ, Klein JC, Schon A, Phan N, Wang L, Biorn AC, Cocklin S, Chaiken I, Freire E, Smith AB, 3rd, Sodroski JG. Proc Natl Acad Sci U S A. 2004;101:5036–5041. doi: 10.1073/pnas.0307953101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Melillo B, Liang S, Park J, Schon A, Courter JR, LaLonde JM, Wendler DJ, Princiotto AM, Seaman MS, Freire E, Sodroski J, Madani N, Hendrickson WA, Smith AB., 3rd ACS Med Chem Lett. 2016;7:330–334. doi: 10.1021/acsmedchemlett.5b00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richard J, Veillette M, Brassard N, Iyer SS, Roger M, Martin L, Pazgier M, Schon A, Freire E, Routy JP, Smith AB, 3rd, Park J, Jones DM, Courter JR, Melillo BN, Kaufmann DE, Hahn BH, Permar SR, Haynes BF, Madani N, Sodroski JG, Finzi A. Proc Natl Acad Sci U S A. 2015;112:E2687–2694. doi: 10.1073/pnas.1506755112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yudin AK. Chem Sci. 2015;6:30–49. doi: 10.1039/c4sc03089c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hopkins AL, Groom CR. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 18.Villar EA, Beglov D, Chennamadhavuni S, Porco JA, Kozakov D, Vajda S, Whitty A. Nat Chem Biol. 2014;10:723–731. doi: 10.1038/nchembio.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Villar EA, Beglov D, Vajda S, Whitty A. Abstr Pap Am Chem S. 2013;245 [Google Scholar]
- 20.Rashad AA, Kalyana Sundaram RV, Aneja R, Duffy C, Chaiken I. J Med Chem. 2015;58:7603–7608. doi: 10.1021/acs.jmedchem.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaiken I, Rashad AA. Future Med Chem. 2015;7:2305–2310. doi: 10.4155/fmc.15.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aneja R, Rashad AA, Li H, Kalyana Sundaram RV, Duffy C, Bailey LD, Chaiken I. J Med Chem. 2015;58:3843–3858. doi: 10.1021/acs.jmedchem.5b00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamanna K, Aneja R, Duffy C, Kubinski P, Rodrigo Moreira D, Bailey LD, McFadden K, Schon A, Holmes A, Tuzer F, Contarino M, Freire E, Chaiken IM. Chemmedchem. 2013;8:322–328. doi: 10.1002/cmdc.201200422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Umashankara M, McFadden K, Zentner I, Schon A, Rajagopal S, Tuzer F, Kuriakose SA, Contarino M, Lalonde J, Freire E, Chaiken I. Chemmedchem. 2010;5:1871–1879. doi: 10.1002/cmdc.201000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beno BR, Yeung KS, Bartberger MD, Pennington LD, Meanwell NA. Journal of Medicinal Chemistry. 2015;58:4383–4438. doi: 10.1021/jm501853m. [DOI] [PubMed] [Google Scholar]
- 26.Meanwell NA. Journal of Medicinal Chemistry. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 27.Gopi H, Cocklin S, Pirrone V, McFadden K, Tuzer F, Zentner I, Ajith S, Baxter S, Jawanda N, Krebs FC, Chaiken IM. J Mol Recognit. 2009;22:169–174. doi: 10.1002/jmr.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gopi H, Umashankara M, Pirrone V, LaLonde J, Madani N, Tuzer F, Baxter S, Zentner I, Cocklin S, Jawanda N, Miller SR, Schon A, Klein JC, Freire E, Krebs FC, Smith AB, Sodroski J, Chaiken I. Journal of Medicinal Chemistry. 2008;51:2638–2647. doi: 10.1021/jm070814r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Rosa M, Unge J, Motwani HV, Rosenquist A, Vrang L, Wallberg H, Larhed M. J Med Chem. 2014;57:6444–6457. doi: 10.1021/jm500434q. [DOI] [PubMed] [Google Scholar]
