

Abstract
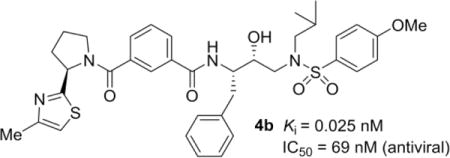
Based upon molecular insights from the X-ray structures of inhibitor-bound HIV-1 protease complexes, we have designed a series of isophthalamide-derived inhibitors incorporating substituted pyrrolidines, piperidines and thiazolidines as P2-P3 ligands for specific interactions in the S2–S3 extended site. Compound 4b has shown an enzyme Ki of 0.025 nM and antiviral IC50 of 69 nM. An X-ray crystal structure of inhibitor 4b-HIV-1 protease complex was determined at 1.33 Å resolution. We have also determined X-ray structure of 3b-bound HIV-1 protease at 1.27 Å resolution. These structures revealed important molecular insight into the inhibitor–HIV-1 protease interactions in the active site.
Keywords: HIV-1 protease, Drug-resistance, X-ray structure, Synthesis, Isophthalamide
Graphical abstract
1. Introduction
Protein structure-based molecular design has had tremendous impact in modern drug design and medicinal chemistry efforts.1,2 The design and development of HIV-1 protease inhibitors and their combination with reverse transcriptase inhibitors, marked the beginning of a new era of treatment of HIV infection and AIDS.3,4 The combination antiretroviral therapies (ART) significantly improved life expectancy and dramatically improved the course of HIV management in developing countries.5,6 Therapeutic protease inhibitors (PIs) block HIV-1 replication by inhibiting viral HIV-1 protease and generate morphologically immature and noninfectious virions. However, selective drug pressure blocks replication of viruses that are susceptible to the drug and generates drug-resistant HIV-1 variants.7,8 A growing number of HIV/AIDS patients are harboring multidrug-resistant HIV-1 variants that can be readily transmitted. These drug-resistant viruses are difficult to treat and raise a serious concern for the long-term treatment options of HIV management.9,10 Therefore, one of the major current therapeutic objectives is to design and develop novel PIs that display broad-spectrum activity against drug-resistant HIV-1 variants and show improved drug-like properties.3,11
In our continuing studies towards the design and development of PIs to combat drug-resistance, we reported a series of novel nonpeptide PIs that showed very potent enzyme inhibitory and antiviral activity. Also, these PIs exhibited potent antiviral activity3,12,13 against a panel of multidrug-resistant HIV-1 variants.
One of our major design strategies is to incorporate cyclic ether or polyether-like templates where ether oxygens are positioned to form specific hydrogen bonds with the active site backbone atoms.14,15 In essence, these ether oxygens mimic the peptide carbonyl group in the active site. This design concept is inspired by the occurrence of numerous bioactive natural products with polyether subunits.16,17 Our laboratory reported a range of exceptionally potent PIs with intriguing molecular features such as TMC-114, TMC126, GRL-06579, GRL-02031, and GRL-519.18,19,20 These inhibitors show broad-spectrum antiviral activity against multidrug-resistant HIV-1 variants and show high genetic barrier to resistance. Darunavir (DRV, 1) has emerged as front-line treatment of HIV/AIDS patients.21,22 In DRV, we have incorporated a stereochemically defined bis-tetrahydrofuran (bis-THF) template to promote hydrogen bonding interactions with the backbone atoms at S2-subsites.17,21 Our structure-based design efforts created numerous intriguing ligands such as 3S-THF, bis-THF, Cp-THF, Tp-THF, and tris-THF.12 All these ligands are involved in interactions in the protease active site, especially extensive hydrogen bonding with the protease backbone atoms.
Recently, we have explored a number of substituted benzoic acid-based acyclic and macrocyclic inhibitors with good to excellent potency.23,24 These inhibitors were designed based upon the P2-ligand of FDA approved nelfinavir and P1′ and P2′ ligands inherent to DRV (1) and TMC-126 (2).17,25 In particular, a series of PIs containing isophthalamide-based P2-P3 ligands showed very potent enzyme inhibitory and antiviral activity.26 One of the important features of these PIs is that they contain only two chiral centers and structural variants of these PIs can be synthesized readily for structure-activity studies. Furthermore, some of these ligand moieties have been utilized in the design and synthesis of inhibitors of β-secretase (BACE-1), an aspartic acid protease target for the treatment of Alzheimer’s disease.27–30 Our initial efforts led us to determine X-ray structures of HIV-1 protease complexes with a number of isophthalamide based inhibitors.26 These X-ray structural studies provided molecular insights into the ligand-binding site interactions in the HIV-1 protease active site. We have now further explored these structural insights and herein we report the design, synthesis, and biological evaluation of a series of PIs incorporating isophthalamide-based P2-P3 ligands with substituted pirrolidines, piperidines and thiazolidines for specific interactions in the S2-S3 extended site.
2. Results and discussion
2.1. Synthesis
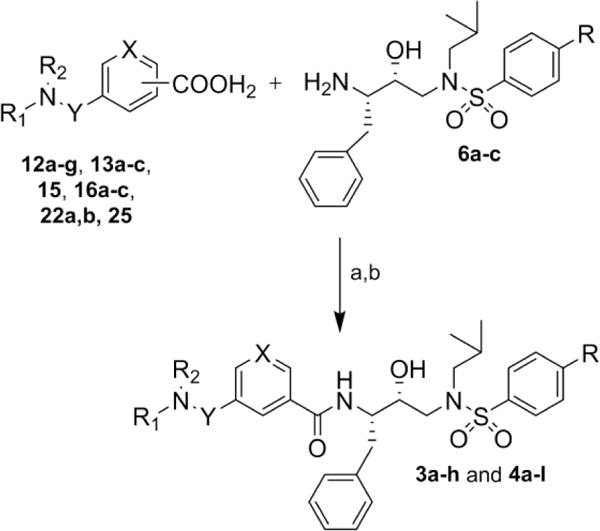
Our general strategy for the synthesis of HIV-1 protease inhibitors 3a–h and 4a-l is illustrated in Scheme 1. We planned to synthesize these inhibitors by amide coupling of the appropriate isophthalic, phthalic or terephthalic acid derivatives 5 with the amines belonging to hydroxyethylamine sulfonamide isosteres (6a–c).19,28 The required acid derivatives were obtained by coupling of readily available isophthalic (7a–d), phthalic (8) or terephthalic (9) monoacids with amines 10a–d or cyclic amines 11a–h.28,31,32
Scheme 1.
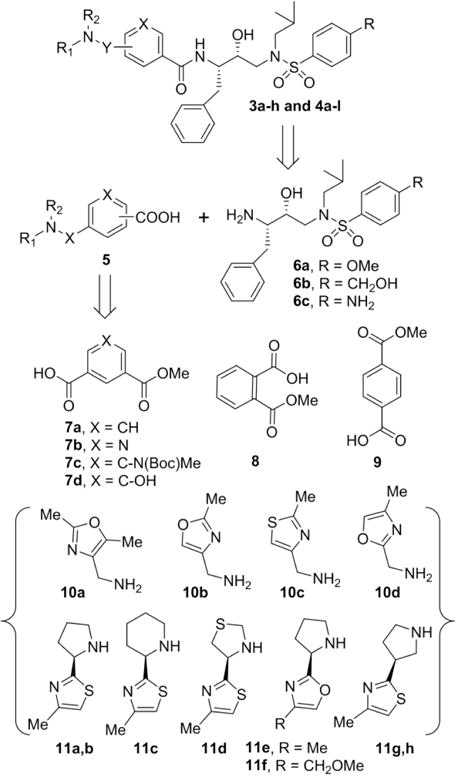
General synthetic approach for inhibitors 3a–h and 4a–l.
The synthesis of oxazole- and thiazole-based P2-P3 ligands 12a–g is shown in Scheme 2. The known28 amine 10a was coupled with isophthalic acid derivatives 7c,d using EDC, HOBt in the presence of diisopropylethylamine to provide the corresponding amide derivatives. The resulting amides were N-methylated with NaH and MeI in THF. Saponification of the resulting methyl ester with aqueous lithium hydroxide afforded isophthalic acid derivatives 12a,b. Acid derivatives 12c–f were prepared using similar reaction sequences using suitable commercially available amines 10b,c and oxazolylmethylamine 10d26 and carboxylic acids 7a, 8 or 9. The resulting amides were N-methylated and subsequent saponification of the methyl ester functionality afforded acids 12c–f.
Scheme 2.
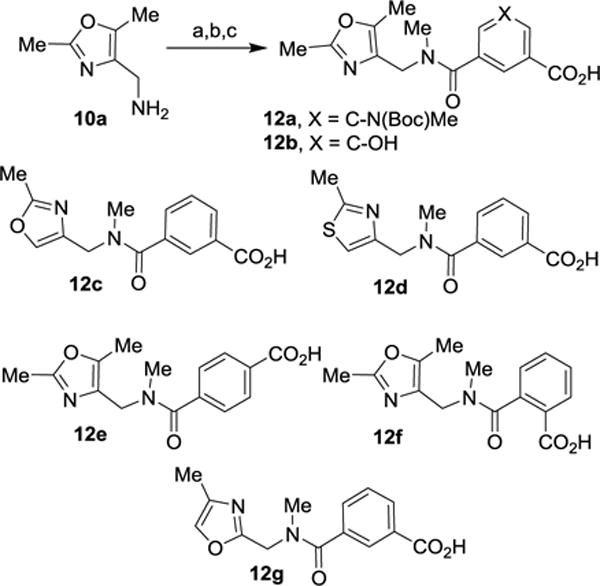
Reagents and conditions: (a) 7c or 7d, EDCI, HOBt, i-Pr2NEt, CH2Cl2, 25 °C, 16 h, 80%; (b) MeI, NaH, dry THF, 25 °C, 1 h, 68%; (c) LiOH.H2O, THF/MeOH/H2O (3:1:1), 25 °C, 16 h, 99%.
The synthesis of various methylthiazolylpyrrolidine-derived isophthalic acids is shown in Scheme 3. As shown, amines 11a,b and isophthalic acid monomethyl esters 7a or 7b were coupled together using standard EDC and HOBt coupling. The resulting esters were saponified with aqueous lithium hydroxide to provide carboxylic acids 13a–c. Compound 13a was also employed for the synthesis of P2-P3 ligand 15 in which a carbonyl group was converted to a methylene linker by reducing the isophthalate carbonyl group. Thus, treatment of 13a with LiAlH4 in dry THF at reflux provided aminoalcohol 14. The hydroxymethyl group of 14 was oxidized with manganese dioxide to aldehyde. Oxidation33 of this aldehyde with NaClO2 provided acid 15. Acid derivatives 16a–c were prepared by following similar sequence of reactions as compound 13a.
Scheme 3.
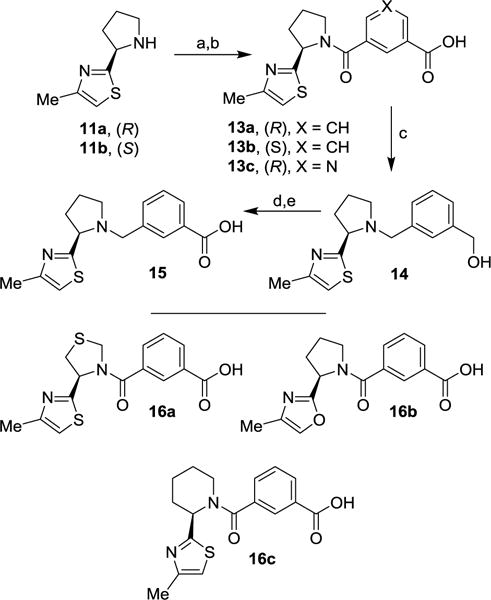
Reagents and conditions: (a) 7a or 7b, EDCI, HOBt, i-Pr2NEt, CH2Cl2, 25 °C, 16 h, 80%; (b) LiOH.H2O, THF/MeOH/H2O (3:1:1), 25 °C, 16 h, 99%; (c) from 13a, LiAlH4, dry THF, reflux, 15 h, 88%; (d) MnO2, CH2Cl2, 25 °C, 5 h; (e) NaClO2, NaH2PO4, t-BuOH/H2O, 2-methyl-2-butene, 25 °C, 4 h, 45% (over 2 steps).
The synthesis of pyrrolidin-3-yl-thiazole amine-derived P2-P3 ligands 11g,h is shown in Scheme 4. (3R)- and (3S)-pyrrolidines were prepared by Michael addition of (R)-α-methylbenzylamine with dimethyl itaconate (17) followed by cyclization of the resulting amino-ester to give an equal mixture of lactams as described previously.34 This mixture was separated by chromatography. Treatment of the resulting esters with benzylalcohol in the presence of PPTS provided benzyl ester derivatives 18a,b. These lactams were treated with Lawesson’s reagent to provide thioamides 19a,b. Reduction of the thioamide functionality by treatment with MeI followed by NaBH4 in EtOH led to derivatives 20a,b. Simultaneous removal of benzyl and α- methylbenzyl groups by catalytic hydrogenation was followed by N-Cbz protection and conversion of the carboxylic functionality to the corresponding amide to furnish carboxamide derivatives 21a,b. Treatment of these carboxamides with Lawesson’s reagent afforded the corresponding thioamides, which underwent cyclization upon treatment with chloroacetone in EtOH. Removal of the Cbz group by exposure to HBr in AcOH, provided the desired amine derivatives 11g,h. These ligands were coupled with the isophthalic acid methyl ester 7a to furnish the corresponding amide derivatives. Saponification of the methyl ester with aqueous lithium hydroxide afforded acids 22a,b.
