

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a tumor with an extremely poor prognosis, predominantly as a result of chemotherapy resistance and numerous somatic mutations. Consequently, PDAC is a prime candidate for the use of sequencing to identify causative mutations, facilitating subsequent administration of targeted therapy. In a feasibility study, we retrospectively assessed the therapeutic recommendations of a novel, evidence‐based software that analyzes next‐generation sequencing (NGS) data using a large panel of pharmacogenomic biomarkers for efficacy and toxicity. Tissue from 14 patients with PDAC was sequenced using NGS with a 620 gene panel. FASTQ files were fed into treatmentmap. The results were compared with chemotherapy in the patients, including all side effects. No changes in therapy were made. Known driver mutations for PDAC were confirmed (e.g. KRAS,TP53). Software analysis revealed positive biomarkers for predicted effective and ineffective treatments in all patients. At least one biomarker associated with increased toxicity could be detected in all patients. Patients had been receiving one of the currently approved chemotherapy agents. In two patients, toxicity could have been correctly predicted by the software analysis. The results suggest that NGS, in combination with an evidence‐based software, could be conducted within a 2‐week period, thus being feasible for clinical routine. Therapy recommendations were principally off‐label use. Based on the predominant KRAS mutations, other drugs were predicted to be ineffective. The pharmacogenomic biomarkers indicative of increased toxicity could be retrospectively linked to reported negative side effects in the respective patients. Finally, the occurrence of somatic and germline mutations in cancer syndrome‐associated genes is noteworthy, despite a high frequency of these particular variants in the background population. These results suggest software‐analysis of NGS data provides evidence‐based information on effective, ineffective and toxic drugs, potentially forming the basis for precision cancer medicine in PDAC.
Keywords: bioinformatics, drug–drug interactions, evidence‐based, NGS, pancreatic cancer
Abbreviations
- 5‐FU
5‐fluorouracil
- ATM
ATM serine/threonine kinase
- BRCA1
breast cancer 1
- CDA
cytidine deaminase
- DPYD
dihydropyrimidine dehydrogenase
- DRDB
Drug Response Database
- FDA
Food and Drug Administration
- FFPE
formalin‐fixed paraffin‐embedded
- FL‐Oxa
fluorouracil leucovorin oxaliplatin
- FOLFIRINOX
fluorouraci leucovorin irinotecan and oxaliplatin
- KRAS
Kirsten rat sarcoma viral oncogene
- MLH1
MutL homolog 1
- MSH
MutS protein homolog 2
- NGS
next‐generation sequencing
- PDAC
pancreatic ductal adenocarcinoma
- RTK
receptor tyrosine kinase
- TP53
tumor protein 53
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer‐related mortality in the USA and Europe (Siegel et al., 2014) and is predicted to become the second by 2030 (Rahib et al., 2014). Death from pancreatic cancer now excedes breast cancer in Europe (Ferlay et al., 2016). Unlike breast or colorectal cancer, pancreatic cancer is always terminal (Löhr, 2006). At diagnosis, approximately 80–90% of pancreatic cancer patients are inoperable with therapy‐resistant locally advanced or metastatic disease. The median survival is approximately 6 months (Bond‐Smith et al., 2012). Even with the best available therapeutic regimens, median survival time does not exceed 10 months (Conroy et al., 2011a,b; Von Hoff et al., 2013). The 5‐year survival rate for all stages of pancreatic cancer has remained close to 5% for the past 25 years and is the lowest for any cancer despite numerous efforts to improve the treatment for PDAC patients (Bond‐Smith et al., 2012; Michl and Gress, 2013; Sohal et al., 2014). PDAC poses one of the greatest unmet medical needs in cancer research and can be regarded as a medical emergency (Löhr, 2014). The lack of treatment response to conventional therapeutic approaches as radiation and chemotherapy is attributable to many factors, including extrinsic or intrinsic resistance (Michl and Gress, 2012; Wang et al., 2011).
Pancreatic cancers may benefit from the developments in precision medicine, which has proven worthy elsewhere. This has been a result of the identification of discriminating tumor markers and the development of targeted therapeutic options, a prime example being hormone receptors and receptor tyrosine‐protein kinase erbB‐2 expression in breast cancer, as well as proto‐oncogene c‐Kit in gastrointestinal stromal tumors. Although these therapies target single biomarkers and PDAC has a heterogeneous mutational landscape, the identification of single biomarkers is a first step in personalized drug combination therapy (Kris et al., 2014). Approximately 5–10% of PDAC patients respond to targeted therapy against vascular endothelial growth factor or rapidly accelerated fibrosarcoma/rat sarcoma viral oncogene homolog kinase (Garrido‐Laguna and Hidalgo, 2015); however, we lack the tools to identify them (Garrido‐Laguna et al., 2015). Because of this dire situation, sequencing has specifically been proposed in pancreatic cancer, allowing mutational cancer analysis to become a prognostic and diagnostic tool readily available to clinicians (Mardis, 2012).
As a result of extensive sequencing efforts, such as the Human Genome Project and The Cancer Genome Atlas, it is becoming clear that identifying singular abnormalities (e.g. mutations in the Kirsten rat sarcoma viral oncogene [KRAS] oncogene or the tumor protein 53 [TP53] tumor suppressor gene from sequencing data) is not sufficient to make therapeutic decisions (Martincorena et al., 2015). Several retrospective studies nevertheless demonstrate convincing explanations for treatment response, or failure, depending on the collective genetic make‐up of the tumor (Gentzler et al., 2014; Kim et al., 2014), which highlights an emerging interest in more complex and sophisticated software tools and algorithms.
The adaptation of next‐generation sequencing (NGS) assays to formalin‐fixed, paraffin‐embedded tissue (FFPE) (Frampton et al., 2013; Holley et al., 2012) and even fine‐needle biopsy material (Young et al., 2013) in pancreatic cancer patients has further facilitated the potential integration of NGS data into clinical practice. To date, only a few studies have prospectively used a sequencing approach and based treatment decisions on the genetic information obtained. These demonstrate a clear survival benefit for the personalized therapy based on pharmacogenomic biomarkers over conventional standard‐of‐care therapy (Kris et al., 2014; Tsimberidou et al., 2014), thus creating a discussion amongst the stakeholders on how to conduct and finance these studies, as well as on how to reimburse personalized cancer medicine in the future (Lewis et al., 2013).
It is therefore necessary to use an integrated analysis of the sequencing data from a given tumor that takes into account the entire body of knowledge on that particular tumor entity and all of the information available for possible treatment options, including their side effects and interactions with other drugs. Furthermore, given the vast amount of data generated, it is clear that the information handed over to the treating physician cannot be raw bioinformatics data and should be presented in an interpreted, clinically relevant and user friendly format (Ellard et al., 2013; Gullapalli et al., 2012). treatmentmap(Molecular Health, Heidelberg, Germany) is such an evidence‐based system for data analysis that provides physicians with tumor profiles based on genome‐sequencing data from a single patient, as well as an objective list of all available scientific and medical data supporting the decision.
In the present study, we aimed to investigate the clinical applicability of using NGS in combination with the software tool (treatmentmap) to generate individualized analysis for a personalized approach for the treatment of pancreatic cancer. We report a feasibility study demonstrating the successful implementation of NGS with treatmentmap into the clinical workflow with the initial results providing a rationale for future studies.
2. Materials and methods
2.1. Patients and study set‐up
This was an open prospective feasibility study aiming to establish NGS in the clinical setting within the framework of our patient‐driven process at the Center for Digestive Diseases, Karolinska University Hospital, with patient recruitment between March 2014 and December 2014. The study was approved by the local ethics committee (EPN; Diarie‐Nr. 2013/2:10). Patients with pancreatic adenocarcinoma who were willing to join the study were provided with information and required to provide informed consent. The tumor material was collected during surgical resection of the tumor, although there was an additional patient included who was not resectable where the tissue was collected from a liver metastases. As a control, either adjacent nontumor tissue (duodenal or gastric) or an EDTA blood sample that was collected at the time of surgery was used.