- 30.Gising J, Odell LR, Larhed M. Org Biomol Chem. 2012;10:2713–2729. doi: 10.1039/c2ob06833h. [DOI] [PubMed] [Google Scholar]
- 31.Larhed M, Hallberg A. Drug Discov Today. 2001;6:406–416. doi: 10.1016/s1359-6446(01)01735-4. [DOI] [PubMed] [Google Scholar]
- 32.Buchstaller HP, Siebert CD, Steinmetz R, Frank I, Berger ML, Gottschlich R, Leibrock J, Krug M, Steinhilber D, Noe CR. J Med Chem. 2006;49:864–871. doi: 10.1021/jm0503493. [DOI] [PubMed] [Google Scholar]
- 33.Hwang KJ, Lee TS, Kim KW, Kim BT, Lee CM, Park EY, Woo RS. Arch Pharm Res. 2001;24:270–275. doi: 10.1007/BF02975090. [DOI] [PubMed] [Google Scholar]
- 34.Sharma VA, Kan E, Sun Y, Lian Y, Cisto J, Frasca V, Hilt S, Stamatatos L, Donnelly JJ, Ulmer JB, Barnett SW, Srivastava IK. Virology. 2006;352:131–144. doi: 10.1016/j.virol.2006.04.030. [DOI] [PubMed] [Google Scholar]
- 35.Lee JH, Ozorowski G, Ward AB. Science. 2016;351:1043–1048. doi: 10.1126/science.aad2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munro JB, Gorman J, Ma X, Zhou Z, Arthos J, Burton DR, Koff WC, Courter JR, Smith AB, 3rd, Kwong PD, Blanchard SC, Mothes W. Science. 2014;346:759–763. doi: 10.1126/science.1254426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munro JB, Mothes W. J Virol. 2015;89:5752–5755. doi: 10.1128/JVI.03187-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Acharya K, Rashad AA, Moraca F, Klasse PJ, Moore JP, Abrams C, Chaiken I. Proteins. 2017;85:843–851. doi: 10.1002/prot.25238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emileh A, Tuzer F, Yeh H, Umashankara M, Moreira DR, Lalonde JM, Bewley CA, Abrams CF, Chaiken IM. Biochemistry. 2013;52:2245–2261. doi: 10.1021/bi400166b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alogheli H, Olanders G, Schaal W, Brandt P, Karlen A. J Chem Inf Model. 2017;57:190–202. doi: 10.1021/acs.jcim.6b00443. [DOI] [PubMed] [Google Scholar]
- 41.Watts KS, Dalal P, Tebben AJ, Cheney DL, Shelley JC. J Chem Inf Model. 2014;54:2680–2696. doi: 10.1021/ci5001696. [DOI] [PubMed] [Google Scholar]
- 42.Caron G, Ermondi G. Drug Discov Today. 2016 doi: 10.1016/j.drudis.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 43.Lioy E, Suarez J, Guzman F, Siegrist S, Pluschke G, Patarroyo ME. Angew Chem Int Ed Engl. 2001;40:2631–2635. doi: 10.1002/1521-3773(20010716)40:14<2631::AID-ANIE2631>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 44.Bastian AR, Ang CG, Kamanna K, Shaheen F, Huang YH, McFadden K, Duffy C, Bailey LD, Sundaram RVK, Chaiken I. Virus Res. 2017;235:33–36. doi: 10.1016/j.virusres.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Louvel J, Carvalho JF, Yu Z, Soethoudt M, Lenselink EB, Klaasse E, Brussee J, Ijzerman AP. J Med Chem. 2013;56:9427–9440. doi: 10.1021/jm4010434. [DOI] [PubMed] [Google Scholar]
- 46.Montefiori DC. Curr Protoc Immunol. 2005:11. doi: 10.1002/0471142735.im1211s64. Chapter 12, Unit 12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.