Scheme 4.
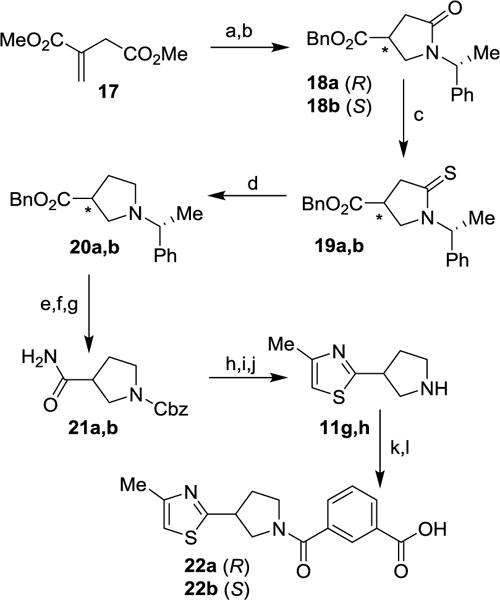
Reagents and conditions: (a) (R)-α-methylbenzylamine MeOH, 115 °C, 18 h, 75%; (b) BnOH, PPTS, toluene, reflux, 24 h, (S) 81%, (R) 73%; (c) Lawesson’s reagent, toluene, 100 °C, 2 h, (S) 99%, (R) 67%; (d) MeI, CH2Cl2, 25 °C, 24 h then NaBH4, EtOH, 0 °C, 1h, (S) 50%, (R) 38%; (e) H2, 10% Pd/C, 25 °C, 20 h, (S) 99%, (R) 99%; (f) CbzCl, 4 N NaOH, 0 °C, 2 h, (S) 51%, (R) 39%; (g) Boc2O, pyridine, NH4HCO3, 1,4-dioxane, 25 °C, 16 h; (h) Lawesson’s reagent, THF, 25 °C, 16 h; (i) chloroacetone, EtOH, 80 °C, 18 h, (S) 42%, (R) 56% (over 3 steps); (j) HBr in AcOH, 25 °C, 1 h, (S) 89%, (R) 88%; (k) 7a, EDCI, HOBt, i-Pr2NEt, CH2Cl2, 25 °C, 16 h, (S) 55%, (R) 68%; (l) LiOH.H2O, THF/MeOH/H2O (3:1:1), 25 °C, 16 h, (S) 55%, (R) 54%.
The synthesis of isophthalic acid derivative 25 is reported in Scheme 5. Ester 23 was treated with LiBH4 in THF at 23 °C to provide alcohol derivative 24. Alkylation of alcohol 24 with MeI and NaH in THF provided the corresponding methyl ether. Removal of the Cbz-group with HBr in acetic acid afforded amine 11f. This amine was coupled with isophthalic acid monomethyl ester 7a using EDC and HOBt in the presence of i-Pr2NEt to provide the corresponding amide derivative. Saponification of the methyl ester with aqueous LiOH afforded acid 25.
Scheme 5.
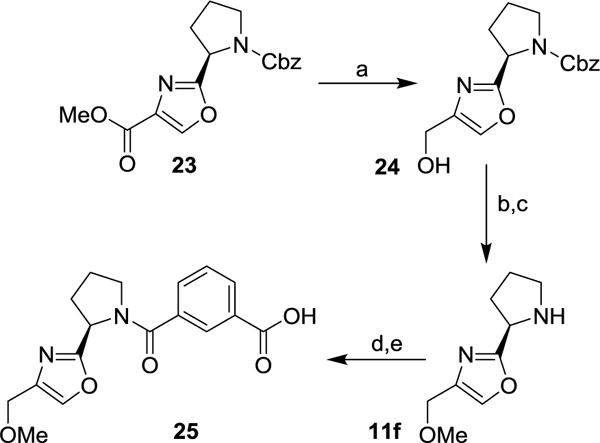
Reagents and conditions: (a) LiBH4, dry THF, 25 °C, 4 h, 42%; (b) MeI, NaH, dry THF, 25 °C, 1 h, 68%; (c) HBr in AcOH, 25 °C, 1 h, 67%; (d) 7a, EDCI, HOBt, i-Pr2NEt, CH2Cl2, 25 °C, 16 h, 96%; (e) LiOH.H2O, THF/MeOH/H2O (3:1:1), 25 °C, 16 h, 98%.
Our syntheses of the final inhibitors are shown in Scheme 6. Synthesis of inhibitors 3a–h and 4a–l were carried out by coupling of acids 12a-g, 13a–c, 15, 16a–c, 22a,b and 25 with hydroxyethylamine sulfonamide isosteres (6a–c) using standard EDC and HOBt coupling. Various isophthalamide-based inhibitors were obtained in good to excellent yields (49–95%).
Scheme 6.
Reagents and conditions: (a) EDCI, HOBt, i-Pr2NEt, CH2Cl2, 25 °C, 16 h (49–95%); (b) for 3e, TFA, CH2Cl2, 25 °C, 2 h, 98%.
2.2. Biological evaluation, structure-activity relationships, and X-ray studies
On the basis of the X-ray studies reported for compounds 3a26 and darunavir-bound HIV-1 protease,35 we first sought to investigate the outcome of a focused series of derivatives bearing isophthalamide as the P2-ligand and various cyclic and acyclic heterocycles as the P3-ligand. An overlay of the X-ray crystal structures of inhibitors 1 (DRV) and 3a in the HIV-1 protease active site is shown in Figure 2. As can be seen, the key interactions in the S1′ and S2′ subsites are very similar. The major differences are in the S2-S3 subsites. The bis-THF of DRV forms two strong hydrogen bonds with Asp-29 and Asp-30 backbone amide NH’s in the S2-subsite. Inhibitor 3a forms a strong hydrogen bond with Asp29 backbone amide NH and forms strong van der Waals interactions with oxazolylmethyl ligand. Based upon this molecular insight, we speculated that cyclic and acyclic heterocycles could form hydrogen bonds with backbone atoms as well as form stronger van der Waals interactions in the active site. In particular, we first sought to investigate the outcome of a focused series of derivatives bearing oxazolylmethyl- or thiazolylmethyl-derived P3-ligands (compounds 3b-h, Table 1). HIV-1 protease inhibitory potency of all inhibitors was first evaluated using the assay protocol reported by Toth and Marshall.6 The antiviral activity of the new inhibitors was determined based on a previously published assay protocol using MT-2 cells exposed to HIV-1 LAI.37 These results are shown in Tables 1 and 2. In compound 3b, we incorporated a 4-methyl oxazole as P3 to promote hydrogen bond through the oxazole nitrogen. However, this compound lost significant potency over compound 3a. Compounds 3c (entry 3) and 3d (entry 4) contained a 2-methyloxazole and a 2-methylthiazole ring, respectively. Derivative 3d displayed 5-fold enhanced protease inhibitory activity over 3c. Nevertheless, the compounds similarly performed in antiviral assay, displaying an over 20-fold improvement with respect to their forerunner 3b (entry 2). The dimethyloxazole moiety was also explored in combination with polar substituents placed at the 3-position of the isophthalamide moiety (entries 5 and 6). As it turned out, phenol derivative 3f overtook its methylamino analogue 3e by 10-fold in terms of protease inhibitory potency. However, the presence of a 3-substituted isophthalamide portion was found detrimental for the antiviral outcome of both compounds. Finally, the exploration of the dimethyloxazole moiety linked to phthalamide- or terephthalamide-based ligands (compounds 3g and 3h, entries 7 and 8, respectively) led to a strong drop in both the enzyme inhibitory and the antiviral activities of the compounds, clearly validating the key role of the isophthalamide moiety for inhibitors’ efficacy. Taking into account the information gained with the first set of compounds we sought to explore a novel series isophthalamide-based extended P2-P3 ligands incorporating substituted pirrolidines, piperidines and thiazolidines as the suitable support for thiazole or oxazole rings (inhibitors 4a–l, Table 2). The incorporation of a 2-thiazolyl pyrrolidine in both (S) or (R) configuration (compounds 4a and 4b, entries 1 and 2, respectively) clearly highlighted the key role of stereochemistry for this series of compounds. Inhibitor 4b with (R)-configuration displayed over 100-fold improved potency over its (S) counterpart 4a. The same trend was also shown for the antiviral outcome of the compounds. The combination of the (R)-2-thiazolyl pyrrolidine moiety with different hydroxyethylamine sulfonamide isosteres (compounds 4c and 4d, entries 3 and 4, respectively) substantially led to a significant drop in both the enzyme inhibitory and the antiviral activities. The increase of the ring size in piperidine-based compound 4e (entry 5) significantly reduced enzyme affinity and antiviral efficacy. Compound 4f (entry 6) bearing a 2-thiazolyl thiazolidine moiety, although characterized by a reduced enzyme inhibitory potency with respect to 4a, displayed the best antiviral activity of this series of compounds (IC50 = 32 nM). Compounds 4g and 4h (entries 7 and 8) containing a 2-oxazolyl pyrrolidine moiety were also explored. In particular, methyloxazole derivative 4h resulted in a 10-fold improved antiviral profile with respect to its methoxymethyloxazole analogue 4g. The exploration of compounds 4i (entry 9) with a methylene replacing one of the isophthalate carbonyl groups, and 4j (entry 10) bearing a pyridyl ring in the isophthalic moiety led to a substantial loss of protease inhibitory activity. The shift of the thiazole ring in 3-position of the pyrrolidine was also investigated for both (R) and (S) configurations (inhibitors 4k and 4l, entries 11 and 12, respectively). Both compounds exhibited comparable protease inhibitory profile and a moderate antiviral activity, indicating in this case a negligible contribution of stereochemistry. To obtain molecular insight into the inhibitor-HIV-1 protease interactions, we have determined the X-ray crystal structures of the inhibitors 3b- and 4b-bound HIV-1 protease complexes.
Figure 2.
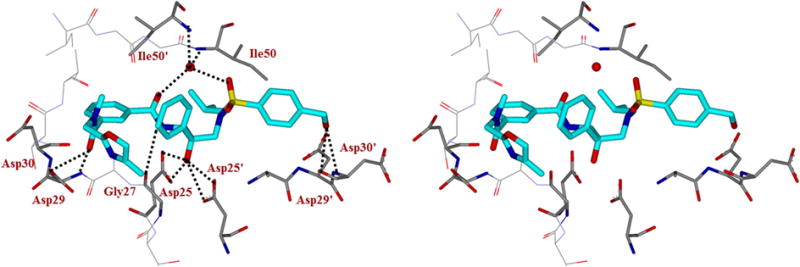
Stereoview of the overlay X-ray structures of inhibitor 3a (green) and darunavir (1, torquois)-bound HIV-1 protease. All strong active site hydrogen bonding interactions of darunavir with HIV-1 protease are shown as dotted lines.
Table 1.
Enzyme inhibitory and antiviral activity of inhibitors 3a–h.
| Entry | Inhibitor structure | Ki | IC50a |
|---|---|---|---|
|
(nM) | |||
| 1 |
|
0.21 | 31 |
| 2 |
|
0.63 | 581 |
| 3 |
|
0.64 | 23 |
| 4 |
|
0.107 | 32 |
| 5 |
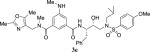
|
5.08 | 600 |
| 6 |
|
0.51 | 290 |
| 7 |
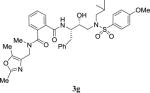
|
629 | > 1000 |
| 8 |
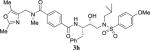
|
70 | > 1000 |
Darunavir (1) exhibited Ki = 16 pM, antiviral IC50 = 3.2 nM
Table 2.
Enzyme inhibitory and antiviral activity of inhibitors 4a–l.
| Entry | Inhibitor structure | Ki | IC50a,b |
|---|---|---|---|
|
(nM) | |||
| 1 |
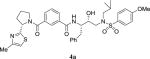
|
30.8 | > 1000 |
| 2 |
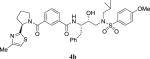
|
0.025 | 69 |
| 3 |
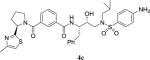
|
1.61 | 189 |
| 4 |
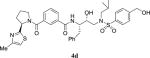
|
0.79 | 164 |
| 5 |
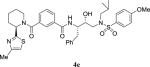
|
0.53 | 221 |
| 6 |
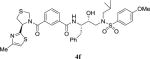
|
0.76 | 32 |
| 7 |
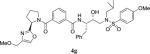
|
0.80 | 327 |
| 8 |
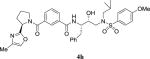
|
0.41 | 33 |
| 9 |
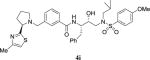
|
23.6 | nt |
| 10 |
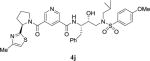
|
2.50 | nt |
| 11 |
|
0.49 | 280 |
| 12 |
|
0.40 | 109 |
Darunavir (1) exhibited Ki = 16 pM, antiviral IC50 = 3.2 nM
nt = not tested.