2.2. DNA extraction and sequencing
Existing hemotoxylin and eosin slides were reviewed by the expert pathologist (CV), assuring the correct histological diagnosis of ductal adenocarcinoma of the pancreas. A block was selected for DNA extraction with a tumor content of at least 20% in line with the prerequisites for NGS and use of the software. DNA extraction was performed with standard protocols using the QIAmp DNA tissue kits (fresh frozen and FFPE tissue samples, as well as blood). DNA was fragmented using Covaris S2 sonicator (Covaris, Woburn, MA, USA) to an average of 100 bp (FFPE tissues) and 300 bp (blood) depending on DNA quality. DNA target enrichment was performed manually using optimized protocols (e.g. prolonged hybridization times, optimized PCR cycles and washing steps) for SureSelectXTall exon V5 Plus (Agilent Technologies Inc., Santa Clara, CA, USA) for whole exome and custom for the SeqCap EZ (NimbleGen, Waldkraiburg, Germany) custom 620 gene panel under study (Table S1). DNA quality control was performed with Life Technologies Qubit Fluorometer and Agilent Bioanalyzer 2100 or an AATI fragment Analyzer at several steps throughout the process. Sequencing was performed using a HiSeq 2500 (Illumina, San Diego, CA, USA) (rapid‐run mode with paired‐end 2 × 100 bp reads) FASTQ generation and demultiplexing was performed using Casava, version 1.8.4 (Illumina). The average fragment length was 200–400 bp. The average coverage achieved was > 100 ×.
2.3. Data processing and software algorithm
An evidence‐based expert system for data analysis was used (treatmentmap). As input information, treatmentmap processes genome‐sequencing data from a single patient, together with basic clinical and demographic patient parameters. This information is then analyzed in three major steps: (i) genome analysis; (ii) evidence mining; and (iii) clinical interpretation, which are further described here.
2.3.1. Step 1: genome analysis
The first major step is the genome data analysis. Here, the system detects genetic alterations in a patient's tumor, based on an analysis of their raw sequencing data. Targeted panel sequencing information is analyzed in a nonpaired fashion and does not include a comparison with the patient's germline reference. The genome analysis pipeline uses a defined set of quality‐controlled, standard analytical applications and reference resource databases that are connected in a controlled workflow. The tools of the pipeline were selected by evaluating sensitivity and precision using synthetic patient data with know variants (R. Bohnert, S. Vivas, & G. Jansen, re‐submitted).
In terms of detailed steps, the genome analysis pipeline takes raw sequence data as input (FASTQ format), together with associated clinical data (i.e. patient diagnosis, age, sex, ethnicity). The genome analysis pipeline has to align the sequence data with the ancestry specific reference genomes. The generated BAM (binary alignment map) files are then processed through the respective algorithm for variant calling, which can detect gene fusion, indels and single nucleotide variants. Tumor‐ and germline‐specific genomic alterations are then mapped to unique reference proteins using Ensembl DB homo_sapiens_core (http://www.ensembl.org) and UniProt (http://www.uniprot.org). The system determines the longest best protein isoform as reference sequence for mapping to the information in the proprietary Nucleus knowledgebase that is part of the software.
2.3.2. Step 2: evidence mining
Once the tumor has been analyzed, the next step of the treatmentmap analytical workflow is to automatically identify all previously published knowledge about the clinical implications of genetic alterations. Accordingly, the treatmentmap system screens all genotype information against the reference information on genes, pathways, biological pathways, variants, treatments, clinical trials, etc., in the Nucleus knowledgebase. In the core of this information is a manually curated database of biomarker information: the so‐called Drug Response Database (DRDB). To aid this quality assured process, the biomedical curation team uses text data mining algorithms and manually classifies pharmacogenomic biomarkers according to three levels of clinical validity (Table 1).
Table 1.
Levels for grading evidence based on treatmentmap.
| Quality level 1: Clinically endorsed pharmacogenetic FDA‐approved biomarkers: Highest relevance information |
| Quality level 2: Clinically observed biomarkers (i.e. observations stemming from clinical data but not yet FDA‐approved: High relevance information |
| Quality level 3: Translational level biomarkers characterized in preclinical studies and/or predicted by bioinformatics algorithms: Information of low or unclear relevance |
Such quality and relevance measures are not only important to this analysis, but also are reported directly in the treatmentmap report, ensuring that they are explicitly clear about how clinically actionable a pharmacogenomic biomarker finding might be for their patient. Other essential information captured during the curation process is also included: (i) The variant (i.e. the type of genomic aberration: SNP, Insertion or Deletion etc.); (ii) the drug or treatment used; (iii) the effect of the variant on treatment (i.e. response, resistance or toxicity); (iv) the quantity of effect (e.g. strong, medium, weak); (v) the observation context (i.e. the disease/disease stage or model system); and (vi) a link to the source information and a grading of its reliability.
The DRDB database includes information about any form of genomic aberration including single nucleotide variants, copy number variations, fusion proteins, insertions and deletions, and combinations thereof. The lineage of the mutation is also captured; for example, whether it is a germline or somatic mutation. Similarly, the database includes information about the drug or treatment associated with a pharmacogenomic (i.e. genomic aberration) being reported, as well as the source of the information (e.g. seen in model systems or patients), and includes MeSH terms (Medical Subject Headings) and other hierarchical classifications. Variants were matched against mutations logged in the Human Gene Mutation Database (HGMD®Professional) (http://www.biobase-international.com/hgmd) from BIOBASE Corporation (http://www.hgmd.org) (Stenson et al., 2009).
The information contained within the DRDB patient and/or tumor mutation profile serves to determine a patient's likelihood of response to therapy, likelihood of resistance to therapy and likelihood of toxicity.
2.3.3. Step 3: clinical interpretation
treatmentmap provides analytical results and access to biomedical resources for a reliable evidence‐based clinical interpretation of the genetic alterations via a web‐based user interface. This online report displays the genetic alterations detected in the tumor genome and the potential effects of these alterations on (i) drug efficacy (i.e. whether the detected genotype confers likelihood of response or resistance to cancer drug treatments) and (ii) drug toxicity (i.e. increased likelihood that the patient might experience adverse drug effects). In addition to the established pharmacogenomic biomarker information, further metrics are provided, such as automated assessments of the importance of a gene in a particular cancer type using a new method referred to as oncoscoring, in addition to a prediction of the functional impact of the aberration on gene/protein function, referred to as ‘functional impact scoring’. Known public functional impact scoring tools (https://omictools.com/functional-predictions-category) are commonly prediction tools using machine‐learning approaches. By contrast, the functional impact scoring system that is implemented in treatmentmap is basing on an evidence‐associated weighted sum of features scoring.
The oncoscore method is a programmed tool that relies on multidimensional data types summarizing real‐time evidence about clinical and molecular importance with respect to specific cancer indications. Features include: gene/protein pathway inclusion facts, drug targets, disease association and interaction neighborhood, as well as indication‐specific protein and targetable attributes. Applying these parameters across individual cancer indications allows a prioritization of the functionally most important genes associated with each cancer type. To understand the impact of aberrations, we contextualized structural, functional, drug response and safety information to provide a novel approach to the prediction of functionally important aberrations. The oncoscore serves to rationally prioritize genes and their specific variants with respect to the disease.
PharmGKB, another drug–drug interaction and pharmacogenetic database based on the Food and Drug Administration (FDA) adverse event reporting system was used for cross‐reference and validation (Thorn et al., 2010; Whirl‐Carrillo et al., 2012). PharmGKB's prediction of the drugs most likely to cause adverse drug reactions to the patients was compared with the treatmentmap data.
