

Abstract
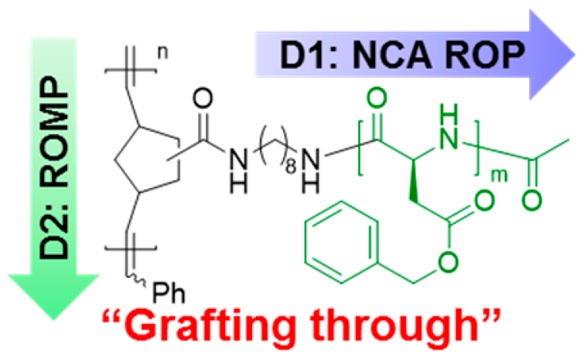
Well-defined molecular brushes bearing polypeptides as side chains were prepared by a “grafting through” synthetic strategy with two-dimensional control over the brush molecular architectures. By integrating N-carboxyanhydride ring-opening polymerizations (NCA ROPs) and ring-opening metathesis polymerizations (ROMPs), desirable segment lengths of polypeptide side chains and polynorbornene brush backbones were independently constructed in controlled manners. The N2 flow accelerated NCA ROP was utilized to prepare polypeptide macromonomers with different lengths initiated from a norbornene-based primary amine, and those macromonomers were then polymerized via ROMP. It was found that a mixture of dichloromethane and an ionic liquid were required as the solvent system to allow for construction of molecular brush polymers having densely-grafted peptide chains emanating from a polynorbornene backbone, poly(norbornene-graft-poly(β-benzyl-l-aspartate)) (P(NB-g-PBLA)). Highly efficient postpolymerization modification was achieved by aminolysis of PBLA side chains for facile installment of functional moieties onto the molecular brushes.
Molecular brushes are a class of cylindrical polymeric materials, in which linear flexible or rigid side chains are densely attached and packed along a linear polymer backbone.1 With high rigidity and large persistence length, versatile chemical structure variability, and molecular nanoscopic dimension, molecular brushes have gathered intense research attention with a potential for utilization in a wide range of applications, including the fields of biomedicine and microelectronics.2−4 For instance, with densely-packed side chains, the molecular brush architecture has been shown to improve the efficiency of delivery systems and enhance the protection of therapeutic loads when acting as a therapeutic carrier.4−7 Recently, Zhang and co-workers reported a novel brush polymer/DNA conjugation system providing DNA with nanoscopic steric selectivity, when DNA was attached onto one end of a brush backbone.4 The hybridization with complementary DNA remained unaffected, due to the small size of complementary DNA, while the interactions with proteins were limited owing to the steric hindrance from the densely-packed side chains, resulting in enhanced resistance of the carried DNA against nucleases and improved in vivo distribution with longer blood circulation. A great number of molecular brushes have contained flexible side chains, such as polyolefins, polyesters, and polyethers.8−10 Molecular brushes containing rigid side chains would render brushes with unique properties, while strategies for such materials with controlled molecular weights (MWs) and narrow molecular weight distributions (MWDs) still remain as a key synthetic challenge.11−13
Polypeptides consisting of α-amino acids with covalent linkages share significant similarities to the primary structure of natural proteins, and recent synthetic methodology advances have facilitated their utilization in biomedicine as scaffolds for tissue engineering, matrices for drug and gene delivery, and responsive materials for biosensors.14−16 Polypeptides have been recently employed as rigid polymers due to their capability to form well-defined secondary structures, including α-helices and β-sheets.2,11,12 Thus, incorporating polypeptides with well-defined structures into molecular brush architectures can provide topologies with unique properties.17−19 The most common approach to the syntheses of polypeptides involves ring-opening polymerizations (ROPs) of amino acid N-carboxyanhydrides (NCAs). In the last decades, several controlled NCA ROPs have been reported to prepare well-defined polypeptides by minimizing side reactions that usually decrease the activity of the terminal amine during the polymerizations.20−27 Our group previously reported a straightforward method to enhance the polymerization rate while maintaining the controlled features of NCA ROP by applying continuous N2 flow to remove CO2, constituting a facile route to the syntheses of polypeptides with controllable polymerization rates, predictable MWs, and narrow MWDs.25