The crystal structures of wild type HIV-1 protease in complex with the inhibitors 3b (GRL-0518A) and 4b (GRL-1118A) were determined and refined at 1.27 Å and 1.33 Å resolution, respectively. Both crystal structures contain the protease dimer and inhibitor bound in a single orientation. The overall structures are comparable to the structure of HIV-1 protease and darunavir35 with low root mean square differences (RMSD) of 0.18 Å and 0.21 Å, respectively, for Cα atoms. Most differences between corresponding Cα atoms are less than 0.4 Å. Only residues 47′–51′ exhibit root mean square differences of about 0.5–0.6 Å, likely due to displacement by the P2 group. For residues 78′–81′, the larger RMSD of about 0.6–0.9 Å occurs due to the opposite effect since the 80’s loop moves in to generate more contacts with the single smaller isobutyl group in P1′ position of these two complexes. The third larger RMSD of about 0.7–0.8 Å in residue 4 may be due to the rotation of its side chain. Both inhibitors form hydrogen bonding interactions of their urethane NH with the carbonyl oxygen of Gly27. In addition, water-mediated interactions connect the inhibitor carbonyl oxygen and sulfonamide oxygen with the amides of Ile50 and 50′ in the flaps. Similar interactions are shared by the protease-darunavir complex and are considered responsible for the high affinity of darunavir for HIV protease and its potency against drug resistant HIV-1 variants.35
Inhibitor 3b contains a 4-methyloxazolyl-2-methylamide as the P3-ligand and a stereoview of the active site interactions is shown in Figure 3. The P2-isophthalamide carbonyl group of the inhibitor maintains a strong hydrogen bonding with backbone amide and side chain of Asp29. However, the oxazole ring flipped toward the S2-extended site and was unable to form any hydrogen bond in the active site. The isophthalamide group in P2 is sandwiched in a cavity formed by the hydrophobic side chains of Ile50′, Ala28, Val32, Ile47 and Ile84. The extended P3 group (4-methyl-2-oxazole) in inhibitor 3b forms hydrophobic interactions with the side chains of Val82’. In comparison, the 2,5-dimethyl-4-oxazole in in inhibitor 3a fits in two conformations with occupancy ratio of 50:50.26 The ligand-binding site interactions of inhibitor 3b in the S1, S1′ and S2′ subsites are similar to daruanvir. The hydroxymethyl group on the P2′-ligand form a strong hydrogen bond with the backbone amide NH of Asp30’ in the S2’-site.
Figure 3.
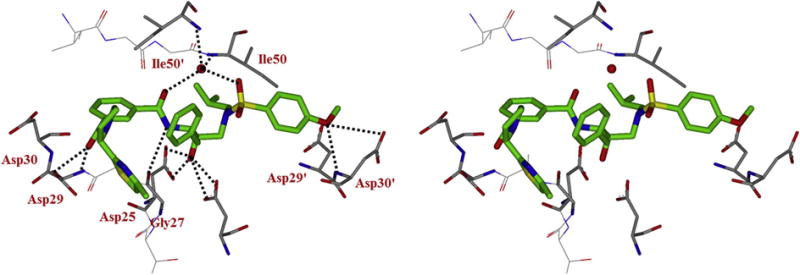
Stereoview of the X-ray structure of inhibitor 3b (torquois)-bound HIV-1 protease (PDB code: 5UPZ). All strong active site hydrogen bonding interactions of inhibitor 3b with HIV-1 protease are shown as dotted lines.
Inhibitor 4b contains a thiazolylpyrrolidine moiety as the P3-liagand and a stereoview of the active site interactions is shown in Figure 4. Like compound 3b, the carbonyl group of inhibitor 4b in the P2 position maintains polar hydrogen bonding interactions with the amide and side chain of Asp29’ with interatomic distances of 2.9 Å. The pyrrolidine group and the larger sulfur atom in thiazoline of inhibitor 4b may constrain the conformation of the extended P3 group (methyl-thiazole) to form π-π stacking interactions with the guanidine group of Arg8’. These extended interactions are expected to stabilize the inhibitor in the binding site. Furthermore, the P2 isophthalamide phenyl ring forms van der Waals contacts with Ile50’, Val32, Ile47 and Ile84.38
Figure 4.
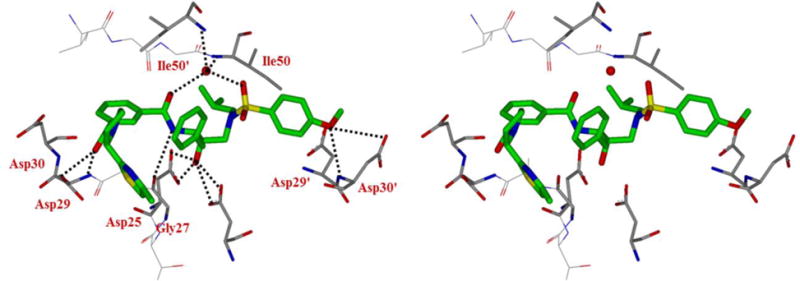
Stereoview of the X-ray structure of inhibitor 4b (green)-bound HIV-1 protease (PDB code: 5UOV). All strong active site hydrogen bonding interactions of inhibitor 4b with HIV-1 protease are shown as dotted lines.
3. Conclusion
In summary, we have designed and synthesized potent HIV-1 protease inhibitors incorporating isophthalamide derivatives as the P2-P3 ligands for the HIV-1 protease active site. These inhibitors were specifically designed to interact with the extended S2 subsite, particularly to form hydrogen bonds with the backbone atoms as well as to promote van der Waals interactions with residues in the S3 subsite. In this context, we have synthesized and evaluated a range of heterocyclic derivatives of isophthalamides to occupy the extended S2-subsite. In particular, we have investigated the suitability of oxazolyl and thiazolyl isophthalamides as well as pyrrolidinyl thiazolyl and pyrrolidinyl oxazole amide derivatives of isophthalamides with donor and acceptor groups to serve as P2-P3 ligands. A number of compounds showed very potent enzyme inhibitory and antiviral activity. Inhibitor 4b with a pyrrolidinyl thiazole isophthalamide derivative showed very potent enzyme inhibitory activity (Ki = 25 pM) and antiviral IC50 value of 69 nM in MT-2 cells exposed to HIV-1 LAI. Further optimization efforts led to inhibitor 4h with a pyrrolidinyl oxazole isophthalamide derivative which showed an improved antiviral IC50 value of 33 nM. We have determined X-ray structures of inhibitor 3b-bound HIV-1 protease and inhibitor 4b-bound HIV-1 protease. These structures provided molecular insights into the key ligand-binding site interactions responsible for inhibitors potency. The isophthalamide P2-P3 ligand amide carbonyl formed very strong hydrogen bonds with Asp29 backbone amide NH and the pyrrolidinylthiazole heterocycles are involved in van der Waals interactions with Leu23’ and Val82’ in the S2 extended site. These ligand-binding site interactions can be further exploited to design more potent and less complex protease inhibitors.
4. Experimental
4.1 General
All moisture-sensitive reactions were carried out in oven-dried glassware under an argon atmosphere unless otherwise stated. Anhydrous solvents were obtained as follows: Diethyl ether and tetrahydrofuran were distilled from sodium metal/benzophenone under argon. Toluene and dichloromethane were distilled from calcium hydride under argon. All other solvents were reagent grade. Column chromatography was performed using Silicycle SiliaFlash F60 230−400 mesh silica gel. Thin-layer chromatography was carried out using EMD Millipore TLC silica gel 60 F254 plates. 1H NMR and 13C NMR spectra were recorded on a Varian INOVA300 or a Bruker ARX400. Low-resolution mass spectra were collected on a Waters 600 LCMS or by the Purdue University Campus-Wide Mass Spectrometry Center. High-resolution mass spectra were collected by the Purdue University Campus-Wide Mass Spectrometry Center.
4.2 Experimental procedures
4.2.1. 3-((tert-Butoxycarbonyl)(methyl)amino)-5-(((2,5-dimethyloxazol-4-yl)methyl)(methyl)carbamoyl)benzoic acid (12a)
To a solution of 7c (45 mg, 0.215 mmol) in dry CH2Cl2 (2 mL), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI) (62 mg, 0.322 mmol), and 1-hydroxybenzotriazole hydrate (HOBt) (50 mg, 0.322 mmol) were added followed by amine 10a (27 mg, 0.215 mmol) and diisopropylethylamine (225 μL, 1.290 mmol). The reaction mixture was stirred at 25 °C for 16 h then CH2Cl2 was added and the resulting solution was washed with saturated aqueous NH4Cl and water. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc/n-hexane 1:4) to furnish methyl 3-((tert-butoxycarbonyl)(methyl)amino)-5-(((2,5-dimethyloxazol-4-yl) methyl)carbamoyl)benzoate (80%). 1H NMR (400 MHz, CDCl3) δ 8.13 (t, 1H, J = 1.5 Hz), 8.03–8.00 (m, 1H), 7.93 (t, 1H, J = 1.8 Hz), 7.32 (t, 1H, J = 5.2 Hz), 4.38 (d, 2H, J = 5.3 Hz), 3.87 (s, 3H), 3.24 (s, 3H), 2.32 (s, 3H), 2.30 (s, 3H), 1.42 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 166.0, 165.9, 159.8, 154.3, 145.2, 144.5, 135.2, 130.8, 129.2, 128.5, 124.1, 81.2, 52.5, 37.0, 35.4, 28.3, 13.8, 10.1. To a solution of this latter (35 mg, 0.084 mmol) in dry THF (2 mL), NaH (60% suspension in mineral oil, 7 mg, 0.168 mmol) was added and the reaction mixture was stirred at 25 °C for 1 h. The reaction was quenched by adding water at 0 °C, diluted with CH2Cl2 and washed twice with water. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc/n-hexane 1:5) providing methyl methyl 3-((tert-butoxycarbonyl)(methyl)amino)-5-(((2,5-dimethyloxazol-4-yl) methyl)(methyl)carbamoyl)benzoate (68%). 1H NMR (400 MHz, CDCl3) δ 8.14–7.90 (m, 1H), 7.88–7.72 (m, 1H), 7.53 (s, 1H), 4.50 (s, 1H), 4.22 (s, 1H), 3.90 (s, 3H), 3.27 (s, 3H), 3.04 (s, 3H), 2.46–2.22 (m, 6H), 1.43 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 166.1, 159.4, 154.3, 146.3, 144.3, 137.0, 130.8, 130.4, 128.6, 124.7, 81.1, 52.5, 42.4, 37.8, 37.1, 28.4, 14.0, 10.2.
To a solution of the methyl ester (17 mg, 0.039 mmol) in a in THF/MeOH/H2O mixture (1.5 mL: 0.5 mL: 0.5 mL) at 0 °C was added LiOH·H2O (3 mg, 0.059 mmol). The mixture was stirred at 25 °C for 16 h. THF and MeOH were evaporated under reduced pressure and the residue was diluted with water, acidified with 1N HCl to pH = 2 and extracted with EtOAc. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure providing pure 12a (99%). 1H NMR (400 MHz, CDCl3) δ 8.07–7.92 (m, 1H), 7.80 (m, 1H), 7.53 (s, 1H), 4.50 (s, 1H), 4.22 (s, 1H), 3.27 (s, 3H), 3.04 (s, 3H), 2.44–2.25 (m, 6H), 1.43 (s, 9H).
4.2.2. 3-(((2,5-Dimethyloxazol-4-yl)methyl)(methyl) carbamoyl)-5-hydroxybenzoic acid (12b)
Compound 12b was prepared from 10a and 7d following the procedure described for 12a. 1H NMR (400 MHz, CDCl3) δ 7.61–7.34 (m, 2H), 7.06–6.80 (m, 1H), 4.42 (s, 1H), 4.19 (s, 1H), 2.95–2.84 (m, 3H), 2.41–2.16 (m, 6H).
4.2.3. 3-(Methyl((2-methyloxazol-4-yl)methyl)carbamoyl) benzoic acid (12c)
Compound 12c was prepared from 10b and 7a following the procedure described for 12a.1H NMR (400 MHz, CDCl3) δ 8.54 (t, 1H, J = 1.5 Hz), 8.07 (dt, 1H, J = 7.7 and 1.4 Hz), 8.02–7.97 (m, 1H), 7.52 (s, 1H), 7.40 (q, J = 7.9 Hz, 1H), 4.46 (s, 1H), 4.37 (s, 1H), 2.95 (m, 3H), 2.34 (s, 3H).
4.2.4. 3-(Methyl((2-methylthiazol-4-yl)methyl)carbamoyl) benzoic acid (12d)
Compound 12d was prepared from 10c and 7a following the procedure described for 12a.1H NMR (300 MHz, CDCl3) δ 11.83 (br, 1H), 8.20–8.06 (m, 2H), 7.64 (d, 1H, J = 6.8 Hz), 7.47–7.42 (m, 1H), 6.87 (s, 1H), 5.04 (s, 1H), 4.74 (s, 1H), 3.11–2.94 (m, 3H), 2.40–2.31 (m, 3 H); 13C NMR (75 MHz, CDCl3) δ 170.7, 168.6, 166.1, 151.6, 135.3, 131.7, 131.4, 130.6, 128.7, 128.5, 115.2, 48.3, 37.7, 16.4. LRMS-ESI (m/z): 313.0 [M+Na].