2.4. Follow‐up
Patients were followed according to clinical routines. Previous therapy, side effects from chemotherapy and second line therapies were recorded.
3. Results
The 14 patients received several chemotherapy regimens, in some cases sequential: gemcitabine monotherapy (n = 8); fluorouracil leucovorin oxaliplatin (FL‐Oxa) (n = 3); gemcitabine + abraxane (n = 1); gemcitabine + capcitabine (n = 1); fluorouracil leucovorin irinotecan and oxaliplatin (FOLFIRINOX) (n = 2) (Table 2). The disease duration (i.e. from the time when surgery was performed until the genetic analysis) was 0.5–24 months (median 12 months). The median survival time after the date of diagnosis was 19.5 months, with three patients still alive at the time of data closure.
Table 2.
Clinicopathogical characteristics and molecular profile of the 14 patients
| Sample ID | Sex | Age | TNM stage | Resection | Systemic treatments | Survival time | Recommended treatments | Treatments to avoid | Increased risk of toxicity |
|---|---|---|---|---|---|---|---|---|---|
| EUP100 | F | 63 | T3N1M0 | Total pancreatectomy, splenectomy, vascular resection (SMV + CHA), R1 | Gemcitabine FL‐Oxa | 18 months, 17 days | Trametinib + Docetaxel AKT inhibitor MEK + PK inhibitor IGF‐1R antibody + Temsirolimus | Single agent RTK‐inhibitors Everolimus | Cisplatin Cyclophosphamide |
| EUP101 | M | 70 | T3N1M0 | Whipple, R1 | Gemcitabine FL‐Oxa | 10 months, 17 days | Trametinib + Docetaxel AKT inhibitor MEK + PK inhibitor | Single agent RTK‐inhibitors Everolimus Imatinib | Capecitabine Cisplatin Cyclophosphamide |
| EUP102 | F | 65 | T3N0M0 | Whipple, R1 | Gemcitabine | 36 months, 2 daysa | Trametinib + Docetaxel AKT inhibitor MEK + PK inhibitor | Single agent RTK‐inhibitors Everolimus | Trastuzumab Capecitabine Cisplatin Cyclophosphamide Paclitaxel |
| EUP103 | F | 67 | T3N1M0 | Whipple, vascular resection (PV), R1 | Gemcitabine Irinotecan + capecitabine | 10 months, 22 days | Trametinib + Docetaxel AKT inhibitor MEK + PK inhibitor | Single agent RTK‐inhibitors Imatinib Everolimus | Paclitaxel Trastuzumab Capecitabine |
| EUP104 | F | 81 | T3N1M0 | Whipple, R1 | None | 14 months, 23 days | Trametinib AKT inhibitor MEK + PK inhibitor IGF‐1R antibody + Temsirolimus | Single agent RTK‐inhibitors Everolimus | Cisplatin Mercaptopurine Thioguanine Trastuzumab |
| EUP105 | M | 65 | T3N1M0 | Whipple, R0 | None | 16 months, 1 day | Trametinib AKT inhibitor MEK + PK inhibitor IGF‐1R antibody + Temsirolimus | Single agent RTK‐inhibitors Everolimus | Capecitabine Cisplatin Cyclophosphamide |
| EUP106 | M | 59 | T3N1M1 | Whipple, R0 | Gemcitabine | 6 months, 18 days | Paclitaxel Trametinib + Docetaxel AKT inhibitor MEK + PK inhibitor IGF‐1R antibody + Temsirolimus Epirubicin | Single agent RTK‐ inhibitors Everolimus Gefitinib | Capecitabine Cisplatin Cyclophosphamide |
| EUP107 | K | 66 | T3N1M0 | Total pancreatectomy, R1 | Gemcitabine | 15 months, 23 days | Trametinib + Docetaxel AKT inhibitor MEK + PK inhibitor | Single agent RTK‐inhibitors Everolimus | Capecitabine |
| EUP108 | M | 50 | T3N1M0 | Whipple, R1 | Gemcitabine FL‐Oxa | 9 months, 1 day | Trametinib + Docetaxel AKT inhibitor MEK + PK inhibitor | Single agent RTK‐inhibitors Everolimus | Capecitabine |
| EUP109 | F | 75 | T3N1M0 | Whipple, R1 | None | 6 months, 7 days | Trametinib + Docetaxel | Single agent RTK‐inhibitors Everolimus | Cisplatin Cyclophosphamide |
| EUP110 | M | 62 | T3N1M0 | Whipple, R1 | Gemcitabine GemCap | 32 months, 24 days | Trametinib + Docetaxel Imatinib Epirubicin | Single agent RTK‐inhibitors Everolimus Doxorubicin | Capecitabine Mercaptopurine Thioguanine Trastuzumab Cisplatin Cyclophosphamide |
| EUP067 | F | 42 | T4N1M1 | None | FOLFIRINOX Gemcitabine/Abraxane | 14 months, 22 days | Lapatinib + Docetaxel | None | Capecitabine |
| EUP122 | M | 64 | T4N0M0 | Distal pancreatectomy, splenectomy, gastrectomy, vascular resection (CHA), R0 | GemCap | 34 months, 1 daya | None | None | Paclitaxel Methotrexate Tamoxifen |
| EUP186 | M | 51 | T3N0M0 | Distal pancreatectomy, R1 | GemCap FOLFIRINOX | 47 months, 17 daysa | FOLFOX FOLFIRINOX MEK inhibitors HSP90 inhibitors PI3K/AKT inhibitors | Singbgble agent RTK Inhibitors Everolimus | Paclitaxel |
Alive as of October 2016.
For all 14 tumor samples analyzed, the median of target coverage ranged from 123 to 212. For the control samples, the median of target coverage ranged from 41 to 223. The tumor samples showed a ≥ 100 × depth in 71.1–94.2% of targeted sites and the control samples showed a ≥ 100 × depth in 2.5–81.9% of targeted sites. The turnaround time for NGS was 10 days with respect to software analysis and reporting took 2 days on average.
The somatic genetic changes identified in these 14 patients are well in line with known driver mutations in pancreatic cancer (Kamisawa et al., 2016) (Table 3): 13 had KRAS mutations (9 × G12D), four patients had additional TP53 mutations, 10 had additional EGFR (including one with wild‐type TP53) and three had additional SMAD4 (mothers against decapentaplegic homolog 4) mutations. In a subset of patients (n = 10) where sufficient quality sequencing data were available, analysis of germline and somatic mutations in the gene panel could be performed (Table S2). Besides the germ line mutations in drug metabolizing enzymes (e.g. dihydropyrimidine dehydrogenase; DPYD), a number of germ line variants could be found in breast cancer 1 (BRCA1) (n = 6), MutS protein homolog 2 (MSH) 2/6 (n = 5), ATM serine/threonine kinase (ATM) (n = 4), MutL homolog 1 (MLH1) (n = 4) and mismatch repair endonuclease PMS2 (PMS2) (n = 4) (Tables S2 and S3); however, these particular variants have a high prevalence in the background population and are presumably without clinical significance. There was no record of a positive family history for pancreatic cancer in these patients (Table S2).
Table 3.
