

Abstract
We previously analyzed the Fab-1:VEGF (vascular endothelial growth factor) system described in this work, with both native top-down mass spectrometry and bottom-up mass spectrometry (carboxyl-group or GEE footprinting) techniques. This work continues bottom-up mass spectrometry analysis using a Fast Photochemical Oxidation of Proteins (FPOP) platform to map the solution binding-interface of VEGF and a fragment antigen-binding region of an antibody (Fab-1). In this study, we use FPOP to compare the changes in solvent accessibility by quantitating the extent of oxidative modification in the unbound versus bound states. Determining the changes in solvent accessibility enables the inference of the protein binding sites (epitope and paratopes) and a comparison to the previously published Fab-1:VEGF crystal structure. Using this method, we investigated peptide-level and residue-level changes in solvent accessibility between the unbound proteins and bound complex. Mapping these data onto the Fab-1:VEGF crystal structure enabled successful characterization of both the binding region and regions of remote conformation changes. These data, coupled with our previous higher order structure (HOS) studies, demonstrate the value of a comprehensive toolbox of methods for identifying the putative epitopes and paratopes for biotherapeutic antibodies.
Graphical abstract
Introduction
Monoclonal antibodies (mAb) are among the most common protein therapeutics used to alleviate disease by specifically binding a foreign target (antigen) through a high-affinity interaction between the antibody paratope and antigen epitope interface. Antibodies and their antigen-binding fragments (i.e., Fab-1) are being developed as therapeutics because they have highly specialized functions[1]. Driven by the need to expedite the development of antibody therapeutics and a better understanding of their mechanism of action, fast and sensitive approaches for epitope mapping are required.
Protein therapeutics typically have molecular weights spanning over 50 kDa [2], and unlike traditional small-molecule therapeutics (< 500 Da), they can be difficult to characterize using the traditional high-resolution tools of NMR and X-ray crystallography. Such macromolecules undergo a complex folding process to form higher order structures (HOS) which are the basis for their activity and function. Misfolding of mAbs and other changes in the HOS may result in loss of biologic or therapeutic activity, and/or could potentiate immunogenicity and toxicity [3]. Thus, more rapid and sensitive structural characterization approaches are needed to complement traditional biophysical techniques.
Mass spectrometry (MS)-based techniques such as native top-down MS and bottom-up MS (protein footprinting) provide structural information with high sensitivity, fast turnaround, and small sample consumption. These protein MS tools also have the advantage of elucidating structural features located at the binding interface and distal from the direct interaction, revealing conformational mechanisms that may not be resolved by static techniques alone. Furthermore, such methods are also very powerful for understanding the overall conformational stability of the therapeutic in question, especially when high- resolution structures are unachievable or time-consuming to obtain. To understand better the value of both native top-down and bottom-up MS technologies for the structural characterization and epitope/paratope mapping of biotherapeutics, we have been systematically evaluating multiple MS technologies by using the well-characterized Fab-1:VEGF complex, for which a high resolution structure and comprehensive alanine- scanning are available [4].
Using native top-down MS with electron capture dissociation (ECD), we previously determined that the binding stoichiometry is 2:1 Fab-1:VEGF, as seen in the crystal structure [5]. In addition, native top- down MS coupled with multiple types of fragmentation identified highly flexible regions in VEGF that were not found in crystal structure. Thus, we rationalized that certain regions with high flexibility can hamper crystallization and, therefore, may be purposely truncated to facilitate crystallization [5]. In parallel, we utilized the bottom-up MS approach of carboxyl-group footprinting for epitope mapping, which uses carbodiimide/glycine ethyl ester (GEE) to label solvent-accessible carboxylic acid moieties found in proteins (e.g., aspartic acid (D), glutamic acid (E) and the C-terminus) [6]. With carboxyl-group footprinting, we identified multiple D and E residues involved in the Fab-1:VEGF binding interface [7], and the results agree with observations obtained by alanine-scanning and crystallography [4].