4.2.5. 4-(((2,5-Dimethyloxazol-4-yl)methyl)(methyl) carbamoyl)benzoic acid (12e)
Compound 12e was prepared from 10a and 9 following the procedure described for 12a. 1H NMR (400 MHz, CDCl3) δ 8.09 (d, 2H, J = 8.2 Hz), 7.69–7.63 (m, 1H), 7.49 (d, 1H, J = 7.2 Hz), 4.56 (s, 1H), 4.27 (s, 1H), 3.01–2.97 (m, 3H), 2.51–2.27 (m, 6H).
4.2.6. 2-(((2,5-Dimethyloxazol-4-yl)methyl)(methyl) carbamoyl)benzoic acid (12f)
Compound 12f was prepared from 10a and 8 following the procedure described for 12a. 1H NMR (400 MHz, CDCl3) δ 8.08–8.01 (m, 1H), 7.54 (q, 1H, J = 7.9 Hz), 7.46–7.36 (m, 1H), 7.28–7.22 (m, 1H), 4.61 (s, 1H), 4.06 (s, 1H), 2.79–2.72 (m, 3H), 2.45–2.34 (m, 6H).
4.2.7. 3-(Methyl((4-methyloxazol-2-yl)methyl)carbamoyl) benzoic acid (12g)
Compound 12g was prepared from 10d and 7a following the procedure described for 12a.1H NMR (400 MHz, CDCl3) δ 9.82 (br, 1H), 8.23 (s, 1H), 8.14–8.09 (m, 1H), 7.72–7.67 (m, 1H), 7.49 (t, 1H, J = 7.7 Hz), 7.40 (s, 1H), 4.89 (s, 1H), 4.55 (s, 1H), 3.18–3.03 (m, 3H), 2.19 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.9, 159.6, 136.2, 135.4, 135.2, 131.9, 131.4, 130.3 (2), 128.8, 128.6, 60.4, 37.9, 14.1.
4.2.8. 3-[(R)-2-(4-Methylthiazol-2-yl)pyrrolidine-1-carbonyl]benzoic acid (13a)
To a solution of isophthalic acid mono methyl ester 7a (133 mg, 0.74 mmol), EDCI (283 mg, 1.48 mmol) and HOBt (200 mg, 1.48 mmol) in anhydrous CH2Cl2 (8 mL) was added a solution of (R)-2-(4-methylthiazol-2-yl)pyrrolidine 11a (124 mg, 0.74 mmol) and diisopropylethylamine (0.77 mL, 4.44 mmol) in anhydrous CH2Cl2 (2 mL) at 0 °C under argon atmosphere and it was allowed to stir for 16 h at 23 °C. The reaction mixture was quenched with water and extracted with CH2Cl2. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH 9:1) to furnish 3-[(R)-2-(4-methylthiazol-2-yl)pyrrolidine-1-carbonyl]benzoic acid methyl ester (80%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 8.26 (s, 1H), 8.13 (d, 1H, J = 7.5 Hz), 7.80 (d, 1H, J = 7.5 Hz), 7.53 (t, 1H, J = 7.5 Hz), 6.80 (s, 1H), 5.68 (dd, 1H, J = 7.2 and 5.4 Hz), 3.94 (s, 3H), 3.82–3.74 (m, 1H), 3.57–3.49 (m, 1H), 2.51–2.35 (m, 2H), 2.45 (s, 3H), 2.17–2.03 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 171.5, 169.0, 166.1, 152.4, 136.6, 131.6, 131.0, 130.0, 128.5, 128.1, 112.7, 58.6, 52.1, 50.0, 32.1, 24.9, 17.0.
To a stirring solution of 3-[(R)-2-(4-methylthiazol-2-yl)pyrrolidine-1-carbonyl]benzoic acid methyl ester (190 mg, 0.58 mmol) in a THF/MeOH/H2O mixture (12 mL: 4 mL: 4 mL) at 0 °C was added solid LiOH·H2O (36 mg, 0.86 mmol), and the resulting solution was stirred at 23 °C for 16 h. After this period, the reaction mixture was evaporated until 1 mL and the mixture was extracted with toluene to remove organic impurities. The aqueous layer was cooled to 0 °C, acidified with 1N HCl to pH = 3, extracted with EtOAc, and dried over anhydrous Na2SO4. The solvent was evaporated to furnish title compound 13a (99%). 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 8.12 (d, 1H, J = 7.6 Hz), 7.78 (d, 1H, J = 7.6 Hz), 7.48 (t, 1H, J = 7.6 Hz), 6.77 (s, 1H), 5.65 (t, 1H, J = 6.8 Hz), 3.82–3.76 (m, 1H), 3.53–3.47 (m, 1H), 2.49–2.39 (m, 2H), 2.40 (s, 3H), 2.14–2.02 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 172.4, 169.6, 168.7, 152.3, 136.2, 132.0, 131.8, 130.9, 130.5, 128.6, 113.1, 58.7, 50.5, 32.8, 25.2, 16.6. LRMS-ESI (m/z): 338.9 [M+Na].
4.2.9. 3-[(S)-2-(4-Methylthiazol-2-yl)pyrrolidine-1-carbonyl] benzoic acid (13b)
Compound 13b was prepared from 11b and 7a following the procedure described for 13a. 1H NMR and 13C NMR data are identical to those reported for compound 13a.
4.2.10. (R)-5-(2-(4-methylthiazol-2-yl)pyrrolidine-1-carbonyl) nicotinic acid (13c)
Compound 13c was prepared from 11a and 7b following the procedure described for 13a. 1H NMR (400 MHz, CD3OD) δ 9.22 (s, 1H), 8.90 (s, 1H), 8.54 (s, 1H), 6.88 (s, 1H), 5.58–5.54 (m, 1H), 3.90–3.82 (m, 1H), 3.60–3.54 (m, 1H), 2.52–2.40 (m, 4H), 2.25–2.08 (m, 2H), 2,02–1.96 (m, 1H); 13C NMR (100 MHz, CD3OD) δ 173.5, 172.9, 168.6, 153.8, 153.3, 152.3, 137.8, 137.1, 115.5, 114.6, 60.6, 51.7, 34.2, 26.3, 17.7. LRMS-ESI (m/z): 340.0 [M+Na].
4.2.11. (R)-(3-((2-(4-methylthiazol-2-yl)pyrrolidin-1-yl) methyl)phenyl)methanol (14)
To a solution of 13a (146 mg, 0.442 mmol) in dry THF (8 mL) LiAlH4 (42 mg, 1.106 mmol) was added portionwise at 0 °C and the reaction mixture was stirred at reflux temperature for 15 h. The reaction was quenched with MeOH and filtered through a Celite® pad. The filtrate was concentrated and the residue was purified by silica gel column chromatography (EtOAc/n-hexane 1:1) affording compound 14 (88%). 1H NMR (300 MHz, CDCl3) δ 7.38–7.27 (m, 4H), 6.83 (s, 1H), 4.72 (s, 2H), 4.08–3.97 (m, 2H), 3.41–3.36 (m, 1H), 3.10–3.05 (m, 1H), 2.44 (s, 3H), 2.38–2.28 (m, 2H), 1.95–1.79 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 177.5, 152.1, 141.2, 139.0, 128.2, 127.6, 127.1, 125.5, 113.2, 65.6, 64.7, 58.6, 53.2, 34.2, 22.9, 16.9.
4.2.12. (R)-3-((2-(4-methylthiazol-2-yl)pyrrolidin-1-yl)methyl) benzoic acid (15)
To a solution of alcohol 14 (112 mg, 0.388 mmol) in dry CH2Cl2 (4 mL) MnO2 (1.18 g, 12.86 mmol) was added at 0 °C and the rection mixture was stirred for 5 h at 25 °C. The reaction mixture was then filtered through a Celite® pad. The filtrate was concentrated and the residue was purified by silica gel column chromatography (EtOAc/n-hexane 1:3) affording (R)-3-((2-(4-methylthiazol-2-yl)pyrrolidin-1-yl)methyl)benzaldehyde. 1H NMR (300 MHz, CDCl3) δ 9.97 (s, 1H), 7.84 (s, 1H), 7.71 (d, 1H, J = 7.6 Hz), 7.62 (d, 1H, J = 7.6 Hz), 7.43 (t, 1H, J = 7.6 Hz), 6.76 (s, 1H), 4.09–3.97 (m, 2H), 3.43–3.39 (m, 1H), 3.03–2.98 (m, 1H), 2.37 (s, 3H), 2.36–2.25 (m, 2H), 1.92–1.74 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 192.4, 176.8, 152.4, 140.4, 136.4, 134.6, 129.6, 128.9, 128.4, 113.2, 66.1, 58.3, 53.4, 34.2, 23.1, 17.1.
To a solution of the aldehyde (72 mg, 0.251 mmol) in 3-methyl-2-butene (4 mL) and t-BuOH (2 mL), a solution of NaClO2 (284 mg, 3.140 mmol) and NaH2PO4 (208 mg, 1.5 mmol) in water (1 mL) was added. The reaction mixture was stirred for 4 h at 25 °C then acidified to pH = 3 with 10% HCl and extracted with EtOAc. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc/ n-hexane 1:1) providing compound 15 (45% over 2 steps). 1H NMR (300 MHz, CDCl3) δ 8.13 (s, 1H), 8.03 (d, 1H, J = 7.5 Hz), 7.67 (d, 1H, J = 7.5 Hz), 7.66–7.42 (m, 2H), 6.86 (s, 1H), 4.19–4.10 (m, 2H), 3.51 (d, 1H, J = 12.9 Hz), 3.16–3.09 (m, 1H), 2.49–2.32 (m, 5H), 1.96–1.87 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 177.8, 170.7, 152.3, 139.4, 133.5, 130.6, 130.2, 128.9, 128.4, 113.6, 65.8, 58.6, 53.5, 34.4, 23.2, 17.0.
4.2.13. (R)-3-(4-(4-methylthiazol-2-yl)thiazolidine-3-carbonyl) benzoic acid (16a)
Compound 16a was prepared from 11d and 7a following the procedure described for 13a 1H NMR (400 MHz, CDCl3) δ 8.79 (br, 1H), 8.28–8.05 (m, 2H), 7.80–7.32 (m, 2H), 6.86 (s, 1H), 6.10 (br, 1H), 4.84 (d, 2H, J = 8.9 Hz), 3.66–3.56 (m, 2H), 2.59–2.30 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 168.9, 166.0, 153.0, 135.8, 131.7, 130.5, 128.8, 128.3, 113.8, 52.3, 39.6, 17.0.
4.2.14. (R)-3-(2-(4-methyloxazol-2-yl)pyrrolidine-1-carbonyl) benzoic acid (16b)
Compound 16b was prepared from 11e and 7a following the procedure described for 13a. 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 8.13–8.00 (m, 1H), 7.77 (d, 1H, J = 7.5 Hz), 7.48 (t, 1H, J = 7.7 Hz), 7.34–7.20 (m, 2H), 5.36 (t, 1H, J = 6.5 Hz), 3.89–3.83 (m, 1H), 3.56–3.50 (m, 1H), 2.46–2.39 (m, 1H), 2.34–2.04 (m, 5H), 1.99–1.91 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 169.3, 168.4, 164.8, 136.2, 135.9, 131.7, 130.8, 130.0, 128.7, 128.4, 127.4, 54.8, 50.1, 31.2, 25.3, 10.9.
4.2.15. (R)-3-(2-(4-methylthiazol-2-yl)piperidine-1-carbonyl) benzoic acid (16c)
Compound 16c was prepared from 11c and 7a following the procedure described for 13a. 1H NMR (300 MHz, CDCl3) δ 11.45 (br, 1H), 8.24 (s, 1H), 8.12 (d, 1H, J = 7.5 Hz), 7.57–7.48 (m 1H), 6.90 (s 1H), 6.27 (br, 1H), 5.16 (br, 0.5H), 4.69 (br, 0.5H), 3.73–3.61 (m, 1H), 3.39–3.25 (m, 1H), 2.66–2.60 (m, 1H), 2.44 (s, 3H), 2.06–2.00 (m, 1H), 1.78–1.57 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 170.2, 169.6, 168.6, 152.3, 135.5, 131.0 (2), 130.2, 128.5, 127.0 113.9, 60.2, 51.3, 44.9, 28.0, 25.4, 19.6, 16.6. LRMS-ESI (m/z): 353.0 [M+Na].
4.2.16. Benzyl (R)-5-oxo-1-((R)-1-phenylethyl)pyrrolidine-3-carboxylate (18a) and benzyl (S)-5-oxo-1-((R)-1-phenylethyl)pyrrolidine-3-carboxylate (18b)
To a solution of dimethyl itaconate 17 (3 g, 18.97 mmol) in MeOH (4 mL) was added (R)-α-methylbenzylamine (3.14 mL, 26.66 mmol). The solution was stirred for 1 h at 80 °C and for 18 h at 115 °C. The reaction mixture was then concentrated under reduced pressure and water was added. The aqueous solution was adjusted to pH = 2 with 10% HCl and extracted with CH2Cl2. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc/pentane 3:7 to 1:1) providing isolated diastereoisomers methyl (R)-5-oxo-1-((R)-1-phenylethyl)pyrrolidine-3-carboxylate (32%) and methyl (S)-5-oxo-1-((R)-1-phenylethyl)pyrrolidine-3-carboxylate (43%).