Pharmacogenetic biomarkers and mutations identified
| Gene | Frequency in these 14 patients | Pathway/action | Potential target therapy | FDA status of targeted therapies | Drug‐biomarker targeted clinical trials | References |
|---|---|---|---|---|---|---|
| AKT | 5 | PI3K/AKT/mTOR | AKT inhibitors. Akt inhibition may modulate platinum‐based therapy resistance | None approved | MK‐2206 in phase II clinical trials, alone, and in combination with platinum‐based chemotherapies. RX‐0201 plus gemcitabine in phase II in advanced PDAC | Cheaib et al. (2015), Nitulescu et al. (2016) |
| APC | 8 | Wnt/β‐catenin | WNT inhibitors | FDA‐approved, PDAC off‐label indication. Potentiation of chemotherapy agents by celecoxib and sulindac | Celecoxib with chemotherapy regimines in phase III trials in PDAC | Lesko et al. (2014), Lipton et al. (2010), Pino et al. (2009) |
| ATM | 7 | Protein kinase. Cell cycle, DNA repair, apoptosis | Associated with either increased or decreased survival when treated with Gemcitabine, depending on variant. Susceptibility to PARP inhibitors. Metformin pathway | Olaparib Metformin | No ATM inhibitors currently in clinical development. Olaparib and rucaparib in phase III trials in PDAC. Phase III trials metformin in combination with chemotherapy | Johnson et al. (2016), Mateo et al. (2015), Soo et al. (2012) |
| AURKA | 3 | Mediates mitosis | Aurora kinase inhibitors, potentiated by other microtubule‐targeting chemotherapies | None approved | Barasertib and danusertib in phase II clinical trials, SNS‐314 in phase I | Bavetsias and Linardopoulos (2015), Meulenbeld et al. (2013), Steeghs et al. (2009), VanderPorten et al. (2009) |
| BRAF | 3 | RAF/MEK/ERK signaling, MAPK | BRAF inhibitors, MEK inhibitors | FDA‐approved, PDAC off‐label indication. Vemurafenib and dabrafenib; trametinib. Sorafenib | Sorafenib with erlotinib and sorafenib with gemcitabine plus cisplatin failed phase II trials in advanced PDAC. | Cardin et al. (2014), Wilhelm et al. (2006) |
| BRCA1 | 10 | DNA repair | Increased susceptability to PARP inhibitors and platinum‐based chemotherapies | FDA‐approved, PDAC off‐label indication for PARPi. Olaparib. Cisplatin och oxaliplatin are FDA‐approved for PDAC, not in monotherapy | Olaparib and rucaparib in phase III trials in PDAC. Veliparib in phase II trials as monotherapy and with gemcitabine and cisplatin | Bendell et al. (2015), Kaufman et al. (2015), Lowery et al. (2011) |
| CDA | 1 | pyrimidine salvaging, deamination of gemcitabine | Increased gemcitabine toxicity | Hung et al. (2012), Nakano et al. (2007), Soo et al. (2012) | ||
| CDKN2A | 6 | Cell cycle | CDK4/6 inhibitor | Palbociclib. FDA‐approved, PDAC off‐label indication | Palbociclib in phase I | Witkiewicz et al. (2015a,b) |
| DPYD | 9 | Inactivation of 5‐FU | Capecitabine, 5‐FU toxicity | FDA‐approved biomarker for adverse drug reaction | Lee et al. (2005), Stoehlmacher and Lenz (2003) | |
| EGFR (ERBB‐1) | 10 | MAPK, JNK, PI3K/AKT/mTOR | Predictive role of EGFR intron length and response to anti‐EGFR therapies shown in other cancer types | Afatinib, cetuximab, panitumumab, temsirolimus. Erlotinib is FDA‐approved for use in PDAC, others off‐label in PDAC | Cetuximab in phase III trials no increase in overall survival. Afatinib phase II (togther with MEK inhibitor selumetinib) | Burtness et al. (2016), Soo et al. (2012) |
| ERBB2 | 4 | MAPK, PKC, JAK/STAT, PI3K/AKT/mTOR, phospolipase Cγ | Her 2/3 inhibitors and antibodies | Afatinib, lapatinib, pertuzumab, (ado‐)trastuzumab emtansine;temsirolimus. Everolimus is FDA‐approved for use in PDAC, others off‐label in PDAC | Trastuzumab with either gemcitabine or capcitabine showed no improval over gemcitabine alone in clinical trial in PDAC | Chou et al. (2013), Harder et al. (2012), Safran et al. (2004) |
| FGFR4 | 8 | Tyrosine kinase receptors | Involved in proliferation and differentiation | FDA‐approved for metastatic melanoma with BRAFV600E mutation | Vemurafenib in phase I trials in PDAC | Hyman et al. (2015), Witkiewicz et al. (2015a,b) |
| KRAS | 12 | MAPK | RAF, MEK (KRAS V12 mutation and copy number variations are resistant to MEK inhibitors), PI3K or farnesyl transferase inhibitors. Decreased drug sensitivity to erlotinib | Trametinib; tipifarnib, Pantimumab, cetuximab. Selumetinib (orphan drug designation). FDA‐approved, PDAC off‐label indication | Selumetinib similar efficacy to capecitabine advanced PDAC phase II trials. Tipifarnib in phase III showed no improval over gemcitabine in PDAC. R115777 farnesyl transferase inhibitors failed phase II | Bramhall et al. (2002), Chiorean and Coveler (2015), Van Cutsem et al. (2004) |
| MAP3K1 | 2 | MAPK | MAP3K1 mutation increases sensitivity to platinum‐based chemotherapy and taxanes | None approved | No MAP3K1 modulators currently in clinical development | Hu et al. (2014), Li et al. (2011) |
| MLH1 | 8 | DNA repair | Decreased sensitivity to 5‐FU and doxorubicin with mismatch repair deficient tumors compared with proficient. Potential susceptibility to platinum‐based chemotherapy, PARP inhibitors | FDA‐approved, PDAC off‐label indication for PARP inhibitor. Olaparib. Cisplatin och oxaliplatin are FDA‐approved for PDAC, not in monotherapy | Olaparib and rucaparib in phase III trials in PDAC. Veliparib in phase II trials as monotherapy and with gemcitabine and cisplatin | Kawakami et al. (2015), Nakano et al. (2007) |
| MSH | 10 | DNA repair | Decreased sensitivity to 5‐FU and doxorubicin with mismatch repair deficient tumors compared with proficient. Potential susceptibility to platinum‐based chemotherapy, PARP inhibitors | FDA‐approved, PDAC off‐label indication for PARPi. Olaparib. Cisplatin och oxaliplatin are FDA‐approved for PDAC, not in monotherapy | Olaparib and rucaparib in phase III trials in PDAC. Veliparib in phase II trials as monotherapy and with gemcitabine and cisplatin | Kawakami et al. (2015), Nakano et al. (2007) |
| PALB2 | 4 | DNA repair | PARP inhibitors | FDA‐approved, PDAC off‐label indication for PARPi. Olaparib. Cisplatin och oxaliplatin are FDA‐approved for PDAC, not in monotherapy | Olaparib and rucaparib in phase III trials in PDAC. Veliparib in phase II trials as monotherapy and with gemcitabine and cisplatin | Childs et al. (2016), Salo‐Mullen et al. (2015), Waddell et al. (2015) |
| PMS1 | 10 | DNA repair | Decreased sensitivity to 5‐FU with mismatch repair deficient tumors compared with proficient | Kawakami et al. (2015) | ||
| PMS2 | 2 | DNA repair | Decreased sensitivity to 5‐FU with mismatch repair deficient tumors compared with proficient | Kawakami et al. (2015) | ||
| STK11 | 2 | Regulates polarity, tumor supressor | Metformin pathway | Metformin. FDA‐approved, PDAC off‐label indication | Phase III trials metformin in combination with chemotherapy | Bhaw‐Luximon and Jhurry (2016), Elmaci and Altinoz (2016) |
In all except one patient, drug targets (positive response biomarkers) could be identified with treatmentmap (range 1–8). In all these patients (range 1–4) biomarkers indicating lack of efficacy could be found. In all patients, biomarkers indicating increased toxicity (range 1–5) were found; in six patients, these were FDA‐approved pharmacogenomic biomarkers for toxicity (Fig. 1).
Figure 1.
