

Abstract
PRDM9 is the only mammalian gene that has been associated with speciation. The PR/SET domain 9 (PRDM9) protein is a major determinant of meiotic recombination hot spots and acts through sequence-specific DNA binding via its C2H2 zinc finger (ZF) tandem array, which is highly polymorphic within and between species. The most common human variant, PRDM9 allele A (PRDM9a), contains 13 fingers (ZF1–13). Allele C (PRDM9c) is the second-most common among African populations and differs from PRDM9a by an arginine-to-serine change (R764S) in ZF9 and by replacement of ZF11 with two other fingers, yielding 14 fingers in PRDM9c. Here we co-crystallized the six-finger fragment ZF8–13 of PRDM9c, in complex with an oligonucleotide representing a known PRDM9c-specific hot spot sequence, and compared the structure with that of a characterized PRDM9a-specific complex. There are three major differences. First, Ser764 in ZF9 allows PRDM9c to accommodate a variable base, whereas PRDM9a Arg764 recognizes a conserved guanine. Second, the two-finger expansion of ZF11 allows PRDM9c to recognize three-base-pair-longer sequences. A tryptophan in the additional ZF interacts with a conserved thymine methyl group. Third, an Arg–Asp dipeptide immediately preceding the ZF helix, conserved in two PRDM9a fingers and three PRDM9c fingers, permits adaptability to variations from a C:G base pair (G–Arg interaction) to a G:C base pair (C–Asp interaction). This Arg–Asp conformational switch allows identical ZF modules to recognize different sequences. Our findings illuminate the molecular mechanisms for flexible and conserved binding of human PRDM9 alleles to their cognate DNA sequences.
Keywords: crystallography, DNA recombination, protein conformation, protein-DNA interaction, zinc finger, Arg–Asp conformational switch, PRDM9, recombination hot spots, zinc finger arrays
Introduction
PRDM9 is a DNA-binding protein that is nonuniformly conserved among vertebrates (1). Among mammals, it is a major determinant of meiotic recombination hot spots that acts through sequence-specific DNA binding and trimethylation of histone H3 lysine 4 in neighboring nucleosomes (2–4). PRDM9 comprises three broad regions: (i) an N-terminal Krüppel-associated box (KRAB)2 domain; (ii) a central PR-SET (PRDF1-RIZ-Su(var)3–9/Enhancer of Zeste/Trithorax) domain that trimethylates histone H3 lysine 4 (2, 5–7); and (iii) a C-terminal zinc-finger (ZF) array, which is highly polymorphic both within and between species (8–11) (Fig. 1A). This polymorphism implies variation in DNA-binding specificity, in agreement with the finding that hot spot positions and activities vary within and between mammalian species (11).
Figure 1.

Human PRDM9 allele C. A, PRDM9 contains a C-terminal tandem ZF DNA-binding array comprising 13 fingers (allele A; accession no. Q9NQV7) or 14 fingers (allele C; GU216224.1). B, sequence alignment of fourteen C2H2 fingers of PRDM9c with variations at DNA base-interacting positions −1, −4, −7, and −8. For comparison, the four “canonical” positions of the helix are indicated at the bottom of the sequence (20, 21). C, there are two protein-DNA complexes (colored in cyan and green) plus an additional protein molecule (yellow) in the crystallographic asymmetric unit. D, the yellow molecule (Mol C) mediates protein–protein interactions and is not paired with a DNA molecule. Only the first ZF of the unpaired protein is shown, as no electron density was observed for other fingers. E, superimposition of the two protein-DNA complexes. F, structure of ZF8–13 in complex with an allele C-specific DNA molecule.
More than 40 allelic variants of human PRDM9 have been documented, which display marked differences in recombination profile and crossover frequency (12–16). Allele A of human PRDM9 (PRDM9a) is the most common form of PRDM9, found in ∼86% of European and ∼50% of African populations (13). PRDM9 allele C is the second most common allele in African populations, with a frequency of 12.8% (13, 14). In A/C heterozygotes, PRDM9c-specific hot spots are more frequent (56% versus 44%) and more active than PRDM9a-specific hot spots (4), suggesting a partial dominance of allele C. Alleles C and A differ by both an arginine-to-serine change (R764S) in ZF9 and by replacement of ZF11 with two other fingers, resulting in an extra finger (12, 13, 17) (Fig. 1, A and B). In addition, allele C has one more difference from allele A, a serine-to-threonine substitution in ZF6 (4, 12), although this change does not affect hot spot recognition (see “Discussion”).