The isolated methyl esters (219 mg, 0.886 mmol) were dissolved in dry toluene (1.8 mL). Benzyl alcohol (140 μL, 1.328 mmol) and PPTS (45 mg, 0.179 mmol) were sequentially added and the reaction mixture was stirred under reflux for 24 h under argon atmosphere. The reaction mixture was quenched with saturated aqueous NaHCO3 and extracted with EtOAc. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc/n-hexane 1:2).
18a (73% yield): 1H NMR (300 MHz, CDCl3) δ 7.40–7.27 (m, 10H), 5.55 (q, 1H, J = 6.9 Hz), 5.12 (s, 2H), 3.64–3.55 (m, 1H), 3.33–3–22 (m, 2H), 2.87–2.69 (m, 2H), 1.58 (d, 3H, J = 7.2 Hz); 13C NMR (75 MHz, CDCl3) δ 172.5 172.0, 139.6, 135.3, 128.7 (2), 128.5 (2), 128.3 (2), 127.7 (2), 127.1 (2), 67.1, 49.2, 44.6, 36.2, 34.4, 16.3.
18b (81% yield): 1H NMR (300 MHz, CDCl3) δ 7.48–7.32 (m, 10H), 5.56 (q, 1H, J = 6.9 Hz), 5.22 (s, 2H), 3.65–3.57 (m, 1H), 3.26–3–15 (m, 2H), 2.89–2.70 (m, 2H), 1.58 (d, 3H, J = 7.2 Hz); 13C NMR (75 MHz, CDCl3) δ 172.7, 171.8, 139.8, 135.4, 128.7 (3), 128.4 (2), 127.7 (2), 127.2 (2), 67.2, 49.2, 44.4, 36.2, 34.6, 16.0.
4.2.17. Benzyl (R)-1-((R)-1-phenylethyl)-5-thioxopyrrolidine-3-carboxylate (19a)
To a solution of lactam 18a (2 g, 6.184 mmol) in dry toluene (39 mL) Lawesson’s reagent (2.0 g, 4.945 mmol) was added and the reaction mixture was stirred for 3 h at 100 °C and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc/n-hexane 1:6) providing 19a (67%). 1H NMR (400 MHz, CDCl3) δ 7.39–7.22 (m, 8H), 7.07–6.99 (m, 2H), 6.34 (q, 1H, J = 7.0 Hz), 5.05 (m, 2H), 3.88–3.84 (m, 1H), 3.79–3.74 (m, 1H), 3.55–3.51 (m, 1H), 3.39–3.34 (m, 1H), 3.29–2.20 (m, 1H), 1.59 (d, 3H, J = 7.1 Hz); 13C NMR (100 MHz, CDCl3) δ 198.1, 171.7, 138.0 135.0, 128.8 (2), 128.4 (2), 128.3, 128.0, 127.2, 126.9, 67.1, 54.1, 53.9, 51.2, 47.5, 37.6, 37.4, 15.2.
4.2.18. Benzyl (S)-1-((R)-1-phenylethyl)-5-thioxopyrrolidine-3-carboxylate (19b)
Compound 19b was prepared following the procedure described for 19a (99%). 1H NMR (400 MHz, CDCl3) δ 7.41–7.29 (m, 10H), 6.34 (q, 1H, J = 7.1 Hz), 5.14 (s, 2H), 3.90–3.85 (m, 1H), 3.43–3.32 (m, 3H), 3.19–3.10 (m, 1H), 1.57 (d, 3H, J = 7.0 Hz); 13C NMR (100 MHz, CDCl3) δ 197.7, 171.9, 138.3, 135.1, 128.7 (2), 128.6 (2), 128.5 (2), 128.3 (2), 128.1, 127.1, 67.2, 54.0, 50.9, 47.6, 37.6, 14.7.
4.2.19. Benzyl (R)-1-((R)-1-phenylethyl)pyrrolidine-3-carboxylate (20a)
To a solution of thioamide 19a (1.44 g, 4.242 mmol), in dry CH2Cl2 (21 mL) MeI (2.6 mL, 42.40 mmol) was added and the solution was stirred for 24 h at 25 °C. The reaction mixture was then concentrated and the residue dissolved in EtOH (7 mL). NaBH4 (1.4 g, 37.00 mmol) was added portionwise at 0 °C and the reaction mixture was stirred at 0 °C for 1 h. The reaction was quenched with water and extracted with CH2Cl2. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc/pentane 1:6) providing 20a (38%). 1H NMR (300 MHz, CDCl3) δ 7.58–7.32 (m, 10H), 5.27–5.18 (m, 2H), 3.38–3.31 (m, 1H), 3.20–3.09 (m, 1H), 2.97–2.86 (m, 2H), 2.77–2.72 (m, 1H), 2.29–2.17 (m, 2H), 1.50 (d, 3H, J = 7.2 Hz); 13C NMR (75 MHz, CDCl3) δ 174.9, 145.5, 136.2, 128.7 (2), 128.5 (2), 128.3, 128.2 (2), 127.2 (2), 127.1, 66.4, 65.4, 55.9, 52.4, 42.2, 27.7, 23.3.
4.2.20. Benzyl (S)-1-((R)-1-phenylethyl)pyrrolidine-3-carboxylate (20b)
Compound 20b was prepared following the procedure described for 20a (50%). 1H NMR (300 MHz, CDCl3) δ 7.51–7.31 (m, 10H), 5.25 (s, 1H), 3.35 (q, 1H, J = 6.3 Hz), 3.25–3.15 (m, 1H), 3.11–3.05 (m, 1H), 2.81–2.76 (m, 1 H), 2.72–2.54 (m, 2H), 2.27–2.12 (m, 2H), 1.50 (d, 3H, 6.3 Hz); 13C NMR (75 MHz, CDCl3) δ 175.0, 145.4, 136.2, 128.7 (2), 128.5 (2), 128.3, 128.2 (2), 127.2 (2), 127.1, 66.5, 65.5, 55.6, 52.7, 42.2, 27.7, 23.3.
4.2.21. Benzyl (R)-3-carbamoylpyrrolidine-1-carboxylate (21a)
To a solution of 20a (500 mg, 1.616 mmol) in MeOH (13 mL) 10% Pd/C (179 mg) was added and the mixture was allowed to stir for 20 h at 25 °C under H2 atmosphere. The reaction was filtered on Celite® and the filtrate was concentrated under reduced pressure quantitatively providing (R)-pyrrolidine-3-carboxylic acid submitted to the next step without further purification. To a solution of this latter (460 mg, 3.995 mmol) in 2N NaOH (2 mL) benzylchloroformate (740 μL, 5.194 mmol) and 4N NaOH (1.5 mL) were added and the mixture was stirred at 0 °C for 2 h. The reaction was extracted with diethyl ether. The aqueous layer was adjusted to pH = 2 with 10% HCl at 0 °C and extracted with EtOAc. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH 9:1) affording (R)-1-((benzyloxy)carbonyl)pyrrolidine-3-carboxylic acid (39%). 1H NMR (300 MHz, CDCl3) δ 9.17 (br, 1H), 7.44–7.32 (m, 5H), 5.18 (s, 2H), 4.73 (s, 1H), 3.73–3.47 (m, 3H), 3.19–3.09 (m, 1H), 2.24–2.13 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 177.0, 155.0, 136.6, 129.6, 128.6, 128.1, 127.6, 127.1, 67.2, 48.3/47.9, 45.7/45.3, 43.1/42.3, 28.8/28.2.
To a solution of this latter (307 mg, 1.231 mmol) in 1,4-dioxane (4 mL) pyridine (62 μL, 0.763 mmol), di-tert-butyldicarbonate (370 mg, 1.695 mmol) and NH4HCO3 (127 mg, 1.606 mmol) were sequentially added. The reaction mixture was stirred at 25 °C for 16 h and then concentrated under reduced pressure. The residue was diluted with EtOAc. The organic layer was washed with water, 5% H2SO4 and brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue containing 21a was used for the next step without further purification. 1H NMR (300 MHz, DMSO-d6) δ 7.43 (m, 1H), 7.34 (m, 4H), 7.30 (br, 1H), 6.94 (br, 1H), 5.04 (s, 1 H), 3.51–3.21 (m, 4 H), 2.93–2.87 (m, 1 H), 2.00–1.88 (m, 2 H).
4.2.22. Benzyl (S)-3-carbamoylpyrrolidine-1-carboxylate (21b)
Compound 21b was prepared following the procedure described for 21a 1H NMR data are identical to those reported for compound 21a.
4.2.23. (R)-4-methyl-2-(pyrrolidin-3-yl)thiazole (11g)
To a solution of amide 21a (254 mg, 1.023 mmol) in THF (5 mL) Lawesson’s reagent (228 mg, 0.564 mmol) was added portionwise at 0 °C and the reaction mixture was allowed to stir at 25 °C for 16 h. The reaction was concentrated under reduced pressure, the residue was diluted with saturated aqueous NaHCO3 and extracted with diethyl ether. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue containing (R)-benzyl 3-carbamothioylpyrrolidine-1-carboxylate was used for the next step without further purification. Accordingly, this latter (109 mg, 0.412 mmol) was dissolved in EtOH (2 mL), chloroacetone (49 μL, 0.618 mmol) was added at 25 °C and the resulting solution was allowed to stir at 80 °C for 18 h. The reaction mixture was the cooled to 25 °C and saturated aqueous NaHCO3 was added. EtOH was evaporated under reduced pressure and the aqueous layer was extracted with EtOAc. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc/ n-hexane 1:3) affording benzyl (R)-3-(4-methylthiazol-2-yl)pyrrolidine-1-carboxylate. 1H NMR (400 MHz, CDCl3) δ 7.35–7.26 (m, 5H), 6.74 (s, 1H), 5.16–5.09 (m, 2H), 3.95–3.89 (m, 1H), 3.76–3.57 (m, 3H), 3.51–3.44 (m, 1H), 2.39 (s, 3H), 2.37–2.33 (m, 1H), 2.26–2.15 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 169.6, 154.5, 152.5, 136.7, 136.6, 128.4, 127.9, 127.8 (2), 112.6, 66.8, 51.9/51.4, 45.7/45.3, 42.5/41.7, 32.8/32.0, 17.0. This latter was submitted to Cbz deprotection as described for amine 11f providing compound 11g. 1H NMR (400 MHz, CD3OD) δ 6.96 (s, 1H), 3.82–373 (m, 1H), 3.49–3.44 (m, 1H), 3.32–3.26 (m, 1H), 3.19–3.11 (m, 2H), 2.53–2.44 (m, 4H), 2.19–2.06 (m, 1H); 13C NMR (100 MHz, CD3OD) δ 173.3, 152.2, 112.7, 53.6, 46.5, 43.2, 33.9, 16.4. LRMS-ESI (m/z): 169.0 [M+H].
4.2.24. (S)-4-methyl-2-(pyrrolidin-3-yl)thiazole (11h)
1H NMR and 13C NMR data are identical to those reported for compound 11g.
4.2.25. (R)-3-(3-(4-methylthiazol-2-yl)pyrrolidine-1-carbonyl) benzoic acid (22a)
Compound 21a was prepared from 11g and 7a following the procedure described for 13a. 1H NMR (400 MHz, CDCl3) δ 12.0 (br, 1H), 8.20–8.03 (m, 2H), 7.72 (s, 1H), 7.46–7.42 (m, 1H), 6.90–6.83 (m, 1H), 4.20–3.63 (m, 5H), 2.46–2.22 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 169.4, 169.2, 168.9, 152.4, 136.5, 132.1, 131.9, 131.6, 130.1, 128.7, 113.0, 54.6, 45.7, 42.2, 31.3, 16.2. LRMS-ESI (m/z): 339.0 [M+Na].
4.2.26. (S)-3-(3-(4-methylthiazol-2-yl)pyrrolidine-1-carbonyl) benzoic acid (22b)
Compound 22b was prepared from 11h and 7a following the procedure described for 13a 1H NMR and 13C NMR data are identical to those reported for compound 22a.
4.2.27. Benzyl (R)-2-(4-(hydroxymethyl)oxazol-2-yl) pyrrolidine-1-carboxylate (24)
To a solution of ester 23 (1.4 g, 4.238 mmol) in dry THF (10 mL) LiBH4 (277 mg, 12.72 mmol) was added portionwise at 0 °C and the reaction mixture was allowed to stir at 25 °C for 4 h. EtOAc (5 mL) was added and the resulting solution was cooled to °C. 1N HCl (10 mL) and water (20 mL) were then added and the reaction mixture was extracted with EtOAc. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH 97:3) providing compound 24 (42%). 1H NMR (300 MHz, CDCl3) δ 7.56–7.21 (m, 5H), 5.25–5.05 (m, 3H), 4–85–4.80 (m, 1H), 4.61–4.57 (m, 1H), 3.79–3.72 (m, 1H), 3.67–3.55 (m, 1H), 2.32–1.85 (m, 5H); 13C NMR (75 MHz, CDCl3) δ 164.6, 154.5, 140.5, 136.5, 128.5, 128.4, 128.0, 127.9, 127.7, 127.2, 66.9, 55.1, 54.7, 46.9, 32.4, 24.3.