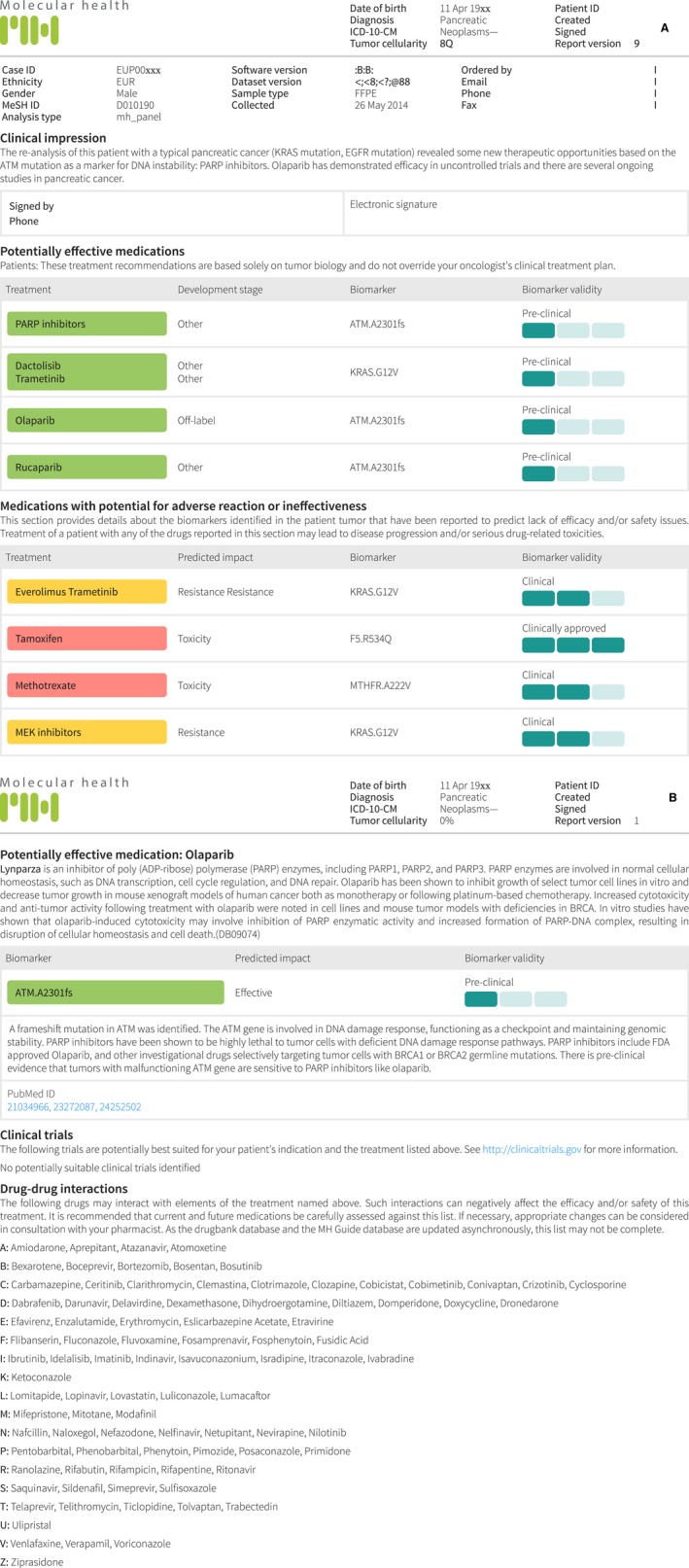
(A) Example of the software‐driven analysis of NGS data resulting in a report that details potentially effective, ineffective or adverse drugs. (B) Example of the more detailed description of a potentially effective drug and drug–drug interactions related to the potentially effective drug.
Of the positive pharmacogenomic biomarkers, only everolimus, erlotinib, cisplatin and oxaliplatin are drugs that are FDA‐approved for use in pancreatic cancer. However, several biomarkers indicated off‐label use for approved drugs, as well as suggesting drugs currently in Phase III studies in pancreatic cancer patients. Eighteen biomarkers in 14 patients indicated at least one to three approved drugs in a given patient: trametinib (ABL1.pK266R; n = 1) plus docetaxel, imatinib (germline KIT.pM541L), lapatinib, cetuximab, IGF‐1R antibody plus temsirolimus (mTOR inhibitor) as a result of the PTPRD.pR995C, Ewing sarcoma, or an AKT‐inhibitor (MK2206) (KRAS.pG12D; n = 13), as well as epirubicin (TP53.pR248Q), paclitaxel (TP53.pR282W) or cisplatin (germline: GSTP1.pI105V). Further drugs in phase III clinical studies that were recommended included PARP inhibitors (olaparib and rucaparib), MEK inhibitors (PD‐0325901 or BAY86‐9766), a PK inhibitor (PF‐05212384), or a combination. Forty‐four biomarkers indicated experimental drugs (pre‐clinical and phase I–III). Because of the KRAS mutations, single agent receptor tyrosine kinase (RTK) inhibitors and erlotinib use was predicted to be ineffective in all but one patient. One patient had a mutation in ERBB4, and the expression of this gene appears to correlate with non‐metastatic pancreatic cancer and a more favorable outcome (Thybusch‐Bernhardt et al., 2001). This mutation was taken as evidence for lapatinib together with docetaxel as a possible treatment option. The combination of lapatinib together with gemcitabine has been studied in pancreatic cancer in a clinical trial that was terminated as a result of ineffectiveness (Safran et al., 2011).
Other drugs predicted to be ineffective because of a KRAS.pG12D mutation are everolimus, gefitinib, imatinib (KRAS.pG12D) and adriablastin/doxorubicin (TP53.pR248Q).
Pharmacogenomic biomarkers with evidence for increased systemic toxicity for a series of drugs were detected (Table S3): cisplatin (ERCC2.pD312N, TPMT.pY240C; n = 8 patients); capecitabine (DPYD.pC29R, FDA‐approved; n = 9); gemcitabine (CDA.pK27Q; n = 1); imatinib (ABL1.pK266R; n = 1), paclitaxel (CYP2C8.pR139K; n = 4); mercaptopurine/thioguanine (TPMT.pA154T, TPMT.pY240C; n = 2); trastuzumab (ERBB2.pI655V; n = 2); and doxorubicin (CBR3.pC4Y; n = 1).
There was a considerable overlap between the drugs suggested by treatmentmap and those recommended by PharmGKB for the patients in the present study. Similarly, there was a strong overlap between the drugs suggested for the entire group by PharmGKB and the drugs that were more probable of demonstrating adverse drug reactions to the patients as individuals based on the treatmentmap data.
4. Discussion
For the first time, in an exploratory way, the present study applied NGS with a panel of 620 genes in combination with a novel evidence‐based software tool in the clinical setting of patients with pancreatic cancer. The turnaround time of 2 weeks will enable application for clinical routine use. The quality and quantity of the DNA extracted from FFPE was sufficient to run NGS with good coverage and sufficient reads. Our mutational analysis found the driver mutations known to be frequently altered in pancreatic adenocarcinoma, namely KRAS, TP53, and SMAD4 (Witkiewicz et al., 2015a,b).
The patients received one of the standard‐of‐care (Seufferlein et al., 2012, 2013) chemotherapy regimens for advanced pancreatic cancer (i.e. gemcitabine monotherapy, combination therapy with capecitabine, erlotinib) (Conroy et al., 2011a,b) or FOLFIRINOX (5‐fluorouracil, folinic acid, irinotecan and oxaliplatin) (Conroy et al., 2011a,b), or gemcitabine in combination with Abraxane (Von Hoff et al., 2011), as first‐line therapy, with varying success (Pelzer et al., 2011; Zaanan et al., 2014). Because this was not an interventional study, no changes in the therapeutic regimen were made based on the NGS/treatmentmap analysis and patients did not receive any of the recommended regimens, with most of them being off‐label uses in pancreatic cancer.