We previously expressed and purified ZF8–12 of PRDM9a and ZF8–13 of PRDM9c and showed that each allele binds with the highest affinity to the respective allele-specific sequence (18), consistent with PRDM9a and PRDM9c acting at entirely different hot spots (4). In addition, PRDM9c has >10-fold higher affinity for the C-specific sequence than the affinity of PRDM9a for the A-specific sequence (18), in agreement with the observation that PRDM9c is partially dominant over PRDM9a (4). This was quite surprising, considering that, of five fingers in PRDM9a or six fingers in PRDM9c, the two proteins share three identical ZFs and one nearly identical ZF with a single amino acid difference. In addition, we studied three additional alleles (L9/L24, L13, and L20) with single amino acid differences from allele A and demonstrated that these alleles possess altered DNA-binding affinity and/or altered DNA sequence specificity (18). Here, to explore the surprisingly different specificities of these PRDM9 proteins, we present the structure of PRDM9c ZF8–13 in complex with allele-specific DNA and compare it with that of PRDM9a (18). In addition, we discuss unique features of PRDM9 that have rarely (if ever) been described for canonical ZF-DNA complexes.
In conventional C2H2 ZF proteins, each finger comprises two β strands and a helix (19). Characteristically, two histidines in the helix together with one cysteine in each β strand coordinate a zinc ion, forming a tetrahedral C2-Zn-H2 structural unit that confers rigidity to fingers. The amino acids occupying four key “canonical” positions of the helix, namely −1, 2, 3, and 6, specify a DNA target sequence of three or four adjacent DNA base pairs (20, 21) (Fig. 1B, bottom). This structure-based numbering scheme refers to the position immediately before the helix (position −1) and positions within the helix (positions 2, 3, and 6). To reduce possible ambiguity, we use the first zinc-coordination His in each finger as reference position 0, with residues before this, at sequence positions −1 (blue), −4 (red), −5 (black), and −7 (green), corresponding to the 6, 3, 2, and −1 of the structure-based numbering (compare top and bottom of Fig. 1B). The new numbering scheme (residues at positions −1, −4, and −7) corresponds to the 5′-middle-3′ of each DNA triplet element.
PRDM9 is unique (relative to other ZF proteins) in that the ZFs are highly repetitive and resulted from sequence duplications (Fig. 1B). This feature provides an opportunity to study identical ZFs (ZF10 and ZF12) or nearly identical fingers with only one amino acid difference (ZF9 and ZF10 at position −4) in response to target sequence variation, along with variable cross-strand base contacts by a position-specific invariant serine residue at position −5, and a “switch” mechanism by Arg–Asp dipeptide at positions −8 and −7 for base recognition. The base-specific interaction by Arg at position −8 (corresponding to the second residue before the helix) has not been considered in the original canonical model, even in a recent large survey of the three-finger DNA-binding landscape (22).
Results
The six-finger fragment of PRDM9c was used for cocrystallization with a 20-bp oligonucleotide derived from the C consensus sequence Motif 1 (16), plus a 5′-overhanging thymine or adenine on the opposite strand. We crystallized the protein-DNA complex in space group P21 and determined the structure to a resolution of 2.4 Å (Table 1). The crystallographic asymmetric unit contains two protein-DNA complexes (Mol A and Mol B in Fig. 1C) and an additional protein that mediates the protein–protein interactions between the two complexes (Mol C in Fig. 1D). In the current model, we only modeled ZF8 of molecule C (Fig. 1D), as no electron density was observed for the rest of the fingers in that molecule (supplemental Fig. S1A). Interestingly, a similar observation of an additional protein molecule (with only one ordered finger) was made in the crystal structure of a designed zinc finger protein bound to DNA (23). (We do not know whether the additional molecule C is biologically relevant, but it does mediate intermolecule contacts in the crystal lattice (supplemental Fig. S1B).) The two protein-DNA complexes (Mol A and Mol B in Fig. 1C) are highly similar, with a root mean square deviation (RMSD) of <0.5 Å when comparing 145 pairs of Cα atoms (Fig. 1E). The DNA molecules are largely B-form (supplemental Table S1) and coaxially stacked, with the overhanging A and T forming a base pair with neighboring DNA molecules, thus forming a pseudocontinuous duplex in the crystal lattice. Here we will only describe the structure of complex A.
Table 1.