In the current work, we expanded on our bottom-up MS approaches by examining the Fab-1:VEGF complex using Fast Photochemical Oxidation of Proteins (FPOP) [8]. This approach utilizes a high-power laser to photolyze hydrogen peroxide into hydroxyl radicals (·OH), facilitating the sub millisecond labeling (oxidation) of proteins. Similar to carboxyl-group footprinting, FPOP is an irreversible labeling technique that, when coupled to protein digestion and LC-MS/MS analysis, enables the identification of solvent accessibility of protein side chains within a protein or protein complexes. However, ·OH react relatively non-specifically, allowing modification of 14 of the different 20 standard amino acids [9] and potentially increasing the resolution of the site-specific labeling compared to carboxyl-group footprinting [7] (limited to D and E residues and the C-terminus). Similar to carboxyl-group footprinting, FPOP is best utilized in a comparative analysis in which the extent of labeling for a given peptide/residue is determined under two or more different conditions such as an unbound vs. a bound state.
Here, we report epitope and paratope mapping by determining the extent of labeling for the unbound Fab-1 and VEGF, and compare this information to the Fab-1:VEGF complex, using our recently improved FPOP format [10]. We observed several tryptic peptides with significantly reduced modification in the bound state and used high-resolution MS2 to obtain residue-level information. When mapped to the crystal structure, much of the observed reduction in solvent accessibility in the complex is indeed located at the predicted epitope and paratopes (on VEGF and Fab-1, respectively) [4]. We also observed residues with reduced modifications that are distal to the binding region, indicative of remote conformational changes that occur in complex. The data for both the binding region and distal region showing a remote conformational change are consistent with our previously published carboxyl-group footprinting results [7]. We also observed that regions with some of the highest levels of modifications in the complex are located at the regions with relatively high flexibility according to our native top-down MS analysis [5], suggesting a correlation between protein flexibility and side-chain solvent accessibility.
Experimental
Chemicals and Materials
H2O2 (30%), trifluoroacetic acid (TFA), formic acid (FA), L-glutamine, L-methionine, cytochrome c, Trizma base, catalase, dithiothreitol (DTT), sodium iodoacetate (IAA), and all HPLC-grade solvents (H2O and acetonitrile, ACN) were obtained from Sigma-Aldrich (St. Louis, MO) at the highest purity available.
Protein Expression, Fab-1:VEGF Preparation and Purification
VEGF and Fab-1 were produced in E. coli, purified, and formulated as bulk drug substances. A 2:1 molar ratio of Fab-1:VEGF was incubated at 37 °C for 15 min. The complex and the individual proteins were purified using a Tosoh Bioscience size-exclusion chromatography (SEC) TSKgel G2000SWXL (7.8×300 mm, 5 µm) column on an Agilent 1100 HPLC system equipped with a fraction collector. Separation was performed using an isocratic run time of 30 min at 0.5 mL/min using the mobile phase buffer (0.2 M K2HPO4, 0.25 M KCl, pH 6.2) at ambient temperature. All fractionated samples were purified to >98% purity as previously described [7], and were concentrated to approximately 40 g/L. Samples were buffer exchanged into an arginine-succinate buffer.
FPOP Labeling
FPOP was performed as described previously [10]. A 248 nm KrF excimer laser (GAM Laser Inc., Orlando, FL) adjusted to approximately 30 mJ/pulse was used to irradiate the flowing sample solution. The laser was focused through a convex lens (Edmunds Optics, Barrington, NJ) onto a 150 µm i.d. fused-silica tubing (Polymicro Technologies, Pheonix, AZ), giving a 2.5-3.0 mm irradiation window. The laser-pulse frequency was controlled by an external pulse generator (B&K Precision, Yorbal Linda, CA). H2O2 and the scavenger (glutamine or the formulation buffer) in a Tris buffer were mixed with the protein solutions in the flow system just prior to the FPOP irradiation window. The mixing involved inserting the silica tubing into a micro-tee mixer (Cobert Associates Lab, St. Louis, MO), giving thorough and rapid mixing by using different i.d. silica tubing in the tee (Polymicro Technologies, Pheonix, AZ) at different flow rates. The oxidatively modified sample was collected in low protein binding tubes containing 10 µL of 50 nM catalase and 200 mM methionine to decompose residual H2O2.