In addition, potential adverse drug reactions yielded several important topical examples where they produced a clear benefit to current treatment recommendations; for example, the DPYD mutations as FDA‐approved biomarkers for toxicity when 5‐fluorouracil (5‐FU) and the oral 5‐FU prodrug capecitabine are used, or cytidine deaminase (CDA) for gemcitabine. Four of the fourteen patients had a predicted toxicity to paclitaxel, which is striking considering that recent studies have shown that the addition of nab‐paclitaxel to standard gemcitabine therapy may provide an additional therapeutic effect in patients with metastatic pancreatic cancer (Von Hoff et al., 2013). Also, two of the patients showed genetic susceptibility to an adverse drug event when using FOLFIRINOX, a regimen that has otherwise shown a significant survival advantage when compared to gemcitabine, despite an increased toxicity that perhaps represents the relative commonness of genetic susceptibility to an adverse drug event (Conroy et al., 2011a,b).
The results of the present study also clearly demonstrate that precision medicine has several hurdles before it can be expected to be regularly utilized in pancreatic oncology practice (Crane, 2013; Knudsen et al., 2015). One such hurdle is the turnover time until the analysis is completed, especially because these patients often have rapid deterioration, as reported from the IMPaCT trial (Chantrill et al., 2015). Two patients in the present study did not receive any chemotherapy as a result of rapid deterioration (Chantrill et al., 2015). Nevertheless, the turnaround time of 2 weeks appears to be clinically sufficient and feasible, especially in patients undergoing surgery with a postoperative recovery time of around 4 weeks. Another lesson from the IMPaCT trial is to include all drugable targets, and not just concentrate on a few of them. With a median of 30 genetic aberrations in PDAC (Waddell et al., 2015) in almost every cellular system and pathway (Jones et al., 2008), all mutations should be taken into account, thus requiring an automated analysis to be fast and feasible for clinical use.
In summary, software‐based approaches that include the genetic susceptibility to an adverse drug event and potential ineffectiveness of a number of treatments are becoming increasingly available to clinicians. Precision medicine analyses as reported in the present study may provide opportunities to reduce the costs and time for drug approval by broadening the use of approved drugs to new applications in cancer therapy, or even repurposing noncancer drugs for use in oncology (Lamb et al., 2015). In the present study, an evidence‐based analysis of the NGS data of a panel of pharmacogenomic biomarkers revealed potential new therapeutic options for pancreatic cancer therapy. However, most recommended chemotherapeutic agents are currently only used for nonpancreatic cancer malignancies. Additionally, the pharmacogenetic diversity identified in these patients could help explain the lack of treatment response to conventional therapeutic approaches used in pancreatic carcinoma (e.g. cetuximab, imatinib, doxorubicin), as well as the toxicity reported in some.
Taken together, NGS in combination with evidence‐based software analysis of the sequence data is feasible in the clinical setting of pancreatic cancer: unraveling novel treatment options and indicating important biomarkers of increased toxicity.
Disclaimer
JML serves as contracting physician, in line with current law to use a medicinal class 1 product (software TreatmentMAP). JML is a consultant to Molecular Health GmbH. AP, KS, CH, SB, RB, MS and DJ are employees at Molecular Health GmbH.
Data Accessibility
Research data pertaining to this article is located at figshare.com: https://dx.doi.org/10.6084/m9.figshare.5311114
Author contributions
The study was designed by JML and HG. Pathological review of the samples and selection of appropriate tissue was carried out by the pathologists (CFM and CSV) who also confirmed the correct histological diagnosis at that time. DNA extraction was performed by RLH and JL. The panel was designed by JL in collaboration with MS, RB, DBJ and SB. Clinical data were provided by LM, SLH, MJM, MGL and MK. Surgery was performed by MDC. Library preparation and NGS analysis was carried out by JL, VW and LE. The software was designed by AP, MS, SB and DBJ. Data analysis was conducted by KS, AP, CH, RB, MS and SB. Data interpretation was performed by LM, SB and JML. The first draft of the manuscript was written by LM and JML. All authors contributed to various forms of the manuscript and approved the final version submitted for publication. This paper comprises part of PhD thesis of LM.
Supporting information
Table S1. List of included genes.
Table S2. Analysis of matched germline and somatic mutations in selected oncogenic cancer syndrome genes (cases with sufficiently good quality sequencing data).
Table S3. Germ line variants.
Acknowledgements
We thank the ClinSeq program for providing the resources and logistics for the sequence analysis. The project was supported by StratCan (to HG) and SLL‐ALF (to JML).
References
- Bavetsias V and Linardopoulos S (2015) Aurora kinase inhibitors: current status and outlook. Front Oncol 5, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendell J, O'Reilly EM, Middleton MR, Chau I, Hochster H, Fielding A, Burke W and Burris H 3rd (2015) Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann Oncol 26, 804–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaw‐Luximon A and Jhurry D (2016) Metformin in pancreatic cancer treatment: from clinical trials through basic research to biomarker quantification. J Cancer Res Clin Oncol 142, 2159–2171. [DOI] [PubMed] [Google Scholar]
- Bond‐Smith G, Banga N, Hammond TM and Imber CJ (2012) Pancreatic adenocarcinoma. BMJ 344, e2476. [DOI] [PubMed] [Google Scholar]
- Bramhall SR, Schulz J, Nemunaitis J, Brown PD, Baillet M and Buckels JA (2002) A double‐blind placebo‐controlled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br J Cancer 87, 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burtness B, Powell M, Catalano P, Berlin J, Liles DK, Chapman AE, Mitchell E and Benson AB (2016) Randomized phase II trial of Irinotecan/Docetaxel or Irinotecan/Docetaxel Plus Cetuximab for metastatic pancreatic cancer: an Eastern Cooperative Oncology Group Study. Am J Clin Oncol 39, 340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin DB, Goff L, Li CI, Shyr Y, Winkler C, DeVore R, Schlabach L, Holloway M, McClanahan P, Meyer K et al (2014) Phase II trial of sorafenib and erlotinib in advanced pancreatic cancer. Cancer Med 3, 572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantrill LA, Nagrial AM, Watson C, Johns AL, Martyn‐Smith M, Simpson S, Mead S, Jones MD, Samra JS, Gill AJ et al (2015) Precision medicine for advanced pancreas cancer: the individualized molecular pancreatic cancer therapy (IMPaCT) trial. Clin Cancer Res 21, 2029–2037. [DOI] [PubMed] [Google Scholar]
- Cheaib B, Auguste A and Leary A (2015) The PI3K/Akt/mTOR pathway in ovarian cancer: therapeutic opportunities and challenges. Chin J Cancer 34, 4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs EJ, Chaffee KG, Gallinger S, Syngal S, Schwartz AG, Cote ML, Bondy ML, Hruban RH, Chanock SJ, Hoover RN et al (2016) Association of common susceptibility variants of pancreatic cancer in higher‐risk patients: a PACGENE Study. Cancer Epidemiol Biomarkers Prev 25, 1185–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiorean EG and Coveler AL (2015) Pancreatic cancer: optimizing treatment options, new, and emerging targeted therapies. Drug Des Devel Ther 9, 3529–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou A, Waddell N, Cowley MJ, Gill AJ, Chang DK, Patch AM, Nones K, Wu J, Pinese M, Johns AL et al (2013) Clinical and molecular characterization of HER2 amplified‐pancreatic cancer. Genome Med 5, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou‐Bourgade S, de la Fouchardiere C et al (2011a) FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364, 1817–1825. [DOI] [PubMed] [Google Scholar]