Molecular brushes are commonly prepared via three synthetic strategies: “grafting onto”, “grafting from”, and “grafting through”, with unique advantages and challenges for each method. “Grafting onto” and “grafting from” strategies had been used to construct polypeptide molecular brushes.2,11−13,28−32 In 2009, Hammond’s group reported molecular brushes by grafting azido-terminated poly(ethylene glycol) (PEG) onto alkyne-functionalized polypeptide backbones.28 With the efficient azide–alkyne cycloaddition click chemistry, greater than 95% grafting efficiency was achieved. Cheng and co-workers reported a one-pot synthesis of polypeptide molecular brushes, starting with ring-opening metathesis polymerization (ROMP) of a norbornene (NB) monomer containing a N-trimethylsilyl (N-TMS) group.12 The N-TMS served not only as a protecting group for the amines during ROMP, but also as initiators for the controlled polymerization of NCA monomers to grow the side chain grafts. However, polypeptide-based molecular brushes were rarely reported by using a “grafting through” synthetic strategy due to the polar and rigid nature of polypeptide materials.6,7
After several failed attempts to build well-defined molecular brushes directly by “grafting-through” polymerizations of polypeptide macromonomers, we recognized that special solution conditions are required to facilitate controlled polymerization of the polypeptides, each as a linear polymer and as a brush architecture. Therefore, we describe, herein, a broadly-applicable “grafting through” synthetic approach for the preparation of well-defined molecular brushes bearing polypeptide side chains by integrating NCA ROPs with ROMPs, which simply takes advantage of ionic liquid cosolvents. Two-dimensional control over the macromolecular architectures was achieved by independently building the desirable segment lengths of polypeptide macromonomer graft chains and brush backbones (Figure 1). The bifunctional initiator (NB-amine, 1) consisted of a primary amine and a NB group, in which the primary amine first initiated NCA ROP to afford poly(β-benzyl-l-aspartate) (PBLA) side chains and then the NB group allowed for ruthenium-catalyzed ROMP to construct the brush backbone. A mixture of dichloromethane (DCM) and an ionic liquid was used in ROMPs to ensure the solubility of polypeptide macromonomers and the intactness of the ruthenium catalyst. Postpolymerization modification was achieved via the aminolysis of PBLA side chains in the densely-packed brush structure.
Figure 1.
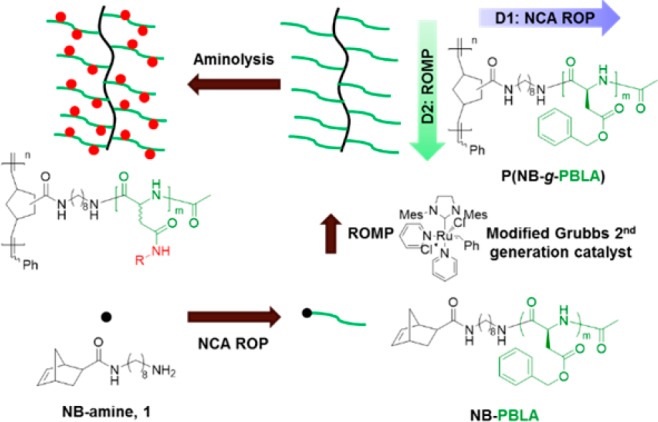
Synthetic design of polypeptide molecular brushes via “grafting through” strategy with postpolymerization modification using aminolysis of PBLA brush side chains.
The polypeptide macromonomer, NB-PBLA, that would ultimately become the brush side chain, was synthesized by controlled ROP of BLA NCA monomer, with 1 as the initiator via our previously reported N2 flow method (Figure 1). The required amounts of BLA NCA monomer and initiator 1 were dissolved in anhydrous N,N-dimethylformamide (DMF), and the reaction was allowed to proceed at room temperature using standard Schlenk techniques.33 To avoid coordination of the chain-end primary amino group of NB-PBLA to the ruthenium catalyst used in the next-step ROMP,34 it was directly modified by amidation with acetic anhydride.35 Without isolation of the polymer after the NCA polymerization had reached a minimum of 95% monomer conversion, acetic anhydride was introduced into the reaction mixture via syringe. The reaction mixture was then precipitated into diethyl ether after another 3 h to isolate N-acetylated NB-PBLA as a white solid product. NB-PBLAs with different chain lengths were synthesized in a similar manner by using monomer to initiator ratios of 10, 20, and 50. The average degree of polymerization (DPn) and number-averaged molecular weight (Mn) of NB-PBLA were determined by comparing the intensity of the NB alkenyl protons resonating at about 6.10 ppm (a in Figure 2a), with the intensity of the PBLA benzyl methylene protons resonating at about 5.03 ppm (c in Figure 2a). Due to the accelerated polymerization rates, NB-PBLAs were synthesized in a rapid manner with well-defined structures, as confirmed by the monomodal distributions and narrow MWDs in size-exclusion chromatography (SEC; Figure S4). Therefore, the N2 flow accelerated NCA ROP provided the first-dimensional control over the brush molecular architecture by building the polypeptide side chains with desirable segment lengths.
