
Figure 1. Two-pronged proteomic workflow using (i) label free quantification for more accurate ratio determination between MSS and control samples and (ii) iTRAQ-based quantification to obtain a deeper proteome coverage.
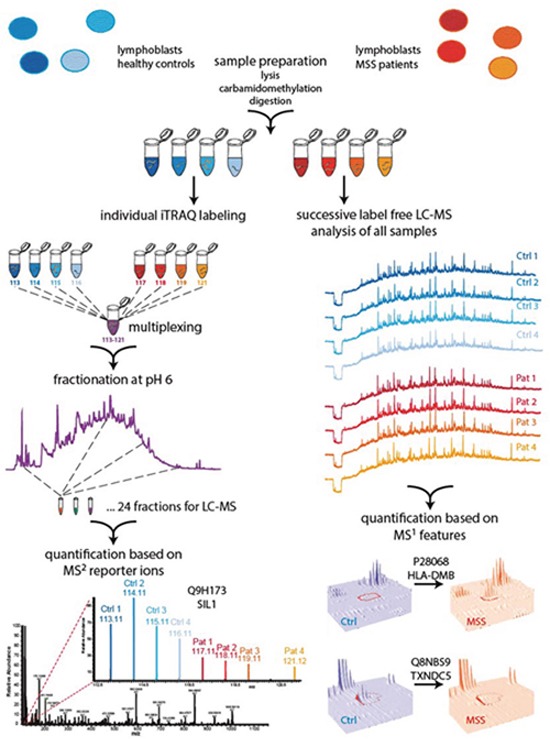
Cells were lysed, proteins carbamidomethylated and digested using trypsin. Generated peptide samples were either iTRAQ 8plex labeled and multiplexed or analysed individually for label free LC-MS analysis followed by quantification with Progenesis and Peptide Shaker. To obtain a deeper coverage the multiplexed iTRAQ sample was fractionated by reversed phase chromatography at pH 6.0 and fractions were analysed by LC-MS, followed by reporter ion quantification using Proteome Discoverer.