Summary of X-ray structural statistics
Values in parenthesis correspond to the highest-resolution shell.
| Protein | Human PRDM9c (ZF8–13) |
|---|---|
| DNA (5′-3′) | TGACCCCAGTGAGCGTTGCCC |
| DNA (3′-5′) | CTGGGGTCACTCGCAACGGGA |
| PDB code | 5V3G |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 63.1, 123.8, 70.2 |
| α, β, γ (degrees) | 90, 116, 90 |
| Beamline | APS 22-ID (SERCAT) |
| Wavelength (Å) | 1.0000 |
| Resolution (Å) | 35.2–2.41 (2.52–2.41) |
| Rmergea | 0.076 (0.721) |
| 〈I/σI〉b | 22.68 (1.3) |
| Completeness (%) | 96.5 (72.3) |
| CC½/CC | (0.541/0.838) |
| Redundancy | 6.5 (2.9) |
| Observed reflections | 233,169 |
| Unique reflections | 35,662 (2,949) |
| Refinement | |
| Resolution (Å) | 2.41 |
| No. of reflections | 35,530 |
| Rworkc/Rfreed | 0.180 / 0.225 |
| No. of atoms | |
| Protein | 2,891 |
| DNA | 1,710 |
| Zinc | 13 |
| Waters | 114 |
| B factors (Å2) | |
| Protein | 79.2 |
| DNA | 65.6 |
| Zinc | 72.4 |
| Waters | 59.9 |
| RMSD | |
| Bond lengths (Å) | 0.06 |
| Bond angles (degrees) | 1.0 |
| All-atom clash score | 8.8 |
| Ramachandran plot (%) | |
| Allowed | 98.3 |
| Additional allowed | 1.7 |
| Cβ deviation | 0 |
a Rmerge = Σ|I − 〈I〉|/ΣI, where I is the observed intensity and 〈I〉 is the averaged intensity from multiple observations.
b 〈I/σI〉 = averaged ratio of the intensity (I) to the error of the intensity (σI).
c Rwork = Σ|Fo − Fc|/Σ|Fo|, where Fo and Fc are the observed and calculated structure factors, respectively.
d Rfree was calculated using a randomly chosen subset (5%) of the reflections not used in refinement.
As was seen in the first structure reported for a three-finger protein Zif268 in complex with DNA (19), the six fingers of PRDM9c interact with DNA exclusively in the major groove (Fig. 1F), primarily recognizing one strand of the double-stranded DNA in a linear, polar fashion from 3′ to 5′ (magenta in Fig. 1F and bottom strand in Fig. 2A), with the corresponding protein sequence proceeding from N to C terminus (ZF8 to ZF13). To our knowledge, this is the largest native tandem ZF array whose structure has been determined in complex with an oligonucleotide, where every finger is involved in DNA sequence-specific interactions. Previously, a designed six-finger zinc finger protein, Aart, was characterized with bound DNA (24) (PDB code 2I13). Pairwise comparison of the two six-finger proteins (Aart versus PRDM9c) yielded an RMSD of ∼2.6 Å over 149 pairs of Cα atoms (supplemental Fig. S2A). In addition, one duplex DNA containing two adjacent sites, bound by two copies of the three-finger Zif268, had been characterized as being equivalent to a six-finger protein (25) (PDB code 1P47). Interestingly, the “relaxed” discontinuous six-finger Zif268 structure is similar (RMSD of ∼2.6 Å) to either the native six-finger PRDM9c or the designed six-finger Aart (supplemental Fig. S2B). More recently, we characterized human CTCF ZF2–7 (a six-finger array) in complex with DNA (26). In CTCF, ZF3 to ZF7 make base-specific contacts, whereas ZF2 continues to follow in the major groove, but the side chains within the DNA-interacting helix were too far away to make base-specific hydrogen bonds (26). Interestingly, only five of the six fingers of CTCF align with either Aart or PRDM9c (supplemental Fig. S2, C and D).
Figure 2.