Proteolysis
Eight identical samples of the proteins of interest (25 µg in 60 µL), were subjected to FPOP labeling, collected and pooled together and concentrated by an MWCO PES cutoff filter (Vivaproducts, Littleton, MA) to ~ 150 µL (200 µg total protein for each digestion). The size of the membrane filter was chosen on the basis of the molecular weight (MW) of the proteins; i.e., 3 kDa for VEGF, and 10 kDa for the Fab-1 and the complex. All samples were run in triplicate as separate FPOP experiments. Trypsin digestion of the proteins was carried out as described previously [7]. Briefly, the concentrated samples following FPOP treatment were diluted with denaturing 360 mM Tris buffer (pH 8.6) containing 6 M guanidinium hydrochloride (GdnCl) and 0.1 M EDTA. Subsequently, the protein samples were reduced with DTT (10 mM at 37°C for 1 h), alkylated with IAA (25 mM at ambient temperature in the dark for 30 min), and quenched with DTT (50 mM at ambient temperature for 5 min). The alkylated samples were then loaded onto a NAP-5 desalting column (GE Healthcare Bio-Sciences, Pittsburgh, PA), eluted with 25 mM Tris buffer, and collected in 1.5 mL, low-binding tubes. The desalted samples were digested at 37 °C for 1.5 h with sequencing grade trypsin (Roche, Indianapolis, IN) at an enzyme:protein ratio of 1:20. The digestion was quenched by adding TFA to a 0.3% final concentration.
Mass Spectrometry Analysis
Tryptic peptides (20 µg) were separated using an Agilent 1200 HPLC with a Waters BEH300 C18 (1.7 µm, 2.1×150 mm) column. Peptide elution was performed using a gradient from 100% solvent A (water, 0.1% trifluoroacetic acid) to 55% solvent B (acetonitrile, 0.08% trifluoroacetic acid) over 45 min at a flow rate of 0.3 mL/min and column temperature of 77 °C. Samples were analyzed on an Orbitrap Elite (Thermo Fisher, Bremen Germany). A full mass scan in the positive-ion mode (60,000 mass resolving power at m/z 400 from m/z 400-2000) was performed using the Orbitrap analyzer. Data-dependent acquisition (DDA) MS2 analysis was performed in the ion trap (IT) analyzer for the product-ion analysis of the six most abundant ions.
Peptide Identification and Data Analysis
The .raw data files were converted to .mgf using MassMatrix Mass Spectrometric Data File Conversion Tools (v 3.9)[11], submitted to Mascot (Matrix Science, London, UK), and then searched against a custom-built database containing sequences of both VEGF and Fab-1 for peptide identification. For the search, all known oxidation-induced modifications[12] were considered as variable modifications. The search criteria include peptide MS1 and MS2 mass tolerances of 10 ppm and 0.8 Da, respectively. The Mascot search was also performed against a decoy (reverse) sequence, and ambiguous identifications were rejected.
For quantitative analysis of oxidative modification at the peptide level, only the intensities of the Extracted Ion Chromatogram (XIC) that were identified as corresponding to either unmodified peptides or related modified peptides from VEGF or Fab-1 were used to report extent of modification [10].
All the assignments were based on MS2 verification. In a few cases, MS2 assignment of a modification at a single residue was ambiguous owing to co-eluted peptides or incomplete fragmentation information. In such cases, the extent of modification was assigned to a range of residues.
Results and Discussion
Normalization of the Oxidative Potential by Varying Scavenger and Protein Amounts
In a typical FPOP experiment, the lifetime of the highly reactive ·OH is primarily controlled by the addition of a scavenger (e.g., simple amino acid), but ·OH radicals are also consumed by the target protein, recombination (or self-quenching) and by potentially any other excipients in solution (e.g., buffer salts, cryo-protectants, surfactants). Thus, the total oxidative potential of the solution should be kept constant for all samples tested, which is typically achieved by using an inert buffer system such as phosphate-buffered saline (PBS) with no other components but scavenger and protein. However, biotherapeutic proteins are typically formulated for therapeutic use and typically contain many components that may affect the oxidative potential of the solution. Therefore, a direct comparison of two different buffer systems may be difficult if the oxidative potentials are not consistent. To investigate the feasibility to normalize the oxidative potential through sample preparation conditions, we compared the global oxidation of our current conditions of 2.8 mM Arg in 10 mM Tris buffer, to that of previous FPOP conditions, which used 20 mM Gln in PBS [10]. We were able to achieve the same level of global oxidation with our previously reported FPOP results by using 20 mM Gln in PBS[10] (previous data = 73.7 ± 0.6%), as compared with our current condition of 2.8 mM Arg in 10 mM Tris buffer (current data = 75.7 ± 0.7%) (Figure S1). In addition, the same level of oxidation is observed for different charge states (Figure S2) indicating normalization of oxidative potential may be an appropriate strategy for comparing two different buffer systems.
