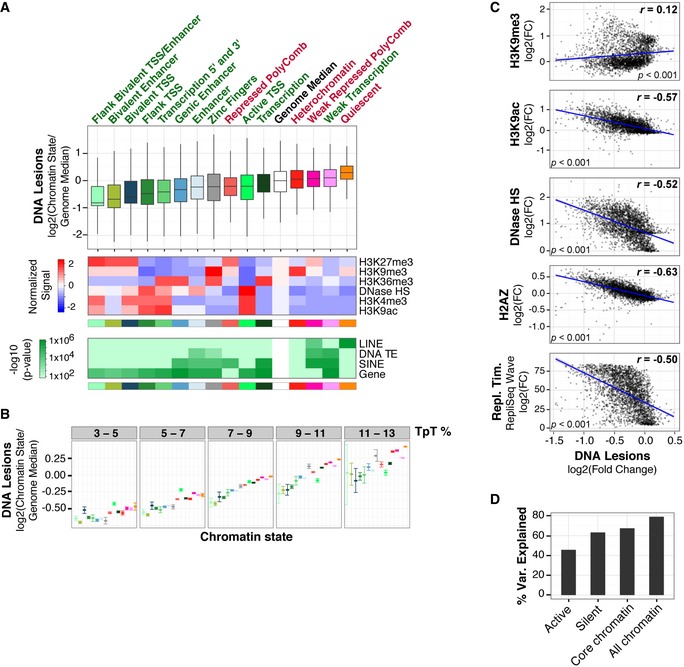
Top panel: Boxplots of DNA lesion abundance within 15 previously defined chromatin states in IMR90 cells (Roadmap Epigenomics Consortium
et al,
2015). Middle quartiles are represented by boxes, top and bottom quartiles are represented by whiskers. Gene‐rich chromatin states are labeled with green text, and gene‐depleted states are labeled with red text. Results are shown as abundance of DNA lesions within individual chromatin states divided by whole‐genome median. Whole‐genome distribution is shown as white box and was determined by pooling DNA lesion abundances from each chromatin state. Statistical outliers are omitted. Flanking and transcriptional start site are abbreviated as Flank and TSS, respectively. Middle panel, mean [log
2(fold change/genome fold change average)] of representative histone modifications and DNase I hypersensitivity (HS) for chromatin states. Bottom panel: Enrichment significance of genes and repetitive features, previously defined (ENCODE Project Consortium
et al,
2012), within each chromatin state. DNA transposable element, long interspersed nuclear element, and short interspersed nuclear element are abbreviated as TE, LINE, and SINE, respectively.
P‐values are based on a hypergeometric distribution (refer to
Materials and Methods for details).