Figure 2.

1H NMR spectrum of (a) NB-PBLA10, (b) P(NB-g-PBLA10)50, and (c) P(NB-g-PABEDA10)50.
In order to synthesize the polypeptide molecular brush, poly(norbornene-graft-poly(β-benzyl-l-aspartate)) (P(NB-g-PBLA)), via the “grafting through” approach with controlled brush backbone length, ROMP was utilized to polymerize the NB groups at the α-chain end of NB-PBLAs with the modified Grubbs second generation catalyst (Figure 1). The Ru-catalyzed ROMP is a robust and well-established polymerization technique to prepare polymeric materials in a controlled manner with high tolerance of functional groups, such as carboxylic acids and alcohols.36 Due to the poor solubility of NB-PBLA and P(NB-g-PBLA) in low-polarity solvents, and issues with coordination of several common high polarity solvents, such as DMF and N,N-dimethylacetamide (DMAc), with the Ru center of the modified Grubbs second generation catalyst,12 it was important to identify an appropriate solvent system in which to conduct the ROMP “grafting-through” reactions. Ionic liquids are a class of organic compounds consisting of ionic species that, when choosing the appropriate ion pair, can fulfill the requirement of high polarity and low coordinating tendency in ROMP.37,38 A solvent mixture of 1-butyl-3-methylimidazolium tetrafluoroborate ([bmim][BF4]) with DCM was used in ROMP of NB-PBLA, due to the limited solubility of the polypeptide in neat [bmim][BF4]. During our initial polymerization attempt, 87% conversion of NB-PBLA10 occurred in 5 vol % [bmim][BF4]/DCM to give a P(NB-g-PBLA) molecular brush having a narrow MWD (Đ = 1.22; entry 1 in Table 1). Upon releasing ring strain during the polymerization, the proton resonance at about 6.10 ppm corresponding to the NB alkene disappeared in the 1H NMR spectrum (a in Figure 2a) and was replaced by a broad peak at about 5.20 ppm for the alkenyl protons along the backbone of P(NB-g-PBLA) (a′ in Figure 2b). As the content of the ionic liquid increased in the solvent mixture, the conversion of NB-PBLA was depressed, even with elongated reaction time (entries 1–3 in Table 1). For example, the conversion of NB-PBLA10 decreased from 87% to 67% when the content of [bmim][BF4] increased from 5 vol % to 20 vol %. At 10 vol % [bmim][BF4], with increased concentration of NB-PBLA during the polymerization, the macromonomer conversion was improved and the well-defined structure of the obtained brush polymer was maintained, as indicated by Đ < 1.3 (entries 2, 5, and 6 in Table 1). Therefore, further ROMPs were conducted under the optimized reaction conditions by using 10 vol % ionic liquid in DCM with the concentration of NB-PBLA at 100 mg/mL.
Table 1. Reaction Condition Optimization of ROMPs for P(NB-g-PBLA) Syntheses.
| entrya | solvent | concn (mg/mL) | time (h) | convb (%) | Mnc (kDa) | DPnc (expected) | Đc |
|---|---|---|---|---|---|---|---|
| 1 | DCM+5 vol % [bmim][BF4] | 40 | 3 | 87 | 117.6 | 49 (44) | 1.22 |
| 2 | DCM+10 vol % [bmim][BF4] | 40 | 3 | 82 | 95.7 | 40 (41) | 1.20 |
| 3 | DCM+20 vol % [bmim][BF4] | 40 | 5 | 67 | 91.2 | 38 (34) | 1.25 |
| 4 | DCM+25 vol % [bmim][BF4] | 20 | 5 | 44 | 88.8 | 37 (22) | 1.22 |
| 5 | DCM+10 vol % [bmim][BF4] | 70 | 3 | 91 | 128.7 | 54 (46) | 1.28 |
| 6 | DCM+10 vol % [bmim][BF4] | 100 | 3 | 96 | 121.5 | 51 (48) | 1.19 |
All polymerizations were performed with a macromonomer (NB-PBLA10) to catalyst ratio at 50 at rt.