- Conroy T, Gavoille C and Adenis A (2011b) Metastatic pancreatic cancer: old drugs, new paradigms. Curr Opin Oncol 23, 390–395. [DOI] [PubMed] [Google Scholar]
- Crane CH (2013) Is personalization of care coming to pancreatic oncology? Ann Surg Oncol 20, 355–356. [DOI] [PubMed] [Google Scholar]
- Ellard S, Patrinos GP and Oetting WS (2013) Clinical applications of next‐generation sequencing: the 2013 human genome variation society scientific meeting. Hum Mutat 34, 1583–1587. [DOI] [PubMed] [Google Scholar]
- Elmaci I and Altinoz MA (2016) A metabolic inhibitory cocktail for grave cancers: metformin, pioglitazone and lithium combination in treatment of pancreatic cancer and glioblastoma multiforme. Biochem Genet 54, 573–618. [DOI] [PubMed] [Google Scholar]
- Ferlay J, Partensky C and Bray F (2016) More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol 55, 1158–1160. [DOI] [PubMed] [Google Scholar]
- Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall‐Levin M, White J, Sanford EM, An P et al (2013) Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31, 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido‐Laguna I and Hidalgo M (2015) Pancreatic cancer: from state‐of‐the‐art treatments to promising novel therapies. Nat Rev Clin Oncol 12, 319–334. [DOI] [PubMed] [Google Scholar]
- Garrido‐Laguna I, Tometich D, Hu N, Ying J, Geiersbach K, Whisenant J, Wang K, Ross JS and Sharma S (2015) N of 1 case reports of exceptional responders accrued from pancreatic cancer patients enrolled in first‐in‐man studies from 2002 through 2012. Oncoscience 2, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentzler RD, Yentz SE, Johnson ML, Rademaker AW and Patel JD (2014) The changing landscape of phase II/III metastatic NSCLC clinical trials and the importance of biomarker selection criteria. Cancer 120, 3853–3858. [DOI] [PubMed] [Google Scholar]
- Gullapalli RR, Desai KV, Santana‐Santos L, Kant JA and Becich MJ (2012) Next generation sequencing in clinical medicine: challenges and lessons for pathology and biomedical informatics. J Pathol Inform 3, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder J, Ihorst G, Heinemann V, Hofheinz R, Moehler M, Buechler P, Kloeppel G, Rocken C, Bitzer M, Boeck S et al (2012) Multicentre phase II trial of trastuzumab and capecitabine in patients with HER2 overexpressing metastatic pancreatic cancer. Br J Cancer 106, 1033–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley T, Lenkiewicz E, Evers L, Tembe W, Ruiz C, Gsponer JR, Rentsch CA, Bubendorf L, Stapleton M, Amorese D et al (2012) Deep clonal profiling of formalin fixed paraffin embedded clinical samples. PLoS One 7, e50586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, Huang Q, Li Z, Wu X, Ouyang Q, Chen J and Cao Y (2014) Silencing MAP3K1 expression through RNA interference enhances paclitaxel‐induced cell cycle arrest in human breast cancer cells. Mol Biol Rep 41, 19–24. [DOI] [PubMed] [Google Scholar]
- Hung SW, Mody HR and Govindarajan R (2012) Overcoming nucleoside analog chemoresistance of pancreatic cancer: a therapeutic challenge. Cancer Lett 320, 138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, Wolf J, Raje NS, Diamond EL, Hollebecque A et al (2015) Vemurafenib in multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med 373, 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B, Vanderwalde A, Javadi N, Feldman R and Reddy SB (2016) Molecular profiling of a case of advanced pancreatic cancer identifies an active and tolerable combination of targeted therapy with backbone chemotherapy. J Gastrointest Oncol 7, E6–E12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A et al (2008) Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisawa T, Wood LD, Itoi T and Takaori K (2016) Pancreatic cancer. Lancet 388, 73–85. [DOI] [PubMed] [Google Scholar]
- Kaufman B, Shapira‐Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A et al (2015) Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 33, 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami H, Zaanan A and Sinicrope FA (2015) Microsatellite instability testing and its role in the management of colorectal cancer. Curr Treat Options Oncol 16, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Guntupalli SR, Lee SJ, Behbakht K, Theodorescu D, Lee JK and Diamond JR (2014) Retrospective analysis of survival improvement by molecular biomarker‐based personalized chemotherapy for recurrent ovarian cancer. PLoS ONE 9, e86532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen ES, O'Reilly EM, Brody JR and Witkiewicz AK (2015) Genetic diversity of pancreatic ductal adenocarcinoma and opportunities for precision medicine. Gastroenterology 150, 48–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba II, Varella‐Garcia M, Franklin WA, Aronson SL, Su PF et al (2014) Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 311, 1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez‐Outschoorn UE, Sotgia F and Lisanti MP (2015) Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget 6, 4569–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Lockhart AC, Kim RB and Rothenberg ML (2005) Cancer pharmacogenomics: powerful tools in cancer chemotherapy and drug development. Oncologist 10, 104–111. [DOI] [PubMed] [Google Scholar]
- Lesko AC, Goss KH and Prosperi JR (2014) Exploiting APC function as a novel cancer therapy. Curr Drug Targets 15, 90–102. [DOI] [PubMed] [Google Scholar]
- Lewis JR, Lipworth WL, Kerridge IH and Day RO (2013) The economic evaluation of personalised oncology medicines: ethical challenges. Med J Aust 199, 471–473. [DOI] [PubMed] [Google Scholar]
- Li Y, Sun Z, Cunningham JM, Aubry MC, Wampfler JA, Croghan GA, Johnson C, Wu D, Aakre JA, Molina J et al (2011) Genetic variations in multiple drug action pathways and survival in advanced stage non‐small cell lung cancer treated with chemotherapy. Clin Cancer Res 17, 3830–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton A, Campbell‐Baird C, Witters L, Harvey H and Ali S (2010) Phase II trial of gemcitabine, irinotecan, and celecoxib in patients with advanced pancreatic cancer. J Clin Gastroenterol 44, 286–288. [DOI] [PubMed] [Google Scholar]
- Löhr M (2006) Is it possible to survive pancreatic cancer? Nat Clin Pract Gastroenterol Hepatol 3, 236–237. [DOI] [PubMed] [Google Scholar]
- Löhr JM (2014) Personal view: pancreatic cancer should be treated as a medical emergency. BMJ 349, g5261. [DOI] [PubMed] [Google Scholar]
- Lowery MA, Kelsen DP, Stadler ZK, Yu KH, Janjigian YY, Ludwig E, D'Adamo DR, Salo‐Mullen E, Robson ME, Allen PJ et al (2011) An emerging entity: pancreatic adenocarcinoma associated with a known BRCA mutation: clinical descriptors, treatment implications, and future directions. Oncologist 16, 1397–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER (2012) Applying next‐generation sequencing to pancreatic cancer treatment. Nat Rev Gastroenterol Hepatol 9, 477–486. [DOI] [PubMed] [Google Scholar]
- Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM et al (2015) Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez‐Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N et al (2015) DNA‐repair defects and olaparib in metastatic prostate cancer. N Engl J Med 373, 1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulenbeld HJ, Bleuse JP, Vinci EM, Raymond E, Vitali G, Santoro A, Dogliotti L, Berardi R, Cappuzzo F, Tagawa ST et al (2013) Randomized phase II study of danusertib in patients with metastatic castration‐resistant prostate cancer after docetaxel failure. BJU Int 111, 44–52. [DOI] [PubMed] [Google Scholar]