4.2.28. (R)-4-(methoxymethyl)-2-(pyrrolidin-2-yl)oxazole (11f)
To a solution of alcohol 24 (138 mg, 0.456 mmol) in dry THF (4 mL) NaH (60% suspension in mineral oil, 33 mg, 0.825 mmol) and MeI (34 μL, 0.547 mmol) were sequentially added at 0 °C and the reaction mixture was allowed to stir at 25 °C for 1 h. The reaction was quenched with saturated aqueous NH4Cl at 0 °C and extracted with EtOAc. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc/n-hexane 3:2) affording benzyl (R)-2-(4-(methoxymethyl)oxazol-2-yl)pyrrolidine-1-carboxylate (68%). 1H NMR (400 MHz, CDCl3) δ 7.50 (s, 1H), 7.41 (s, 1H), 7.40–7.29 (m, 3H), 7.23–7.13 (m, 1H), 5.16–4.94 (m, 3H), 4.31 (d, 2H, J = 17.4 Hz), 3.70–3.65 (m, 1H), 3.58–3.43 (m, 1H), 3.37 (s, 3H), 2.27–2.00 (m, 3H), 1.93–1.88 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 164.7, 154.7/154.3, 137.7, 136.6/136.4, 136.0/135.7, 128.3, 128.2, 127.8, 127.6, 127.5, 66.9/66.7, 66.3/66.2, 58.4, 54.9/54.6, 46.8/46.4, 32.3/31.2, 24.1/23.3.
This latter (98 mg, 0.310 mmol) was dissolved in a solution of HBr in acetic acid (1.0 mL) and the resulting mixture was stirred at 25 °C for 1 h. The addition of ether (2 mL) caused the amine salt to precipitate. The solvent was drawn off and the resulting residue retained. To the etheral solution was added petroleum ether (4 mL) to give a further precipitate. The solvent was evaporated under reduced pressure and the two batches of solid were combined and dried to provide amine 11f (67%). 1H NMR (400 MHz, CDCl3) δ 7.48 (s, 1H), 4.28 (m, 3H), 3.35 (s, 3H), 3.09–3.03 (m, 1H), 2.96–2.90 (m, 1H), 2.54 (br, 1H), 2.16–2.07 (m, 1H), 2.02–1.94 (m, 1H), 1.89–1.71 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 166.7, 137.2, 136.0, 66.1, 58.3, 55.4, 46.7, 30.7, 25.3.
4.2.29. (R)-3-(2-(4-(methoxymethyl)oxazol-2-yl)pyrrolidine-1-carbonyl)benzoic acid (25)
Compound 25 was prepared from 11f and 7a following the procedure described for 13a. 1H NMR (400 MHz, CDCl3) δ 9.39 (br, 1H), 8.13–8–08 (m, 1H), 8.00 (s, 1H), 7.79–7.75 (m, 1H), 7.57 (s, 1H), 7.49–7.45 (m, 1H), 5.40 (t, 1H, J = 6.3 Hz), 4.44 (s, 2H), 3.93–3.78 (m, 2H), 3.56–3.51 (m, 1H), 3.37 (s, 3H), 2.44–2.36 (m, 1H), 2.28–2.12 (m, 2H), 1.98–1.90 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 169.2, 168.6, 164.8, 137.5, 136.5, 136.1, 132.0, 131.7, 131.0, 130.3, 128.7, 66.0, 58.4, 54.8, 50.1, 31.0, 25.2.
4.2.30. N1-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl) sulfonamido)-1-phenylbutan-2-yl)-N3-methyl-N3-((2-methyloxazol-4-yl)methyl)isophthalamide (3c)
1H NMR (400 MHz, CDCl3) δ 8.08 (m, 1H), 8.17–8.04 (m, 1H), 8.01–7.91 (m, 1H), 7.77 (d, 1H, J = 7.8 Hz,), 7.73–7.61 (m, 3H), 7.43 (t, J = 7.7 Hz, 2H), 7.47–7.04 (m, 4H), 7.34–7.02 (m, 2H), 7.10–6.75 (m, 2H), 4.56 (s, 1H), 4.46 (d, J = 5.4 Hz, 1H), 4.42–4.21 (m, 2H), 4.07–3.94 (m, 1H), 3.89–3.78 (m, 3H), 3.21–2.70 (m, 5H), 2.49 2.35 (m, 2H), 2.10 (br, 1H), 1.92–1.78 (m, 2H), 1.66–1.50 (m, 1H), 0.84 (dd, J = 6.4, 1.9 Hz, 6H). LRMS-ESI (m/z): 685.5 [M+Na]. HRMS-ESI (m/z) [M+Na]+ calculated for C35H42N4O7SNa 685.2672, found 685.2650.
4.2.31. N1-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-N3-methyl-N3-((2-methylthiazol-4-yl)methyl)isophthalamide (3d)
1H NMR (300 MHz, CDCl3) δ 8.07 (s, 1H), 7.99 (s, 1H), 7.96 (s, 1H), 7.77–7.59 (m, 4H), 7.49–7.40 (m, 1H), 7.27–7.16 (m, 3H), 6.97–6.82 (m 3H), 6.54 (d, 1H, J = 7.8 Hz), 4.96 (s, 1H), 4.88 (d, 1H, J = 5.4 Hz), 4,38–4.18 (m, 2H), 4.03–3.97 (m, 1H), 3.86 (s, 3H), 3.17–2.98 (m, 4H), 2.87 (d, 2H, J = 6.6 Hz), 2.47–2.39 (m, 2H), 1.91–1.83 (m, 1H), 0.86 (dd, J = 6.4, 1.9 Hz, 6H). LRMS-ESI (m/z): 701.4 [M+Na]. HRMS-ESI (m/z) [M+Na]+ calculated for C35H42N4O6S2Na 701.2444, found 701.2438.
4.2.32. N1-((2,5-dimethyloxazol-4-yl)methyl)-N3-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-N1-methyl-5-(methylamino)isophthalamide (3e)
1H NMR (400 MHz, CDCl3) δ 7.81–7.60 (m, 3H), 7.50 (s, 1H), 7.38–7.03 (m, 5H), 6.96 (d, 2H, J = 8.7 Hz,), 6.85–6.61 (m, 1H), 6.38 (br, 1H), 4.93 (s, 1H), 4.49 (s, 1H), 4.42–4.15 (m, 2H), 4.16–3.90 (m, 3H), 3.86 (s, 3H), 3.41–2.66 (m, 8H), 2.44–2.24 (m, 6H), 2.16 (s, 3H), 1.99–1.80 (m, 1H), 0.86 (dd, 6H, J = 12.9 and 6.8 Hz). LRMS-ESI (m/z): 728.5 [M+Na]. HRMS-ESI (m/z) [M+Na]+ calculated for C37H47N5O7SNa 728.3096, found 728.3088.
4.2.33. N1-((2,5-dimethyloxazol-4-yl)methyl)-5-hydroxy-N3-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl) sulfonamido)-1-phenylbutan-2-yl)-N1-methylisophthalamide (3f)
1H NMR (400 MHz, CDCl3) δ 8.01 (s, 1H), 7.77–7.61 (m, 3H), 7.36–6.85 (m, 8H), 6.72 (br, 1H), 4.64–3.92 (m, 2H), 3.84 (s, 3H), 3.36–2.71 (m, 12H), 2.52–2.24 (m, 3H), 2.20–1.68 (m, 2H), 0.97–0.72 (m, 6H). LRMS-ESI (m/z): 715.5 [M+Na]. HRMS-ESI (m/z) [M+Na]+ calculated for C36H44N4O8SNa 715.2778, found 715.2773.
4.2.34. N1-((2,5-dimethyloxazol-4-yl)methyl)-N2-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-N1-methylphthalamide (3g)
1H NMR (400 MHz, CDCl3) δ 7.72 (d, 2H, J = 8.8 Hz,), 7.61–7.23 (m, 7H), 7.23–7.12 (m, 2H), 7.03 (d, 1H, J = 8.3 Hz,), 6.94 (d, 2H, J = 8.7 Hz), 4.55 (br, 1H), 4.49–4.28 (m, 2H), 4.22 (br, 1H), 4.06 (m, 2H), 3.88–3.79 (m, 3H), 3.24–3.01 (m, 3H), 3.01–2.82 (m, 4H), 2.72 (s, 3H), 2.38 (s, 3H), 2.12–1.80 (m, 2H), 0.91–0.79 (m, 6H). LRMS-ESI (m/z): 699.6 [M+Na]. HRMS-ESI (m/z) [M+H]+ calculated for C36H45N4O7S 677.3009, found 677.2994.
4.2.35. N1-((2,5-dimethyloxazol-4-yl)methyl)-N4-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-N1-methylterephthalamide (3h)
1H NMR (400 MHz, CDCl3) δ 7.65 (d, 2H, J = 8.9 Hz), 7.62–7.48 (m, 2H), 7.35 (d, 1H, J = 7.8 Hz), 7.31–7.21 (m, 4H), 7.21–7.12 (m, 1H), 6.91 (d, 2H, J = 8.9 Hz), 6.75–6.57 (m, 1H), 4.49 (s, 1H), 4.42–4.25 (m, 2H), 4.13 (s, 1H), 3.98 (dd, 1H, J = 7.5 and 3.6 Hz,), 3.83 (s, 3H), 3.23–2.89 (m, 6H), 2.84 (d, 2H, J = 7.5 Hz), 2.35 (m, 5H), 2.02 (s, 1H), 1.90–1.80 (m, 1H), 0.83 (dd, 6H, J = 6.5 and 3.3 Hz). LRMS-ESI (m/z): 699.6 [M+Na]. HRMS-ESI (m/z) [M+Na]+ calculated for C36H44N4O7SNa 699.2829, found 699.2820.
4.2.36. N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-3-((S)-2-(4-methylthiazol-2-yl)pyrrolidine-1-carbonyl)benzamide (4a)
To a solution of 3-[(S)-2-(4-methylthiazol-2-yl)pyrrolidine-1-carbonyl]benzoic acid 13b (40 mg, 0.13 mmol), EDCI (37.4 mg, 0.20 mmol) HOBt (26.4 mg, 0.20 mmol) in anhydrous CH2Cl2 (3 mL) a solution of amine 6a (52 mg, 0.13 mmol) and diisopropylethylamine (0.14 mL, 0.78 mmol) in anhydrous CH2Cl2 (1 mL) were added at 0 °C under argon atmosphere and it was allowed to stir for 16 h at 23 °C. The reaction mixture was quenched with water and extracted with CH2Cl2. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH 9:1) to furnish inhibitor 4a (95%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.81 (s, 1H), 7.64–7.59 (m, 3H), 7.36 (t, 1H, J = 7.7 Hz), 7.29–7.09 (m, 7H), 6.91 (d, 2H, J = 8.7 Hz), 6.72 (s, 1H), 5.59 (dd, 1H, J = 7.4 and 4.8 Hz), 4.38–4.32 (m, 2H), 4.02–3.97 (m, 1H), 3.81 (m, 4H), 3.67–3.62 (m, 1H), 3.43–3.37 (m, 1H), 3.24–3.19 (m, 1H), 3.15–2.98 (m, 3H), 2.92–2.80 (m, 2H), 2.38 (s, 3H), 2.35–2.27 (m, 2H), 2.08–1.98 (m, 1H), 1.90–1.81 (m, 2H), 0.83 (dd, 6H, J = 6.5 and 3.2 Hz); 13C NMR (100 MHz, CDCl3) δ 171.7, 171.1, 169.2, 167.1, 162.8, 152.5, 138.0, 136.7, 134.3, 130.2, 129.8, 129.3 (2), 129.2, 128.6, 128.4 (2), 126.4, 125.8, 114.3 (2), 112.9, 72.6, 58.8, 58.5, 55.5, 54.7, 53.2, 50.1, 34.8, 32.4, 27.0, 24.9, 20.0, 19.9, 17.1. LRMS-ESI (m/z): 727.6 [M+Na]. HRMS-ESI (m/z) [M+H]+ calculated for C37H45N4O6S2 705.2781, found 705.2784.