PRDM9c ZF8–13 form base-specific interactions. A, schematic representation of ZF8–13 interactions with DNA. The top line indicates the 18-bp consensus sequence. The second line indicates the base pair positions (positions 0–19). The third and the fourth lines are the sequence of the double-stranded oligonucleotide used for crystallization, shown with the top strand matching the consensus sequence. Amino acids of each finger interact specifically with the DNA bases, as shown below. B, DNA base-specific interactions involve a particular residue of each ZF. Atoms are colored dark blue for nitrogen and red for oxygen, and carbon atoms are decorated with finger-specific colors according to Fig. 1F. The numbers indicate the interatomic distance in angstroms (between protein and DNA). For clarity, the H-bond distances for Watson–Crick base pairs are not shown. w, water molecules (small red spheres). C, His733 interaction with G2. D, Arg736 interaction with G3. E, Arg757 interaction with G4. F, His761 interaction with G5. G, Ser764 is positioned too far away from T6. H, Asp786 interaction with C7. I, Asn789 interaction with A8. J, Ser792 forms a weak H-bond with C9. K, Trp814 forms a π–methyl interaction with the T10 methyl group. L, Val817 forms a van der Waals contact with C11. M, Arg820 interaction with G12. N, Asp842 and Ser844 interaction with the G13:C13 base pair. O, Asn845 interaction with A14. P, Ser848 is too far away from A15. Q, Asn870 and Ser872 are positioned too far away from the G16:C16 base pair. R, His873 interaction with G17. S, Arg876 interaction with G18.
The convention that we used for numbering nucleotides and amino acids is shown in Fig. 2A. Base pairs of the crystallization oligonucleotide are numbered 0–19, with the allele C-specific consensus hot spot sequence motif as the “top” strand (Fig. 2A). Like classic C2H2 ZF proteins (20, 21, 27), each finger interacts with a “triplet” element consisting of three adjacent DNA base pairs. In the case of PRDM9c, it is the opposite “bottom” strand that is being recognized by the ZF array. To keep the nomenclature of the consensus sequence, ZF8 interacts with the three base pairs of 5′ sequence (ACC), ZF9 with the second triplet (CCA), ZF10 with the third triplet (GTG), ZF11 with the fourth triplet (AGC), ZF12 with the fifth triplet (GTT), and ZF13 with the 3′ sequence (GCC). Analysis of the allele C-specific consensus sequence motifs (4, 16) suggested that each triplet contains one variable base (N), at the first position of the first and sixth triplets (N1 and N16), the second position of the fourth triple (N11), or the third position of the second, third, and fifth triplets (N6, N9, and N15 in Fig. 2A). In addition, we used a web server for predicting C2H2 ZF DNA-binding specificity (28), which gave a predicted sequence for PRDM9c in general agreement with the experimentally determined allele C-specific consensus sequence motif, including five variable positions (supplemental Fig. S2). However, there are two notable and reciprocal exceptions; at nucleotide position 4, a conserved C:G base pair in the consensus motif is variable in the prediction, whereas at nucleotide position 11, a variable base in the consensus is strongly predicted to be a G:C base pair.
From the protein side, the side chains from specific amino acids within the N-terminal portion of each helix and the preceding loop make major groove contacts primarily with the bases of the “bottom” recognition strand. Using the first zinc-coordination His in each finger as reference position 0, residues before this, at positions −1 (blue), −4 (red), and −7 or −8 (green) lie on the protein-DNA interface and form hydrogen bonds (H-bonds) with the exposed edges of the DNA bases in the major groove (Figs. 1B and 2A). Variations at the four positions (−8, −7, −4, and −1) among zinc fingers correspond to the varied sequences each finger recognized. For example, ZF9 and ZF10 (or ZF12) differ only at the −4 position, with His in ZF9 and Asn in ZF10 and ZF12 (red shading in Fig. 1B).
The conserved G-Arg, G-His, and A–Asn interactions
The seven conserved C:G base pairs in the consensus sequence (Fig. 2A) are recognized primarily by H-bonds between the guanines of the “bottom” recognition strand and arginine or histidine residues of ZF8, ZF9, ZF11, and ZF13. The terminal Nη1 and Nη2 groups of arginine residues 736 of ZF8, 757 of ZF9, 820 of ZF11, and 876 of ZF13 donate H-bonds to the O6 and N7 atoms of guanines at base pair positions 3, 4, 12, and 18, respectively (Fig. 2, D, E, M, and S), a bonding pattern specific to guanine (29, 30). Depending on side chain rotomer conformation, the Nϵ2 group of histidine residues 733 of ZF8, 761 of ZF9, and 873 of ZF13 donate one H-bond to either guanine O6 or guanine N7 (Fig. 2R), the adjacent ring Cϵ1 atom makes a C–H … N type of hydrogen bond (Fig. 2F) or a water-mediated interaction (Fig. 2C).