- Michl P and Gress TM (2012) Improving drug delivery to pancreatic cancer: breaching the stromal fortress by targeting hyaluronic acid. Gut 61, 1377–1379. [DOI] [PubMed] [Google Scholar]
- Michl P and Gress TM (2013) Current concepts and novel targets in advanced pancreatic cancer. Gut 62, 317–326. [DOI] [PubMed] [Google Scholar]
- Nakano Y, Tanno S, Koizumi K, Nishikawa T, Nakamura K, Minoguchi M, Izawa T, Mizukami Y, Okumura T and Kohgo Y (2007) Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br J Cancer 96, 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitulescu GM, Margina D, Juzenas P, Peng Q, Olaru OT, Saloustros E, Fenga C, Spandidos D, Libra M and Tsatsakis AM (2016) Akt inhibitors in cancer treatment: the long journey from drug discovery to clinical use (Review). Int J Oncol 48, 869–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelzer U, Schwaner I, Stieler J, Adler M, Seraphin J, Dorken B, Riess H and Oettle H (2011) Best supportive care (BSC) versus oxaliplatin, folinic acid and 5‐fluorouracil (OFF) plus BSC in patients for second‐line advanced pancreatic cancer: a phase III‐study from the German CONKO‐study group. Eur J Cancer 47, 1676–1681. [DOI] [PubMed] [Google Scholar]
- Pino MS, Milella M, Gelibter A, Sperduti I, De Marco S, Nuzzo C, Bria E, Carpanese L, Ruggeri EM, Carlini P et al (2009) Capecitabine and celecoxib as second‐line treatment of advanced pancreatic and biliary tract cancers. Oncology 76, 254–261. [DOI] [PubMed] [Google Scholar]
- Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM and Matrisian LM (2014) Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 74, 2913–2921. [DOI] [PubMed] [Google Scholar]
- Safran H, Iannitti D, Ramanathan R, Schwartz JD, Steinhoff M, Nauman C, Hesketh P, Rathore R, Wolff R, Tantravahi U et al (2004) Herceptin and gemcitabine for metastatic pancreatic cancers that overexpress HER‐2/neu. Cancer Invest 22, 706–712. [DOI] [PubMed] [Google Scholar]
- Safran H, Miner T, Bahary N, Whiting S, Lopez CD, Sun W, Charpentier K, Shipley J, Anderson E, McNulty B et al (2011) Lapatinib and gemcitabine for metastatic pancreatic cancer. A phase II study. Am J Clin Oncol 34, 50–52. [DOI] [PubMed] [Google Scholar]
- Salo‐Mullen EE, O'Reilly EM, Kelsen DP, Ashraf AM, Lowery MA, Yu KH, Reidy DL, Epstein AS, Lincoln A, Saldia A et al (2015) Identification of germline genetic mutations in patients with pancreatic cancer. Cancer 121, 4382–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seufferlein T, Bachet JB, Van Cutsem E and Rougier P (2012) Pancreatic adenocarcinoma: ESMO‐ESDO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol 23(Suppl 7), vii33–40. [DOI] [PubMed] [Google Scholar]
- Seufferlein T, Porzner M, Becker T, Budach V, Ceyhan G, Esposito I, Fietkau R, Follmann M, Friess H, Galle P et al (2013) S3‐guideline exocrine pancreatic cancer. Z Gastroenterol 51, 1395–1440. [DOI] [PubMed] [Google Scholar]
- Siegel R, Ma J, Zou Z and Jemal A (2014) Cancer statistics, 2014. CA Cancer J Clin 64, 9–29. [DOI] [PubMed] [Google Scholar]
- Sohal DP, Walsh RM, Ramanathan RK and Khorana AA (2014) Pancreatic adenocarcinoma: treating a systemic disease with systemic therapy. J Natl Cancer Inst 106, dju011. [DOI] [PubMed] [Google Scholar]
- Soo RA, Yong WP and Innocenti F (2012) Systemic therapies for pancreatic cancer–the role of pharmacogenetics. Curr Drug Targets 13, 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeghs N, Eskens FA, Gelderblom H, Verweij J, Nortier JW, Ouwerkerk J, van Noort C, Mariani M, Spinelli R, Carpinelli P et al (2009) Phase I pharmacokinetic and pharmacodynamic study of the aurora kinase inhibitor danusertib in patients with advanced or metastatic solid tumors. J Clin Oncol 27, 5094–5101. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS and Cooper DN (2009) The human gene mutation database: 2008 update. Genome Med 1, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoehlmacher J and Lenz HJ (2003) Implications of genetic testing in the management of colorectal cancer. Am J Pharmacogenomics 3, 73–88. [DOI] [PubMed] [Google Scholar]
- Thorn CF, Klein TE and Altman RB (2010) Pharmacogenomics and bioinformatics: PharmGKB. Pharmacogenomics 11, 501–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thybusch‐Bernhardt A, Beckmann S and Juhl H (2001) Comparative analysis of the EGF‐receptor family in pancreatic cancer: expression of HER‐4 correlates with a favourable tumor stage. Int J Surg Investig 2, 393–400. [PubMed] [Google Scholar]
- Tsimberidou AM, Wen S, Hong DS, Wheler JJ, Falchook GS, Fu S, Piha‐Paul S, Naing A, Janku F, Aldape K et al (2014) Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: validation and landmark analyses. Clin Cancer Res 20, 4827–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, Schoffski P, Post S, Verslype C, Neumann H et al (2004) Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol United States.22, 1430–1438. [DOI] [PubMed] [Google Scholar]
- VanderPorten EC, Taverna P, Hogan JN, Ballinger MD, Flanagan WM and Fucini RV (2009) The Aurora kinase inhibitor SNS‐314 shows broad therapeutic potential with chemotherapeutics and synergy with microtubule‐targeted agents in a colon carcinoma model. Mol Cancer Ther 8, 930–939. [DOI] [PubMed] [Google Scholar]
- Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN et al (2013) Increased survival in pancreatic cancer with nab‐paclitaxel plus gemcitabine. N Engl J Med 369, 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, Korn RL, Desai N, Trieu V, Iglesias JL et al (2011) Gemcitabine plus nab‐paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 29, 4548–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K et al (2015) Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Li Y, Ahmad A, Banerjee S, Azmi AS, Kong D and Sarkar FH (2011) Pancreatic cancer: understanding and overcoming chemoresistance. Nat Rev Gastroenterol Hepatol 8, 27–33. [DOI] [PubMed] [Google Scholar]
- Whirl‐Carrillo M, McDonagh EM, Hebert JM, Gong L, Sangkuhl K, Thorn CF, Altman RB and Klein TE (2012) Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther 92, 414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, Schwartz B, Simantov R and Kelley S (2006) Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 5, 835–844. [DOI] [PubMed] [Google Scholar]
- Witkiewicz AK, Borja NA, Franco J, Brody JR, Yeo CJ, Mansour J, Choti MA, McCue P and Knudsen ES (2015a) Selective impact of CDK4/6 suppression on patient‐derived models of pancreatic cancer. Oncotarget 6, 15788–15801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A et al (2015b) Whole‐exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 6, 6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young G, Wang K, He J, Otto G, Hawryluk M, Zwirco Z, Brennan T, Nahas M, Donahue A, Yelensky R et al (2013) Clinical next‐generation sequencing successfully applied to fine‐needle aspirations of pulmonary and pancreatic neoplasms. Cancer Cytopathol 121, 688–694. [DOI] [PubMed] [Google Scholar]
- Zaanan A, Trouilloud I, Markoutsaki T, Gauthier M, Dupont‐Gossart AC, Lecomte T, Aparicio T, Artru P, Thirot‐Bidault A, Joubert F et al (2014) FOLFOX as second‐line chemotherapy in patients with pretreated metastatic pancreatic cancer from the FIRGEM study. BMC Cancer 14, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of included genes.
Table S2. Analysis of matched germline and somatic mutations in selected oncogenic cancer syndrome genes (cases with sufficiently good quality sequencing data).
Table S3. Germ line variants.
Data Availability Statement
Research data pertaining to this article is located at figshare.com: https://dx.doi.org/10.6084/m9.figshare.5311114