In addition to buffer components, the amount of protein in solution will contribute to the overall oxidation potential. Previous work using gamma-ray radiolysis of water demonstrated that increasing protein concentration (from 11 µM to 44 µM) resulted in a subsequent decrease in total protein oxidation [13]. This is because at higher protein concentration there is less total oxidative events per protein molecule. However, unlike FPOP, gamma-ray radiolysis of water does not utilize the addition of a scavenging reagent to control the lifetime of the generated hydroxyl radicals. The chemical kinetics of FPOP relies on the principles that the dominating chemistry for controlling the lifetime of the radical is the scavenger. Therefore, to investigate if total protein oxidation dependent on protein concentration, we compared the oxidative footprint of Fab-1 at our experimental conditions (~0.5 g/L, 10 µM protein) and a 4-fold increase (2 g/L, 40 µM). We observed that protein concentration did not significantly affect total oxidation for the individual peptides of Fab-1 (Figure S3). These data demonstrate that protein concentration does affect the total of oxidative events per mole under conditions similar to that of gamma-ray radiolysis.
Mapping an epitope by FPOP requires identifying peptides with significantly less modification in the bound vs. unbound states. For the current work, we employed an “equal-weight” strategy in which each sample had the same total protein (g/L), scavenger, and H2O2 to ensure further that the a similar oxidative potential for the free proteins (Fab-1 ~50 kDa, VEGF ~40 kDa) and the complex (Fab-1:VEGF ~140 kDa). All of samples have a different molar amount of protein, but they all contain 25 µg/60 µL (0.42 g/L) of footprinted sample (Fab-1 = 8.4 µM, VEGF = 10.5 µM, complex = 4.7 µM) described in the Supporting Information). Based on the results shown in Figure S3, the total amount of protein for each sample in our epitope mapping experiment (~0.5 g/L) does not affect the total number of oxidative events per molecule, and thus Fab-1 and VEGF should be equally reactive in unbound state as the bound state.
Epitope and Paratope Mapping for VEGF and Fab-1 at the Peptide Level
To obtain the epitope and paratope maps, we digested the protein samples in the unbound and bound states after being subjected to hydroxyl radical labeling with trypsin, and analyzed the peptide mixtures by LC-MS/MS. Based on Mascot database searching and manual analysis, we obtained approximately 88%, 90%, and 86% sequence coverage for VEGF, heavy chain (Fab-1 HC), and light chain (Fab-1 LC), respectively. The missing peptides were small and hydrophilic, ranging from one to five amino acids that eluted at or near the void volume. The modification extents of peptides were calculated based on integrating the XIC corresponding to both unmodified and modified peptides, as previously described [11, 12]. All peptides chosen for interpretation satisfied two criteria: accurate mass within 10 ppm and a correct MS2 assignment by manual inspection after evaluation of Mascot results. It is well known that methionine residues, for example, can be oxidized during storage and sample preparation. Such background (non- FPOP) oxidation can be estimated from the analysis of the non-laser control samples, the results of which are background-subtracted from laser-exposed samples. Figure 1 shows an example extracted ion chromatograms for a VEGF-containing tryptic peptide in the unbound and bound state.
Figure 1.

MS (a) and XIC peaks (b) and product-ion (MS/MS) (c) spectra of peptide V17-23 of VEGF, both unmodified (black traces) and modified (blue and red traces). Blue traces in (a) represent MS spectra of oxidized peptide. Blue and red traces in (b) and (c) represent XIC peak or product-ion spectra of peptide oxidation on residue M and Y, respectively.
For VEGF, we identified ten tryptic peptides by LC-MS/MS. The modification extents varied (Figure 2) from ~19% for the N-terminal peptide (V1-16). Comparing the differences in modification extents between the unbound and Fab-1-bound VEGF, we found seven peptides that showed no significant difference (single variable t-test using a 95% confidence interval), indicating that solvent accessibility, and hence conformation, in these regions do not change upon VEGF binding. Three peptides (V17-23, V57-82, and V83-101) of VEGF, however, were significantly less modified upon Fab-1 binding, all three of which contain residues involved in binding, according to alanine-scanning results [4].
Figure 2.

Extent of modification of free VEGF (red bars) compared to Fab-1-bound VEGF (blue bars) at the peptide level (data from in triplicate injections). Regions with significant differences (p < 0.05 between the two states are shown by red asterisks.