Determined by 1H NMR spectroscopy.
Determined by DMF SEC characterization with a dn/dc value of 0.0790 mL/g.
With the optimized reaction conditions, ROMPs of NB-PBLA with different chain lengths at various monomer to catalyst ratios were performed and the results are summarized in Table 2. With an increase of the macromonomer to catalyst ratio, the NB-PBLA conversion decreased correspondingly, which was determined by measuring the intensity of NB alkenyl protons by 1H NMR spectroscopy. For example, when the macromonomer to catalyst ratio increased from 20 to 100, the conversion of NB-PBLA10 decreased from 99% to 78% (entries 1–3 in Table 2). Well-defined structures for the P(NB-g-PBLA)s were indicated by unimodal SEC distribution patterns for all three brush polymers derived from NB-PBLA10 (Figure 3a). As the steric hindrance of the polypeptide macromonomer increased from NB-PBLA10 to NB-PBLA50, the conversion of NB-PBLA decreased markedly at the same monomer to catalyst ratio, especially for NB-PBLA50, which possessed the largest steric hindrance of the three NB PBLA brush side chains (entries 1, 4, and 6 in Table 2).
Table 2. P(NB-g-PBLA) Syntheses via ROMPs with Varying NB-PBLA Block Lengths, Macromonomer to Catalyst Ratios, and Ionic Liquid Species.
| entrya | Ma | ILa | M/Ca | time (h) | convb (%) | Mnc (kDa) | DPnc (expected) | Đc |
|---|---|---|---|---|---|---|---|---|
| 1 | NB-PBLA10 | [bmim][BF4] | 20 | 1 | 99 | 56.7 | 23 (20) | 1.17 |
| 2 | NB-PBLA10 | [bmim][BF4] | 50 | 3 | 96 | 112.1 | 47 (48) | 1.09 |
| 3 | NB-PBLA10 | [bmim][BF4] | 100 | 3 | 78 | 280.5 | 117 (78) | 1.27 |
| 4 | NB-PBLA20 | [bmim][BF4] | 20 | 3 | 92 | 82.6 | 22 (19) | 1.16 |
| 5 | NB-PBLA20 | [bmim][BF4] | 100 | 3 | 64 | 200.7 | 59 (64) | 1.35 |
| 6 | NB-PBLA50 | [bmim][BF4] | 20 | 6 | 74 | 141.0 | 12 (15) | 1.16 |
| 7 | NB-PBLA10 | [bdmim][BF4] | 50 | 3 | 99 | 138.7 | 57 (49) | 1.23 |
| 8 | NB-PBLA10 | [bdmim][BF4] | 100 | 3 | 92 | 251.5 | 105 (92) | 1.36 |
| 9 | NB-PBLA20 | [bdmim][BF4] | 20 | 3 | 99 | 89.2 | 23 (20) | 1.26 |
| 10 | NB-PBLA20 | [bdmim][BF4] | 100 | 3 | 80 | 385.4 | 101 (80) | 1.32 |
All polymerizations were performed under the optimized conditions with the concentration of macromonomer at 100 mg/mL in the solvent mixture with 10 vol % ionic liquid in DCM: M, macromonomer; IL, ionic liquid; C, catalyst.
Determined by 1H NMR spectroscopy.
Determined by DMF SEC characterization with a dn/dc value of 0.0790 mL/g.
Figure 3.
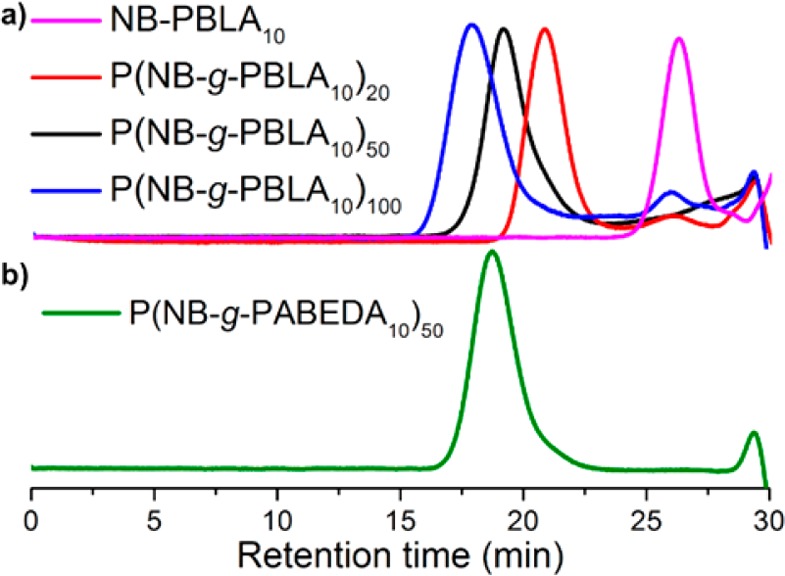
SEC chromatograms of (a) NB-PBLA10 and P(NB-g-PBLA10) with macromonomer repeat units of 20, 50, and 100 and (b) P(NB-g-PABEDA10)50 from P(NB-g-PBLA10)50 aminolysis modification.