4.2.37. N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-3-((R)-2-(4-methylthiazol-2-yl)pyrrolidine-1-carbonyl)benzamide (4b)
1H NMR (400 MHz, CDCl3) δ 7.79 (s, 1H), 7.63 (d, 2H, J = 8.7 Hz), 7.60 (d, 1H, J = 7.7 Hz), 7.35 (t, 1H, J = 7.7 Hz), 7.30–7.11 (m, 6H), 6.90 (d, 2H, J = 8.7 Hz), 6.73 (s, 1H), 5.60 (dd, 1H, J = 7.3 and 4.9 Hz), 4.38–4.31 (m, 1H), 4.01–3.96 (m, 1H), 3.80 (s, 3H), 3.63–3.58 (m, 1H), 3.41–3.35 (m, 1H), 3.20 (dd, 1H, J = 15.0 and 3.0 Hz), 3.12–2.96 (m, 3H), 2.91–2.79 (m, 2H), 2.38 (s, 3H), 2.36–2.25 (m, 2H), 2.07–1.99 (m, 1H), 1.91–1.81 (m, 2H), 0.83 (dd, 6H, J = 6.4 and 2.8 Hz); 13C NMR (100 MHz, CDCl3) δ 171.7, 169.3, 167.1, 162.8, 152.6, 137.9, 136.6, 134.4, 130.1, 129.7, 129.3, 128.6, 128.4, 126.3, 125.7, 114.2, 112.8, 72.7, 58.7, 58.4, 55.5, 54.5, 53.3, 50.1, 35.0, 32.3, 27.0, 24.9, 20.0, 19.9, 17.0. LRMS-ESI (m/z): 705.7 [M+H]. HRMS-ESI (m/z) [M+Na]+ calculated for C37H44N4O6S2Na 727.2600, found 727.2592.
4.2.38. N-((2S,3R)-4-((4-amino-N-isobutylphenyl) sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)-3-((R)-2-(4-methylthiazol-2-yl)pyrrolidine-1-carbonyl)benzamide (4c)
1H NMR (400 MHz, CDCl3) δ 7.79 (s, 1H), 7.62–7.53 (m, 2H), 7–39–7.32 (m, 2H), 7.27–7.12 (m, 6H), 6.88 (d, 1H, J = 8.6 Hz), 6.74 (s, 1H), 6.49 (d, 2H, J = 8.6 Hz), 5.61 (dd, 1H, J = 7.5 and 4.8 Hz), 4.35–4.30 (m, 3H), 4.03–3.97 (m, 1H), 3.63–3.57 (m, 1H), 3.42–3.36 (m, 1H), 3.17–3.08 (m, 2H), 3.00–2.84 (m, 3H), 2.76–2.71 (m, 1H), 2.40–2.24 (m, 5H), 2.06–1.99 (m, 1H), 1.92–1.82 (m, 1H), 0.85 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 171.7, 169.4, 167.0, 152.6, 151.1, 137.9, 136.7, 134.5, 130.0, 129.3 (3), 128.7 (3), 128.4 (2), 126.4, 125.9, 125.0, 113.9 (2), 112.9, 72.8, 58.8, 54.3, 53.6, 50.1, 35.4, 32.4, 29.6, 27.0, 24.9, 20.0 (2), 17.1. LRMS-ESI (m/z): 690.7 [M+H]. HRMS-ESI (m/z) [M+Na]+ calculated for C36H43N5O5S2Na 712.2604, found 712.2601.
4.2.39. N-((2S,3R)-3-hydroxy-4-((4-(hydroxymethyl)-N-isobutylphenyl)sulfonamido)-1-phenylbutan-2-yl)-3-((R)-2-(4-methylthiazol-2-yl)pyrrolidine-1-carbonyl)benzamide (4d)
1H NMR (400 MHz, CDCl3) δ 7.77 (s, 1H), 7.72–7.56 (m, 3H), 7.37–7.14 (m, 9H), 6.95 (d, 1H, J = 8.6 Hz), 6.75 (s, 1H), 5.57 (dd, 1H, J = 7.5 and 4.9 Hz), 4.68–4.58 (m, 2H), 4.31–4.23 (m, 1H), 4.02–3.95 (m, 1H), 3.64–3.57 (m, 1H), 3.42–3.36 (m, 1H), 3.21–2.72 (7H), 2.38–2.24 (m, 5H), 2.06–1.99 (m, 1H), 1.92–1.86 (m, 2H), 0.86 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 171.7, 169.6 167.1, 152.6, 146.9, 137.9, 136.5, 136.4, 134.4, 129.9, 129.3 (2), 128.7 (2), 128.5 (2), 127.3 (2), 126.9 (2), 126.4, 125.9, 112.9, 72.8, 63.6, 58.8, 58.5, 54.4, 53.3, 50.2, 35.3, 32.5, 26.9, 24.9, 20.0 (2), 17.0. LRMS-ESI (m/z): 705.6 [M+H]. HRMS-ESI (m/z) [M+Na]+ calculated for C37H44N4O6S2Na 727.2600, found 727.2595.
4.2.40. N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-3-((R)-2-(4-methylthiazol-2-yl)piperidine-1-carbonyl)benzamide (4e)
1H NMR (400 MHz, CDCl3) δ 7.79 (s, 1H), 7.63 (d, 2H, J = 8.7 Hz), 7.60 (d, 1H, J = 7.7 Hz), 7.35 (t, 1H, J = 7.7 Hz), 7.30–7.11 (m, 6H), 6.90 (d, 2H, J = 8.7 Hz), 6.73 (s, 1H), 5.60 (dd, 1H, J = 7.3 and 4.9 Hz), 4.38–4.31 (m, 1H), 4.01–3.96 (m, 1H), 3.80 (s, 3H), 3.63–3.58 (m, 1H), 3.41–3.35 (m, 1H), 3.20 (dd, 1H, J = 3.0 and 15.0 Hz), 3.12–2.96 (m, 3H), 2.91–2.79 (m, 2H), 2.38 (s, 3H), 2.36–2.25 (m, 3H), 2.07–1.99 (m, 1H), 1.91–1.81 (m, 4H), 0.83 (dd, 6H, J = 6.3 and 2.8 Hz); 13C NMR (100 MHz, CDCl3) δ 171.7, 169.3, 167.1, 162.8, 152.6, 137.9, 136.6, 134.4, 130.1, 129.7, 129.3 (2), 128.6 (2), 128.4 (2), 126.3, 125.7, 114.2 (2), 112.8, 72.7, 58.7, 58.4, 55.5, 54.5, 53.3, 50.1, 35.0, 32.3, 27.0, 25.7, 24.9, 20.0, 19.9, 17.0. LRMS-ESI (m/z): 757.6 [M+K]. HRMS-ESI (m/z) [M+Na]+ calculated for C38H46N4O6S2Na 741.2757, found 741.2749.
4.2.41. N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-3-((R)-4-(4-methylthiazol-2-yl)thiazolidine-3-carbonyl)benzamide (4f)
1H NMR (400 MHz, CDCl3) δ 7.80 (s, 1H), 7.75–7.58 (m, 3H), 7.41 (br, 1H), 7.26–7.21 (m, 4H), 7.19–7.15 (m, 1H), 6.94 (d, 2H, J = 8.6 Hz), 6.82–6.76 (m, 2H), 6.13 (br, 1H), 4.63–4.58 (m, 1H), 4.38–4.24 (m, 2H), 4.05–3.98 (m, 1H), 3.83 (s, 3H), 3.49 (m, 2H), 3.19–3.03 (m, 5H), 2.86 (d, 2H, J = 7.4 Hz), 2.40 (s, 3H), 1.90–1.82 (m, 1H), 0.85 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 168.8, 166.8, 162.9, 153.0, 137.8, 135.8, 134.5, 129.7, 129.3 (3), 129.2 (3), 128.9, 128.5 (2), 126.5, 125.8, 114.3 (2), 113.0, 72.7, 60.3, 58.6, 55.6, 54.5, 53.4, 34.9, 27.1, 21.0, 20.0, 19.9, 17.0. LRMS-ESI (m/z): 745.5 [M+Na]. HRMS-ESI (m/z) [M+Na]+ calculated for C36H42N4O6S3Na 745.2165, found 745.2161.
4.2.42. N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl) sulfonamido)-1-phenylbutan-2-yl)-3-((R)-2-(4-(methoxymethyl)oxazol-2-yl)pyrrolidine-1-carbonyl) benzamide (4g)
1H NMR (400 MHz, CDCl3) δ 7.82 (s, 1H), 7.74–7.69 (m, 3H), 7.52 (s, 1H), 7.39–7.30 (m, 1H), 7.26–7.13 (m, 6H), 6.96–6.85 (m, 3H), 5.37 (dd, 1H, J = 7.3 and 4.9 Hz), 4.36–4.30 (m, 3H), 4.11 (m, 1H), 4.02–3.97 (m, 1H), 3.82 (s, 3H), 3.68–2.62 (m, 1H), 3.36–3.40 (m, 1H), 3.38 (s, 3H), 3.19–2.97 (m, 4H), 2.92–2.80 (m, 2H), 2.38–2.28 (m, 1H), 2.22–2.04 (m, 2H), 1.95–1.83 (m, 2H), 0.84 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 168.9, 167.0, 164.3, 162.9, 137.9, 137.7, 136.5, 136.0, 134.4, 134.2, 130.3, 129.7, 129.3 (3), 128.7, 128.6, 128.4 (2), 126.4, 125.8, 114.2 (2), 72.7, 66.2, 58.6, 58.4, 55.5, 54.6, 54.4, 53.4, 49.9, 35.0, 30.8, 27.0, 25.1, 20.0, 19.9. LRMS-ESI (m/z): 741.7 [M+Na]. HRMS-ESI (m/z) [M+Na]+ calculated for C38H46N4O8SNa 741.2935, found 741.2932.
4.2.43. N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-3-((R)-2-(4-methyloxazol-2-yl)pyrrolidine-1-carbonyl)benzamide (4h)
1H NMR (400 MHz, CDCl3) δ 7.85 (s, 1H), 7.75–7.64 (m, 3H), 7.40–7.18 (m, 8H), 6.97–6.90 (m, 2H), 6.82 (d, 1H, J = 7.0 Hz), 5.36–5.33 (m, 1H), 4.39–4.32 (m, 1H), 4.02–3.97 (m, 1H), 3.84–3.82 (m, 3H), 3.68–3.61 (m, 1H), 3.49–3.44 (m, 1H), 3.24–2.95 (m, 4H), 2.86 (d, 2H, J = 7.1 Hz), 2.36–2.31 (m, 1H), 2.18–2.09 (m, 4H), 1.97 (br, 1H), 1.93–1.82 (m, 2H), 0.85 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 168.9, 167.0, 163.7, 162.9, 137.9 136.6, 136.3, 134.1, 130.3, 129.7 (2), 129.3 (3), 128. 7 (2), 128.5 (3), 126.4, 125.8, 114.2 (2), 72.7, 58.6, 55.5, 54.6, 54.4, 53.4, 49.9, 35.0, 30.9, 27.1, 25.1, 20.0, 19.9, 14.1. LRMS-ESI (m/z): 711.6 [M+Na]. HRMS-ESI (m/z) [M+Na]+ calculated for C37H44N4O7SNa 711.2829, found 711.2833.
4.2.44. N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-3-(((R)-2-(4-methylthiazol-2-yl)pyrrolidin-1-yl)methyl)benzamide (4i)
1H NMR (400 MHz, CDCl3) δ 7.66 (d, 2H, J = 8.8 Hz), 7.60 (s, 1H), 7.51–7.45 (m, 2H), 7.34–7.25 (m, 5H), 7.21–7.17 (m, 1H), 6.92 (d, 2H, J = 8.8 Hz), 6.78 (s, 1H), 6.55 (d, 1H, J = 8.2 Hz), 4.41–4.34 (m, 1H), 4.04–3.96 (m, 3H), 3.84 (s, 3H), 3.36 (d, 1H, J = 13.0 Hz), 3.23–2.98 (m, 5H), 2.89–2.83 (m, 2H), 2.40 (s, 3H), 2.40–2.24 (m, 3H), 1.92–1.76 (m, 4H), 0.86 (dd, 6H, J = 6.5 and 1.8 Hz); 13C NMR (100 MHz, CDCl3) δ 176.9, 168.1 162.9, 152.4, 139.8, 137.8, 133.9, 131.9, 129.7, 129.4 (4), 128.5 (3), 127.0 126.5, 125.5, 114.2 (2), 113.3, 72.8, 66.0, 58.7, 58.4, 55.5, 54.5, 53.4, 34.9, 34.2, 29.6, 27.1, 23.1, 20.0, 19.9, 17.1. LRMS-ESI (m/z): 691.7 [M+H]. HRMS-ESI (m/z) [M+H]+ calculated for C37H47N4O5S2 691.2988, found 691.2983.
4.2.45. N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-5-((R)-2-(4-methylthiazol-2-yl)pyrrolidine-1-carbonyl)nicotinamide (4j)
1H NMR (400 MHz, CDCl3) δ 8.82 (s, 1H), 8.78 (s, 1H), 8.09 (s, 1H), 7.65 (d, 2H, J = 8.7 Hz), 7.23–7.09 (m, 6H), 6.92 (d, 2H, J = 8.7 Hz), 6.76 (s, 1H), 5.64–5.58 (m, 1H), 4.39–4.33 (m, 1H), 4.10–4.06 (1H), 3.82 (s, 3H), 3.71–3.62 (m, 1H), 3.45–3.38 (m, 1H), 3.24–3.05 (m, 3H), 2.96–2.81 (m, 3H), 2.43–2.28 (m, 4H), 2.19–2.01 (m, 2H), 1.97–1.85 (m, 2H), 0.89–0.78 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 171.1, 166.9, 165.2, 162.9, 152.7, 150.1, 149.3, 138.0, 133.9, 131.9, 129.9 129.7, 129.3 (2), 129.2, 128.6, 128.4 (2), 126.4, 114.3 (2), 113.0, 72.7, 59.0, 58.5, 55.6, 54.6, 53.3, 50.1, 32.4, 29.6, 27.0, 24.9, 20.0, 19.9, 17.1. LRMS-ESI (m/z): 728.6 [M+Na]. HRMS-ESI (m/z) [M+H]+ calculated for C36H44N5O6S2 706.2733, found 706.2723.