For Fab-1, we identified ten tryptic peptides from the HC (Figure 3a) and nine peptides for the LC (Figure 3b). We were unable to detect several peptides of less than five amino acids located at the C- terminus of the HC. Four peptides from HC (HC20-38, HC44-65, HC77-87, and HC99-127) and three from LC (LC19-42, LC46-61, and LC62-103) were significantly less modified upon VEGF binding, indicating reduced solvent accessibility or perhaps lower flexibility in the bound state. The crystal structure [4] indicates that three loops from both Fab-1 HC and LC comprise the binding region (CDR1, CDR2 and CDR3) of Fab-1. Peptides LC19-42, LC46-61, and LC62-103 undergo reduced modification in the bound vs. unbound states, and they cover the CDR1, CDR2, and CDR3 regions of Fab-1 LC, respectively. Likewise, for Fab-1 HC, peptides HC20-38, HC44-65, and HC99-127 cover the CDR1, CDR2, and CDR3 regions, respectively. Interestingly, peptide HC77-87 is not associated with the binding interface, as determined by X-ray crystallography, yet also shows reduced solvent accessibility in Fab-1 upon VEGF binding.
Figure 3.

Extent of modification of (a) HC from free (red bars) compared to VEGF-bound Fab-1 (blue bars) and (b) LC from free (red bars) compared to VEGF-bound Fab-1 (blue bars) at peptide level (data in triplicate). Regions with significant differences (p < 0.05) between the two states are shown by red asterisks.
In addition to observing peptides with significantly less modification in the bound vs. unbound state, we observed many peptides in both Fab-1 and VEGF that remain unchanged. These peptides not only indicate regions which are not affected by the binding interactions, but also demonstrate that the overall oxidative potential of bound and unbound samples appears to be equivalent.
Epitope and Paratope Mapping for VEGF and Fab-1 on the Amino-Acid Residue Level
In general, peptide-level analysis provides adequate information to demonstrate the regions of a protein with changes in solvent accessibility from the unbound vs. bound states. However, with high quality MS2 data, residue-level information can be achieved with certain peptides. To demonstrate this, we analyzed all oxidized residues by manually checking all available product-ion spectra of the modified peptides, as demonstrated in Figure 1. We extracted residue-level information from the XICs for various modifications (e.g., +16, peptides usually eluting earlier than the unmodified) and assigned each modification site on the basis of its product-ion spectrum.
For VEGF, three residues (Y21, M81, and M94, from peptides V17-23, V57-82, and V83-101, respectively) show significant decreases in modification upon Fab-1 binding (Figure 4). In fact, mutation of M81 to alanine results in over 70-fold decrease in binding affinity [4]. These three residues lie in regions that contact Fab-1 (Figure 6), and their side chains point toward the binding interface.
Figure 4.

Extent of modification of free VEGF (red bars) compared to Fab-1-bound VEGF (blue bars) at the residue level (data in triplicate). Residues with significant differences (p < 0.05) between the two states are shown by red asterisks.
Figure 6.

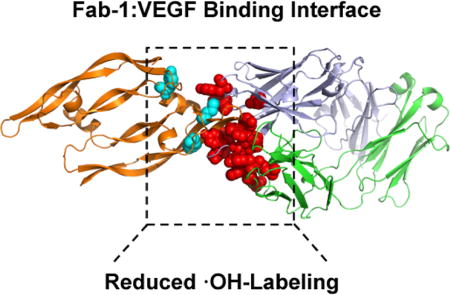
FPOP data mapped onto a previously published X-ray structure (PDB: 1BJ1). (a) Full view of the Fab-1:VEGF complex represented as cartoon structures for VEGF (orange), Fab- 1 HC (blue) and Fab-1 LC (green). (b) Expanded view of the epitope and paratope regions. Residues with positive identification by MS2 with reduced solvent accessibility in the Fab- 1:VEGF complex are represented by sticks in cyan (VEGF) and red (Fab-1 HC and Fab-1 LC). Peptide regions in the Fab-1 HC with reduced solvent accessibility in the Fab-1:VEGF complex in which MS2 could not identify the exact modified residues are represented by sticks in magenta (Y99-103) and orange (H107-Y109). Peptides regions in the Fab-1:VEGF complex identified without changes in solvent accessibility are represented in grey. Residues previously identified with a reduced rate of GEE labeling in the Fab-1:VEGF complex are represented by sticks in brown[7]. Residue regions previously identified as flexible using Native Top-Down MS with ECD are colored in yellow[5]."