In addition to the steric hindrance from the polypeptide macromonomer that could cause the observed decrease in macromonomer conversion in ROMP, the increased polypeptide chain length could cause greater potential for the amides to coordinate with the Ru catalyst, and the ionic liquid could cause adverse effects. Therefore, we revisited the ionic liquid used in the polymerization, to better solvate the molecular brushes during polymerization and also avoid potential effects from the ionic liquid or its degradation products. The [bmim] cation has been reported to degrade into a carbene species via proton abstraction at the 2-position, and this carbene compound could coordinate to the metal to deactivate the catalyst.37−39 In order to avoid this potential negative effect, 1-butyl-2,3-dimethylimidazolium tetrafluoroborate ([bdmim][BF4]), which has a methyl group replacing the proton at the 2-position, was investigated in the ROMP of NB-PBLA (Table 2). For both NB-PBLA10 and NB-PBLA20, the conversions of polypeptide macromonomers were improved over 80%, in which a 16% increase was achieved for NB-PBLA20 at the monomer to catalyst ratio of 100 (entry 10 in Table 2). With the improved conversion, the largest molecular weight of P(NB-g-PBLA) reached about 380.5 kDa, which was a 40% increase in comparison with the brushes synthesized with [bmim][BF4] (entry 3 in Table 2). Therefore, the ROMP of NB-PBLA in a mixture of DCM and an ionic liquid provided the second-dimensional control over the brush molecular architecture by building the brush backbone with desirable segment length.
The aminolysis of PBLA is a facile and efficient approach to attach functional moieties onto the backbone of PBLA without undesired side reactions, such as cleavage of the amide linkages along the polypeptide backbone.40,41 This reaction is found to proceed via the formation of a succinimide intermediate in the polymer backbone, which is converted to polyaspartamide, accompanying the α, β isomerization of the backbone. To investigate the efficiency of the aminolysis of PBLA in the more densely-packed brush structure of P(NB-g-PBLA), N-Boc-ethylenediamine was employed to replace the benzyl ester groups of PBLA side chains. The obtained polyaspartamide product, P(NB-g-PABEDA), was characterized by 1H NMR spectroscopy (Figure 2c), in which the disappearance of the methylene and phenyl protons of the benzyl ester groups (b′ and c′ in Figure 2b) coincided with the observation of the aliphatic and NH proton resonances from installation of the Boc-protected ethylene diamino moiety (g′′, h′′, and j′′ in Figure 2c). Quantification of the proton intensities confirmed the high efficiency (>95%) of the aminolysis of PBLA as a postpolymerization modification approach for P(NB-g-PBLA) brush. In addition, the well-controlled brush structure was maintained during the modification, which was indicated from the monomodal SEC with Đ = 1.23 for the product (Figure 3b).
In summary, the technical details of a “grafting through” synthetic approach integrating NCA ROP with ROMP were investigated to optimize conditions under which well-defined molecular brushes bearing polypeptides as brush side chains may be constructed, with two-dimensional control over the brush molecular architectures. Polypeptide side chains with desirable segment lengths were synthesized by NCA ROPs using the N2 flow method, which provided the first-dimensional control over the brush side chains. By using a solvent mixture of an ionic liquid with DCM, the second-dimensional control was achieved via ROMPs of the polypeptide macromonomers to construct brush backbones with well-controlled structures. Aminolysis of the PBLA side chain esters was demonstrated as a facile and efficient postpolymerization modification method to further install functional groups onto the densely-packed molecular brush structure. Given the numerous types of NCA monomers and NB-incorporated materials, this synthetic approach can be expanded to build various polypeptide hybrid materials.