4.2.46. N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-3-((R)-3-(4-methylthiazol-2-yl)pyrrolidine-1-carbonyl)benzamide (4k)
1H NMR (400 MHz, CDCl3) δ 7.76 (s, 1H), 7.67–7.58 (m, 3H), 7.43–7.38 (m, 1H), 7.26–7.14 (m, 5H), 6.8–6.88 (m, 3H), 6.79–6.70 (m, 2H), 4.40–4.33 (m, 1H), 4.13–4.09 (m, 1H), 4.03–3.97 (m, 1H), 3.85–3.64 (m, 6H), 3.58–3.48 (m, 2H), 3.19–3.03 (m, 4H), 2.87–2.79 (m, 2H), 2.42–2.23 (m, 3H), 1.92–1.84 (m, 2H), 0.93–0.85 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 168.7, 168.5, 167.1, 162.9, 152.7, 137.8, 136.9, 134.3, 130.1, 129.7, 129.3 (2), 129.2 (2), 128.7 (2), 128.5 (2), 126.5, 125.6, 114.3 (2), 112.9, 72.7, 58.7, 55.6, 54.6, 53.4, 45.7, 42.8, 41.2, 34.9, 29.6, 27.1, 20.0, 19.9, 16.9. LRMS-ESI (m/z): 743.6 [M+K]. HRMS-ESI (m/z) [M+Na]+ calculated for C37H44N4O6S2Na 727.2600, found 727.2604.
4.2.47. N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-3-((S)-3-(4-methylthiazol-2-yl)pyrrolidine-1-carbonyl)benzamide (4l)
1H NMR (400 MHz, CDCl3) δ 7.77 (s, 1H), 7.67–7.59 (m, 4H), 7.42–7.37 (m, 1H), 7.27–7.14 (m, 5H), 6.93 (d, 2H, J = 8.8 Hz), 6.80–6.75 (m, 2H), 4.38–4.31 (m, 2H), 4.03–3.98 (m, 1H), 3.90–3.77 (m, 5H), 3.75–3.66 (m, 2H), 3.58–3.49 (m, 1H), 3.19–3.04 (m, 4H), 2.86 (d, 2H, J = 7.4 Hz), 2.47–2.20 (m, 4H), 1.90–1.84 (m, 2H), 0.91–0.84 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 168.7, 168.6, 167.1, 162.9, 152.6, 137.8, 136.9, 134.3, 130.1, 129.7, 129.3 (2), 129.2 (2), 128.7 (2), 128.5 (2), 126.5, 125.6, 114.2 (2), 112.8, 72.7, 58.7, 55.5, 54.6, 53.4, 45.7, 42.8, 41.2, 34.9, 29.6, 27.1, 20.0, 19.9, 16.9. LRMS-ESI (m/z): 727.6 [M+Na]. HRMS-ESI (m/z) [M+Na]+ calculated for C37H44N4O6S2Na 727.2600, found 727.2596.
Supplementary Material
Figure 1.
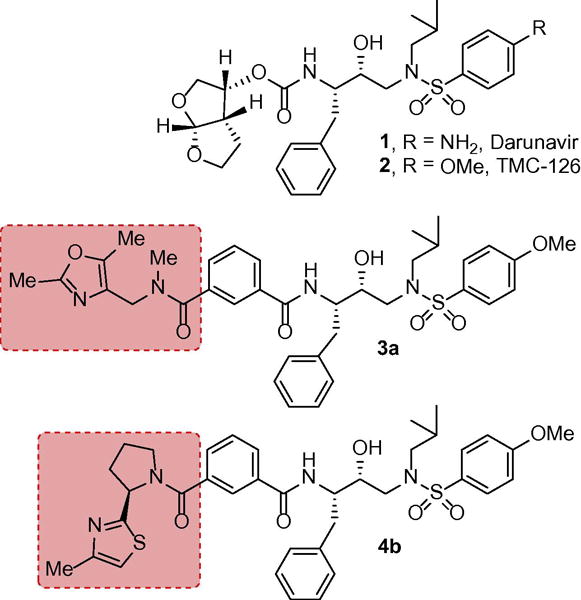
Structures of inhibitors 1,2, 3a and 4b.
Acknowledgments
This research was supported by the National Institutes of Health (Grant GM53386, AKG and Grant GM62920, ITW). X-ray data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22BM at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38. This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health, and in part by a Grant-in-Aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu Kagakusho), a Grant for Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan, and the Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Reemerging Infectious Diseases (Renkei Jigyo) of Monbu-Kagakusho. We thank Mr. Emilio Cardenas (Purdue University) for helpful discussions. The authors would like to thank the Purdue University Center for Cancer Research, which supports the shared NMR and mass spectrometry facilities.
Footnotes
Supplementary data
Supplementary data associated with this article can be found in the online version.
References and notes
- 1.Chapter 15 in: Structure-based Design of Drugs and Other Bioactive Molecules: Tools and Strategies. Wiley-VCH; 2014. Inhibitor, Dabigatran Etexilate, as an Anticoagulant Drug”; pp. 337–354. [Google Scholar]
- 2.Hubbard RE. Structure-based Drug Discovery: An Overview. RSC Publishing; 2006. [Google Scholar]
- 3.Ghosh AK, Osswald HL, Prato G. J Med Chem. 2016;59:5172–5208. doi: 10.1021/acs.jmedchem.5b01697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hue S, Gifford RJ, Dunn D, Fernhill E, Pillay D. J Virol. 2009;83:2645–2654. doi: 10.1128/JVI.01556-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen MS, Chen YQ, McCauley M. N Engl J Med. 2011;365:493–505. doi: 10.1056/NEJMoa1105243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diffenbach CW, Fauci AS. Ann Intern Med. 2011;154:766–771. doi: 10.7326/0003-4819-154-11-201106070-00345. [DOI] [PubMed] [Google Scholar]
- 7.Gupta R, Hill A, Sawyer AW, Pillay D. Clin Infect Dis. 2008;47:712–722. doi: 10.1086/590943. [DOI] [PubMed] [Google Scholar]
- 8.Patel K, Hernán MA, Williams PL, Seeger JD, McIntosh K, Van Dyke RB, Seage GR., III Clin Infect Dis. 2008;46:507–515. doi: 10.1086/526524. [DOI] [PubMed] [Google Scholar]
- 9.Pennigs PS. Infect Dis Rep. 2013;5:21–25. [Google Scholar]
- 10.Wilhelmsson P, Lindgreen PE. The Lancet. 2016;16:512–513. [Google Scholar]
- 11.Cihlar T, Fordyce M. Curr Opin Virol. 2016;18:50–56. doi: 10.1016/j.coviro.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh AK, Anderson DD, Weber IT, Mitsuya H. Angew Chem Int Ed. 2012;51:1778–1802. doi: 10.1002/anie.201102762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh AK. J Med Chem. 2009;52:2163–2176. doi: 10.1021/jm900064c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh AK, Chapsal BD. Design of the anti-HIV protease inhibitor darunavir. In: Ganellin CR, Roberts SM, Jefferis R, editors. From Introduction to Biological and Small Molecule Drug Research and Development. 2013. pp. 355–384. [Google Scholar]
- 15.Ghosh AK, Sridhar PR, Kumaragurubaran N, Koh Y, Weber IT. Chem Med Chem. 2006;1:939–950. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh AK. J Org Chem. 2010;75:7967–7989. doi: 10.1021/jo101606g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh AK, Chapsal B, Mitsuya H. Darunavir, a New PI with Dual Mechanism: From a Novel Drug Design Concept to New Hope Against Drug-Resistant HIV. In: Ghosh AK, editor. Aspartic Acid Proteases as Therapeutic Targets. Wiley-VCH Verlag GmbH & Co KGaA; Weinheim: 2010. pp. 205–243. [Google Scholar]
- 18.Ghosh AK, Chapsal B, Weber IT, Mitsuya H. Acc Chem Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. J Med Chem. 2006;49:5252–5261. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh AK, Xu C-X, Rao KV, Baldridge A, Agniswamy J, Wang Y-F, Weber IT, Aoki M, Miguel SGP, Amano M, Mitsuya H. Chem Med Chem. 2010;5:1850–1854. doi: 10.1002/cmdc.201000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosh AK, Dawson ZL, Mitsuya H. Bioorg Med Chem. 2007;15:756–7580. doi: 10.1016/j.bmc.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang Y-F, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Antimicrob Agent Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh AK, Schiltz GE, Rusere LN, Osswald HL, Walters DE, Amano M, Mitsuya H. Org Biomol Chem. 2014;12:6842–6854. doi: 10.1039/c4ob00738g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh AK, Swanson LM, Cho H, Hussain KA, Leschenko S, Kay S, Walters DE, Mitsuya H. J Med Chem. 2005;48:3576. doi: 10.1021/jm050019i. [DOI] [PubMed] [Google Scholar]
- 25.Kaldor SW, Kalish VJ, Davies JF, Shetty BV, Fritz JE, Appelt K, Burgess JA, Campanale KM, Chirgadze NY, Clawson DK, Dressman BA, Hatch SD, Khalil DA, Kosa MB, Lubbehusen PP, Muesing MA, Patick AK, Reich SH, Su KS, Tatlock JH. J Med Chem. 1997;40:3979–3985. doi: 10.1021/jm9704098. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh AK, Takayama J, Kassekert LA, Ella-Menye JR, Yashchuk S, Agniswamy J, Wang YF, Aoki M, Amano M, Weber IT, Mitsuya H. Bioorg Med Chem Lett. 2015;25:4903–4909. doi: 10.1016/j.bmcl.2015.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh AK, Kumaragurubaran N, Hong L, Kulkarni S, Xu X, Miller HB, Reddy DS, Weerasena V, Turner R, Chang W, Koelsch G, Tang J. Bioorg Med Chem Lett. 2008;18:1031–1036. doi: 10.1016/j.bmcl.2007.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghosh AK, Kumaragurubaran N, Hong L, Kulkarni SS, Xu X, Chang W, Weerasena V, Turner R, Koelsch G, Bilcer G, Tang J. J Med Chem. 2007;50:2399–2407. doi: 10.1021/jm061338s. [DOI] [PubMed] [Google Scholar]
- 29.Horn RK, Gailunas M, Fang LY, Tung JS, Walker DD, Thorsett ED, Jewette NE, Moon JB, Varghese J. J Med Chem. 2004;47:158–164. doi: 10.1021/jm0304008. [DOI] [PubMed] [Google Scholar]
- 30.Beck JP, Mergott DJ. Peptidic, peptidomimetic, and HTS-derived BACE Inhibitors. In: John V, editor. BACE Lead Target for Orchestrated Therapy of Alzheimer’s Disease. Wiley; NJ: 2010. pp. 59–105. Chapter 4. [Google Scholar]
- 31.Ghosh AK, Liu C, Devasamudram T, Lei H, Swanson LM, Ankala SV, Lilly JC, Bilcer GM. Preparation of (3-hydroxy-4-amino-butan-2-yl)-3-[2-(thiazol-2-yl)pyrrolidine-1-carbonyl]benzamide derivatives and related compounds as selective beta-secretase inhibitors. WO2009042694. PCT Int Appl. 2009
- 32.Ghosh AK. Aryldicarboxamide derivatives as non-peptide HIV-1 protease inhibitors and their preparation, pharmaceutical compositions and use in the treatment of HIV, AIDS, and AIDS-related diseases. WO2010002994. PCT Int Appl. 2010
- 33.Lindgren Bengt O, Nilsson Torsten, Husebye Steinar, Mikalsen ØYvind, Leander Kurt, Swahn Carl-Gunnar. Preparation of Carboxylic Acids from Aldehydes (Including Hydroxylated Benzaldehydes) by Oxidation with Chlorite. Acta Chem Scan. 1973;27:888–890. 1973. [Google Scholar]
- 34.Culbertson TP, Domagala JM, Nichols JB, Priebe S, Skeean RW. J Med Chem. 1987;30:1711–1715. doi: 10.1021/jm00393a004. [DOI] [PubMed] [Google Scholar]
- 35.Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. JMol Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 36.Toth MV, Marshall GR. Protease Int J Pept Protein Res. 1990;36:544–550. doi: 10.1111/j.1399-3011.1990.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 37.Koh Y, Amano M, Towata T, Danish M, Leshchenko-Yashchuk S, Das D, Nakayama M, Tojo Y, Ghosh AK, Mitsuya H. J Virol. 2010;84:11961–11969. doi: 10.1128/JVI.00967-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.The coordinates and structure factors for the structure of HIV-1 protease with inhibitors 3b and 4b have been deposited in Protein Data Bank with codes: 5UPZ and 5UOV.
- 39.For details of X-ray studies, please see Supplementary information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.