In addition to mapping the epitope on VEGF, we identified residues which appear to be involved in binding on the Fab-1 paratope. We found four specific residues of Fab-1 HC (H31 and M34 from peptide HC20-38, Y54 from peptide HC44-65, and M83 from peptide HC77-87) and two specific residues of Fab-1 LC (F50 and L54 from peptide LC46-61) that displayed significantly less modification upon VEGF binding (Figure 5). In addition, both the Fab-1 HC and Fab-1 LC contained a single peptide each that may be associated with binding, but our MS2 data was unable to identify the exact modified residue. The modifications for these two peptides are denoted by a range of residues with in the peptide (Y99-Y103 and H107-Y019 for Fab-1 HC99-127, Y32-L33 for Fab-1 LC46-61) (Figure 5).
Figure 5.

Extent of modification of (a) HC from free Fab-1 (red bars) compared to VEGF-bound Fab-1 (blue bars) and (b) LC from free Fab-1 (red bars) compared to VEGF-bound Fab-1 (blue bars) at residue level (data in triplicate). Residues with significant differences (p < 0.05) between the two states are shown with red asterisks.
As expected, the HC-containing residues M34 and Y54 displayed decreases in binding affinity when mutated to alanine (6.3-fold and 9.4-fold, respectively), but surprisingly we found no significant effect on affinity for the LC-containing residues F50 and L54 (loss of 1.4-fold and 0.83-fold, respectively). Although the affinity data from the latter residues are consistent with the notion that LC CDR2 is often not required for binding [14], our data demonstrate these residues are in part, affected by binding.
Three of the four residues of the Fab-1 HC (H31, M34, and Y54) showed significant decreases in modification upon binding, and all lie on the paratope region in the Fab-1:VEGF structural model. Only residue M83 is remote from the paratope region shown in the X-ray structure, and the change in modification may be caused by a remote conformational change upon binding. In addition, we observed several isomeric modified peptides at residues F132 (HC128-139), F50 (LC46-61), F116 and F118 (LC109-126), L154 (LC150-196) and L182 (LC170-183). The isomers of each residue are of comparable abundance and display significantly lower modification levels in the bound state (Figure 5). Peptides in which the MS2 data were unable to identify the exact modified residue are denoted as a range of residues for VEGF (Figure 4) and the Fab-1 HC and Fab-1 LC (Figure 5). Similar denotation is provided for peptides with isobaric modifications in the Fab-1 HC (V2-V5, L18-R19) and Fab-1 LC (P141-R142) in which the MS2 data were insufficient to identify the exact modified residue (Figure 5). Mapping of our FPOP data onto the crystal structure reveals a complete picture for the Fab-1:VEGF binding interface.
Integrative mapping by multiple MS based approaches
Mapping antibody-antigen interfaces and monitoring the HOS of proteins are important applications of MS in biotechnology and therapeutic protein development. Multiple approaches from both top-down and bottom-up MS analysis can provide comprehensive structural information for target systems. We previously showed that native top-down MS with ECD fragmentation can probe the flexible region of Fab- 1:VEGF complexes [5], assisting the identification of protein regions that may hinder crystallization. In addition, we demonstrated that our bottom-up approach using site-specific carboxyl-group labeling successfully identified some of the peptides/residues at the interface between Fab-1:VEGF [7]. Here, we show that FPOP provides higher structural resolution (i.e., more residue-level information) of the binding interface compared to carboxyl-group footprinting and confirms the suspected remote conformational changes within the complex detected by our previous data. Each of these methods has their own utility and methodological benefits for deciphering solution-state structural interactions (described below).
The site-specific carboxyl-group labeling of Fab-1:VEGF is a simpler bench-top footprinting method that does not require the laser platform of FPOP. In addition, the site-specific labeling of carboxyl groups makes data analysis more straightforward than FPOP analysis [7]. However, this method is limited by the location of the carboxyl groups, which may not be in the sites of interest (i.e., binding interfaces), and it produces a slower “snapshop” (longer exposure) than does FPOP. Comparing the two techniques, we previously showed using carboxyl-group footprinting that only one residue (E93) of VEGF and two residues of Fab-1 HC (D28 and E57) in the binding region displayed significantly reduced rates of labeling upon Fab-1 binding[7] (Table 1). These three residues are located in the same peptides that displayed significant reduction in ·OH labeling, demonstrating the complementarity of these methods (Figure 6). In addition, we observed that residue E89 in the Fab-1 HC (HC88-98), which is distal from the Fab-1 CDR, also displayed significant reduction in carboxyl-group rate of labeling in the bound state. If the crystal structure were not available, the conformational effects on E89 may have been mistakenly assigned as involved in binding. Nonetheless, these data correlate with our FPOP data, demonstrating a different Fab-1 HC tryptic peptide (HC77-87) that is also distal to the binding interface, and showing a significantly lower modification in the presence of VEGF. Further residue-level analysis assigned M83 as the primary modified residue in this peptide, and although M83 and E89 are associated with different tryptic peptides, these residues map to a similar region in the presence of VEGF (Figure 6). Thus, we suggest that either of these two complementary footprinting approaches can support structural information about the conformation of a binding interface and also provide evidence for identifying binding-induced changes remote from the interface.