Acknowledgments
This work was financially supported by the National Heart, Lung and Blood Institute of the National Institutes of Health as a Program of Excellence in Nanotechnology (HHSN268201000046C) and the National Science Foundation (DMR-1507429). The Welch Foundation is gratefully acknowledged for support through the W. T. Doherty-Welch Chair in Chemistry (Grant No. A-0001).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmacrolett.7b00603.
Experimental details, including materials, instrumentation, monomer, polymer and brush syntheses, and characterizations, and Figures S1–S4 (PDF).
The authors declare no competing financial interest.
Supplementary Material
References
- Verduzco R.; Li X. Y.; Pesek S. L.; Stein G. E. Structure, function, self-assembly, and applications of bottlebrush copolymers. Chem. Soc. Rev. 2015, 44, 2405–2420. 10.1039/C4CS00329B. [DOI] [PubMed] [Google Scholar]
- Li W.; Zhang X. Q.; Wang J.; Qiao X.; Liu K.; Zhang A. Peptidic molecular brushes with enhanced chirality. J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 4063–4072. 10.1002/pola.26208. [DOI] [Google Scholar]
- Sun G. R.; Cho S. H.; Clark C.; Verkhoturov S. V.; Eller M. J.; Li A.; Pavia-Jimenez A.; Schweikert E. A.; Thackeray J. W.; Trefonas P.; Wooley K. L. Nanoscopic Cylindrical Dual Concentric and Lengthwise Block Brush Terpolymers as Covalent Preassembled High-Resolution and High-Sensitivity Negative-Tone Photoresist Materials. J. Am. Chem. Soc. 2013, 135, 4203–4206. 10.1021/ja3126382. [DOI] [PubMed] [Google Scholar]
- Lu X. G.; Tran T. H.; Jia F.; Tan X. Y.; Davis S.; Krishnan S.; Amiji M. M.; Zhang K. Providing Oligonucleotides with Steric Selectivity by Brush-Polymer-Assisted Compaction. J. Am. Chem. Soc. 2015, 137, 12466–12469. 10.1021/jacs.5b08069. [DOI] [PubMed] [Google Scholar]
- Parelkar S. S.; Chan-Seng D.; Emrick T. Reconfiguring polylysine architectures for controlling polyplex binding and non-viral transfection. Biomaterials 2011, 32, 2432–2444. 10.1016/j.biomaterials.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Blum A. P.; Kammeyer J. K.; Yin J.; Crystal D. T.; Rush A. M.; Gilson M. K.; Gianneschi N. C. Peptides Displayed as High Density Brush Polymers Resist Proteolysis and Retain Bioactivity. J. Am. Chem. Soc. 2014, 136, 15422–15437. 10.1021/ja5088216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum A. P.; Kammeyer J. K.; Gianneschi N. C. Activating peptides for cellular uptake via polymerization into high density brushes. Chem. Sci. 2016, 7, 989–994. 10.1039/C5SC03417E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjichristidis N.; Iatrou H.; Pitsikalis M.; Mays J. Macromolecular architectures by living and controlled/living polymerizations. Prog. Polym. Sci. 2006, 31, 1068–1132. 10.1016/j.progpolymsci.2006.07.002. [DOI] [Google Scholar]
- Peleshanko S.; Tsukruk V. V. The architectures and surface behavior of highly branched molecules. Prog. Polym. Sci. 2008, 33, 523–580. 10.1016/j.progpolymsci.2008.01.003. [DOI] [Google Scholar]
- Su L.; Heo G. S.; Lin Y.-N.; Dong M.; Zhang S.; Chen Y.; Sun G.; Wooley K. L. Syntheses of triblock bottlebrush polymers through sequential ROMPs: Expanding the functionalities of molecular brushes. J. Polym. Sci., Part A: Polym. Chem. 2017, 55, 2966–2970. 10.1002/pola.28647. [DOI] [Google Scholar]
- Zhang B.; Fischer K.; Schmidt M. Cylindrical polypeptide brushes. Macromol. Chem. Phys. 2005, 206, 157–162. 10.1002/macp.200400266. [DOI] [Google Scholar]