Table 1.
Comparison of the Fab-1:VEGF Complex higher order structure data obtained from different techniques
| Regions/Residues Identified by Different Methods | ||||
|---|---|---|---|---|
|
| ||||
| Protein | Peptides | FPOPa | Carboxyl Groupa Footprinting |
Native Top-Down MSb with ECD |
| V1-16 | A1-H12 | |||
| V17-23 | Y21 | |||
| V57-82 | M81 | |||
| VEGF | V83-101 | M94 | E93 | |
|
| ||||
| HC1-19 | E1-G10 | |||
| HC20-38 | H31,M34 | D28 | ||
| HC44-65 | Y54 | E57 | ||
| HC77-87 | M83 | |||
| HC88-98 | E89 | |||
| Y99-Y103 | ||||
| HC | HC99-127 | H107-Y109 | ||
| HC225-228 | S225-K228 | |||
| HC229-231 | T229-L231 | |||
|
| ||||
| LC1-18 | D1-S9 | |||
| LC19-42 | Y32-L33 | |||
| LC | LC46-61 | F50, L54 | ||
By comparing the structural data that we obtained from our previous native top-down MS study on the Fab-1:VEGF complex[5] to the current FPOP data, we can further demonstrate the utility of combining MS-based approaches for biotherapeutic HOS characterization (Table 1). Using native top-down MS with ECD fragmentation, we identified multiple regions of the complexes of VEGF, Fab-1 HC and Fab-1 LC that appear to have conformational flexibility: (1) the N-terminus of VEGF (from A1 to H12), (2) both N- and C-termini of Fab-1 HC (from E1 to G10 and from S225 to L231, respectively), and (3) the N-terminus of Fab-1 LC (D1-S9). We hypothesize that these regions may be more solvent-accessible, and indeed we observed a higher extent of FPOP labeling for the N-terminal peptide of VEGF (V1-16). This peptide was not present in the crystal structure, suggesting on the basis of our native top-down MS data that this peptide was purposely omitted during expression of VEGF to facilitate crystal formation. Interestingly, the N- terminus for both the Fab-1 HC and LC do not appear to be the most solvent accessible compared to the rest of the Fab-1 molecule, even though our native top-down MS analysis indicates these regions may have more conformational flexibility. Therefore, although there does appear to be a correlation between conformational flexibility and solvent accessibility, further experiments are required to strengthen the interpretation of the data yielded by these technologies. This work demonstrates that combining top-down and bottom-up MS-based approaches significantly improves our understanding of the higher order structure of complex protein-protein binding interactions. To assess further our HOS toolbox, we are currently investigating the Fab-1:VEGF complex using hydrogen/deuterium exchange analysis. However, the disulfides bonds found in VEGF have proven to be difficult using our current online reduction strategy.
Conclusion
Hydroxyl radical labeling techniques such as FPOP provide insight about the changes in solvent accessibility of side chains. Using an “equal-weight” strategy, we successfully analyzed two different proteins (with presumably two different rates of radical consumption) in their respective unbound and bound states, to obtain not only solution-state epitope and paratope mapping, but also conformational stability and remote structural changes. We demonstrate that the data generated from FPOP are consistent with the predicted interactions at the binding interface as previous determined by crystallography and alanine-scanning. In addition, measuring the decrease in solvent accessibility by FPOP provides more information on the binding interface between Fab-1 and VEGF than does carboxyl-group footprinting. Nevertheless, we demonstrate the complementarity of FPOP, carboxyl-group footprinting and native top- down MS analysis, and show that combining the data from these approaches can offer a more complete picture about the solution conformation of a protein and protein complex. Thus, we have a continued interest in understanding the complementarity within our HOS toolbox. By applying multiple static-state and solution-state HOS technologies to a single system like the Fab-1:VEGF complex, we intend to demonstrate the value of each of these techniques for biotherapeutic development and characterization.
Supplementary Material
Acknowledgments
This research was supported by the National Institute of General Medical Sciences (8 P41GM103422) of the National Institutes of Health to MLG and by the Protein Analytical Chemistry Department of Genentech, South San Francisco, CA. The authors would like to thank M.S. Kalo and N.K. Garcia for thoughtful discussion and review of this manuscript. This research was financially supported by National Institute of General Medical Sciences Grant 8 P41 GM103422 of the National Institutes of Health to M.L.G., and the Protein Analytical Chemistry Department of Genentech, South San Francisco, CA.
References
- 1.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nature reviews. Drug discovery. 2010;9:767–774. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- 2.Stern M, Herrmann R. Overview of monoclonal antibodies in cancer therapy: Present and promise. Critical reviews in oncology/hematology. 2005;54:11–29. doi: 10.1016/j.critrevonc.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 3.Services, U.D.o.H.a.H., Administration, U.D.o.F.a.D. Immunogenicity assessment for therapeutic protein products. 2014 http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm338856.pdf.
- 4.Muller YA, Chen Y, Christinger HW, Li B, Cunningham BC, Lowman HB, de Vos AM. Vegf and the fab fragment of a humanized neutralizing antibody: Crystal structure of the complex at 2.4 a resolution and mutational analysis of the interface. Structure (London, England : 1993) 1998;6:1153–1167. doi: 10.1016/s0969-2126(98)00116-6. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Cui W, Wecksler AT, Zhang H, Molina P, Deperalta G, Gross ML. Native ms and ecd characterization of a fab–antigen complex may facilitate crystallization for x-ray diffraction. Journal of the American Society for Mass Spectrometry. 2016;27:1139–1142. doi: 10.1007/s13361-016-1398-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang H, Wen J, Huang RY, Blankenship RE, Gross ML. Mass spectrometry-based carboxyl footprinting of proteins: Method evaluation. Int J Mass Spectrom. 2012;312:78–86. doi: 10.1016/j.ijms.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wecksler AT, Kalo MS, Deperalta G. Mapping of fab-1:Vegf interface using carboxyl group footprinting mass spectrometry. Journal of the American Society for Mass Spectrometry. 2015;26:2077–2080. doi: 10.1007/s13361-015-1273-0. [DOI] [PubMed] [Google Scholar]
- 8.Hambly DE, Gross ML. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom. 2005;16:2057–2063. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 9.Wang L, Chance MR. Structural mass spectrometry of proteins using hydroxyl radical based protein footprinting. Analytical chemistry. 2011;83:7234–7241. doi: 10.1021/ac200567u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Rempel DL, Zhang H, Gross ML. An improved fast photochemical oxidation of proteins (fpop) platform for protein therapeutics. J Am Soc Mass Spectrom. 2015;26:526–529. doi: 10.1007/s13361-014-1055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan Y, Chen G, Wei H, Huang RY, Mo J, Rempel DL, Tymiak AA, Gross ML. Fast photochemical oxidation of proteins (fpop) maps the epitope of egfr binding to adnectin. J Am Soc Mass Spectrom. 2014;25:2084–2092. doi: 10.1007/s13361-014-0993-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deperalta G, Alvarez M, Bechtel C, Dong K, McDonald R, Ling V. Structural analysis of a therapeutic monoclonal antibody dimer by hydroxyl radical footprinting. mAbs. 2013;5:86–101. doi: 10.4161/mabs.22964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tong X, Wren JC, Konermann L. Effects of protein concentration on the extent of gamma-ray-mediated oxidative labeling studied by electrospray mass spectrometry. Analytical chemistry. 2007;79:6376–6382. doi: 10.1021/ac070724u. [DOI] [PubMed] [Google Scholar]
- 14.Wilson IA, Stanfield RL. Antibody-antigen interactions: New structures and new conformational changes. Curr Opin Struct Biol. 1994;4:857–867. doi: 10.1016/0959-440x(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 15.Jones LM, J BS, J AC, Gross ML. Fast photochemical oxidation of proteins for epitope mapping. Analytical chemistry. 2011;83:7657–7661. doi: 10.1021/ac2007366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.