- Lu H.; Wang J.; Lin Y.; Cheng J. J. One-Pot Synthesis of Brush-Like Polymers via Integrated Ring-Opening Metathesis Polymerization and Polymerization of Amino Acid N-Carboxyanhydrides. J. Am. Chem. Soc. 2009, 131, 13582–13583. 10.1021/ja903425x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. F.; Yin Q.; Lu H.; Xia H. W.; Lin Y.; Cheng J. J. PEG-Polypeptide Dual Brush Block Copolymers: Synthesis and Application in Nanoparticle Surface PEGylation. ACS Macro Lett. 2013, 2, 809–813. 10.1021/mz4003672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deming T. J. Regenerative Medicine Noodle Gels for Cells. Nat. Mater. 2010, 9, 535–536. 10.1038/nmat2789. [DOI] [PubMed] [Google Scholar]
- Lalatsa A.; Schatzlein A. G.; Mazza M.; Thi B. H. L.; Uchegbu I. F. Amphiphilic poly(l-amino acids) - New materials for drug delivery. J. Controlled Release 2012, 161, 523–536. 10.1016/j.jconrel.2012.04.046. [DOI] [PubMed] [Google Scholar]
- Matson J. B.; Stupp S. I. Self-assembling peptide scaffolds for regenerative medicine. Chem. Commun. 2012, 48, 26–33. 10.1039/C1CC15551B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Lu H.; Kamat R.; Pingali S. V.; Urban V. S.; Cheng J. J.; Lin Y. Supramolecular Polymerization from Polypeptide-Grafted Comb Polymers. J. Am. Chem. Soc. 2011, 133, 12906–12909. 10.1021/ja202268t. [DOI] [PubMed] [Google Scholar]
- Wang J.; Lu H.; Ren Y.; Zhang Y. F.; Morton M.; Cheng J. J.; Lin Y. Interrupted Helical Structure of Grafted Polypeptides in Brush-Like Macromolecules. Macromolecules 2011, 44, 8699–8708. 10.1021/ma201833b. [DOI] [Google Scholar]
- Baumgartner R.; Fu H.; Song Z.; Lin Y.; Cheng J. Cooperative polymerization of α-helices induced by macromolecular architecture. Nat. Chem. 2017, 9, 614–622. 10.1038/nchem.2712. [DOI] [PubMed] [Google Scholar]
- Aliferis T.; Iatrou H.; Hadjichristidis N. Living polypeptides. Biomacromolecules 2004, 5, 1653–1656. 10.1021/bm0497217. [DOI] [PubMed] [Google Scholar]
- Deming T. J. Facile synthesis of block copolypeptides of defined architecture. Nature 1997, 390, 386–389. 10.1038/37084. [DOI] [PubMed] [Google Scholar]
- Dimitrov I.; Schlaad H. Synthesis of nearly monodisperse polystyrene-polypeptide block copolymers via polymerisation of N-carboxyanhydrides. Chem. Commun. 2003, 2944–2945. 10.1039/B308990H. [DOI] [PubMed] [Google Scholar]
- Conejos-Sanchez I.; Duro-Castano A.; Birke A.; Barz M.; Vicent M. J. A controlled and versatile NCA polymerization method for the synthesis of polypeptides. Polym. Chem. 2013, 4, 3182–3186. 10.1039/c3py00347g. [DOI] [Google Scholar]
- Zhao W.; Gnanou Y.; Hadjichristidis N. From competition to cooperation: a highly efficient strategy towards well-defined (co)polypeptides. Chem. Commun. 2015, 51, 3663–3666. 10.1039/C4CC09055A. [DOI] [PubMed] [Google Scholar]
- Zou J.; Fan J.; He X.; Zhang S.; Wang H.; Wooley K. L. A Facile Glovebox-Free Strategy To Significantly Accelerate the Syntheses of Well-Defined Polypeptides by N-Carboxyanhydride (NCA) Ring-Opening Polymerizations. Macromolecules 2013, 46, 4223–4226. 10.1021/ma4007939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.; Cheng J. Hexamethyldisilazane-mediated controlled polymerization of alpha-Amino acid N-carboxyanhydrides. J. Am. Chem. Soc. 2007, 129, 14114–14115. 10.1021/ja074961q. [DOI] [PubMed] [Google Scholar]
- He X.; Fan J. W.; Wooley K. L. Stimuli-Triggered Sol-Gel Transitions of Polypeptides Derived from -alpha-Amino Acid N-Carboxyanhydride (NCA) Polymerizations. Chem. - Asian J. 2016, 11, 437–447. 10.1002/asia.201500957. [DOI] [PubMed] [Google Scholar]
- Engler A. C.; Lee H. I.; Hammond P. T. Highly Efficient ″Grafting onto″ a Polypeptide Backbone Using Click Chemistry. Angew. Chem., Int. Ed. 2009, 48, 9334–9338. 10.1002/anie.200904070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Chen P.; Li Z. B. Molecular Bottlebrushes with Polypeptide Backbone Prepared via Ring-Opening Polymerization of NCA and ATRP. Macromol. Rapid Commun. 2012, 33, 287–295. 10.1002/marc.201100649. [DOI] [PubMed] [Google Scholar]
- Engler A. C.; Bonner D. K.; Buss H. G.; Cheung E. Y.; Hammond P. T. The synthetic tuning of clickable pH responsive cationic polypeptides and block copolypeptides. Soft Matter 2011, 7, 5627–5637. 10.1039/c1sm05064h. [DOI] [Google Scholar]
- Rhodes A. J.; Deming T. J. Soluble, Clickable Polypeptides from Azide-Containing N-Carboxyanhydride Monomers. ACS Macro Lett. 2013, 2, 351–354. 10.1021/mz4001089. [DOI] [PubMed] [Google Scholar]
- Rhodes A. J.; Deming T. J. Tandem Catalysis for the Preparation of Cylindrical Polypeptide Brushes. J. Am. Chem. Soc. 2012, 134, 19463–19467. 10.1021/ja308620h. [DOI] [PubMed] [Google Scholar]
- Fan J.; Li R.; He X.; Seetho K.; Zhang F.; Zou J.; Wooley K. L. Construction of a versatile and functional nanoparticle platform derived from a helical diblock copolypeptide-based biomimetic polymer. Polym. Chem. 2014, 5, 3977–3981. 10.1039/C4PY00628C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compain P. Olefin Metathesis of Amine-Containing Systems: Beyond the Current Consensus. Adv. Synth. Catal. 2007, 349, 1829–1846. 10.1002/adsc.200700161. [DOI] [Google Scholar]
- Kim K. T.; Park C.; Vandermeulen G. W. M.; Rider D. A.; Kim C.; Winnik M. A.; Manners I. Gelation of Helical Polypeptide–Random Coil Diblock Copolymers by a Nanoribbon Mechanism. Angew. Chem., Int. Ed. 2005, 44, 7964–7968. 10.1002/anie.200502809. [DOI] [PubMed] [Google Scholar]
- Bielawski C. W.; Grubbs R. H. Living ring-opening metathesis polymerization. Prog. Polym. Sci. 2007, 32, 1–29. 10.1016/j.progpolymsci.2006.08.006. [DOI] [Google Scholar]
- Csihony S.; Fischmeister C.; Bruneau C.; Horvath I. T.; Dixneuf P. H. First ring-opening metathesis polymerization in an ionic liquid. Efficient recycling of a catalyst generated from a cationic ruthenium allenylidene complex. New J. Chem. 2002, 26, 1667–1670. 10.1039/B205920G. [DOI] [Google Scholar]
- Vygodskii Y. S.; Shaplov A. S.; Lozinskaya E. I.; Filippov O. A.; Shubina E. S.; Bandari R.; Buchmeiser M. R. Ring-opening metathesis polymerization (ROMP) in ionic liquids: Scope and limitations. Macromolecules 2006, 39, 7821–7830. 10.1021/ma061456p. [DOI] [Google Scholar]
- Trnka T. M.; Morgan J. P.; Sanford M. S.; Wilhelm T. E.; Scholl M.; Choi T. L.; Ding S.; Day M. W.; Grubbs R. H. Synthesis and activity of ruthenium alkylidene complexes coordinated with phosphine and N-heterocyclic carbene ligands. J. Am. Chem. Soc. 2003, 125, 2546–2558. 10.1021/ja021146w. [DOI] [PubMed] [Google Scholar]
- Nakanishi M.; Park J. S.; Jang W. D.; Oba M.; Kataoka K. Study of the quantitative aminolysis reaction of poly(beta-benzyl L-aspartate) (PBLA) as a platform polymer for functionality materials. React. Funct. Polym. 2007, 67, 1361–1372. 10.1016/j.reactfunctpolym.2007.08.009. [DOI] [Google Scholar]
- Uchida H.; Itaka K.; Nomoto T.; Ishii T.; Suma T.; Ikegami M.; Miyata K.; Oba M.; Nishiyama N.; Kataoka K. Modulated Protonation of Side Chain Aminoethylene Repeats in N-Substituted Polyaspartamides Promotes mRNA Transfection. J. Am. Chem. Soc. 2014, 136, 12396–12405. 10.1021/ja506194z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.