Triplets 3 and 5 of the consensus sequence include an invariant T:A base pair at positions 8 and 14, which are recognized by Asn789 of ZF10 and Asn845 of ZF12, respectively (Fig. 2, I and O). Juxtaposition of Asn with A is a common mechanism for recognition of this base (29), as the side chain of asparagine donates one H-bond to adenine N7 and accepts one from adenine N6. As mentioned above, ZF10 and ZF12 differ from ZF9 only at position −4; an asparagine (for A) replaces the histidine in ZF9 and changes the base preference (for G). These specific G–Arg, G–His, and A–Asn interactions involve bidentate H-bond interactions. The total of nine invariant base pairs account for half of the recognition sequence (9 of 18). Interestingly, the Arg757–G4 interaction was predicted to involve a variable base pair (supplemental Fig. S3). This may be explained by the fact that Arg757 is located at position −8 of ZF9, which is not part of the canonical model, and thus was not considered in the survey (28).
Interactions with variable base pairs
Interactions with the variable (N) base pairs of the consensus sequence involve water-mediated H-bonds with Asn730 (A1:T1 of triplet 1; Fig. 2B), hydrophobic interaction with Val817 (G11:C11 of triplet 4; Fig. 2L), and a gap in the protein–DNA interface with Asn870 positioned too far away from the base (Fig. 2Q). The smaller serine side chains are also too far away to form interactions: Ser764 of ZF9, Ser792 of ZF10, and Ser848 of ZF12 (dashed lines in Fig. 2, G, J, and P; all located at the −1 position of each finger). Serine and asparagine can each act as both an H-bond donor and acceptor, and they might accommodate alternative base pairs. The participating amino acids may, in other words, alter their conformation so as to interact with different base pairs and, in this way, intimately fit the ZF array to a variety of different sequences. For example, when a T:A base pair occurs to position 1 (A:T in the currently used DNA molecule; Fig. 2B) or 16 (currently G:C; Fig. 2Q), an A–Asn interaction could occur similar to those between asparagine residues at ZF10 and ZF12 that specify the invariant T:A base pairs at positions 8 and 14 (Fig. 2, I and O). We note that this observation (that a serine residue at the −1 position of ZF9, ZF10, or ZF12 could accommodate T, C, or A, whereas arginine at the corresponding −1 position of ZF9, ZF11, and ZF13 recognizes only G) is in agreement with a similar observation made with the designed six-finger protein Aart, where triplets containing 5′ A, C, or T of the recognition strand are typically not specified by direct interaction with the small amino acid (Ala, Ser, Val) in position −1 of the recognition helix (24).
The adaptive interaction with variable sequence by the invariant serine at position −5
Despite complete conservation of Ser at position −5 in each ZF (Fig. 1B), the way in which it interacts with DNA differs from triplet to triplet. Nevertheless, most of the serines interact with the top strand, the nucleotide immediately before and/or the first base pair of the cognate triplet (Fig. 3A). Thus, Ser732 of ZF8 interacts with guanine immediately before its own triplet (G0 in Fig. 3B). Ser760 of ZF9 interacts, via a water molecule, with cytosine of its own triplet (C4 in Fig. 3C). Ser788 of ZF10 interacts with an adenine of the previous triplet (A6 in Fig. 3D). Ser816 (ZF11) joins through a water bridge between a guanine of the previous triplet (G9 in Fig. 3E) and T10 of its own triplet. Ser844 hydrogen-bonds with the first guanine of its own triplet (G13 in Fig. 3F). Finally, Ser872 uses the Cβ carbon atom to make a van der Waals contact with the methyl group of T15 of the previous triplet (Fig. 3G). This “adaptability” stems in part from the ability of serine to act as an H-bond donor or acceptor or both at the same time. This observation also suggests that the cross-strand contact mediated by the small amino acid (such as serine) at position −5, corresponding to position 2 of the structure-based numbering scheme (Fig. 1B, bottom), is generally not a determinant of DNA-binding specificity.
Figure 3.

Adaptive interactions by conserved residues to sequence variations. A, a completely conserved Ser at position −5 in each ZF interacts with DNA that differs from triplet to triplet. B, Ser732 interaction with G0. C, Ser760 interaction with C4 via a water molecule. D, Ser788 hydrogen-bonds with A6. E, Ser816 bridges between G9 and T10 via a water molecule. F, Ser844 interaction with G13. G, Ser872 make a van der Waals contact with T15. H, superimposed ZF9 (cyan) and ZF10 (green). The Arg of the conserved RD dipeptide of ZF9 makes a DNA guanine base interaction (see Fig. 2E), whereas the corresponding Arg of ZF10 makes a DNA-phosphate interaction (I). I, Arg785 of the RD dipeptide of ZF10 contacts a DNA backbone phosphate group. J, superimposition of ZF9 (cyan) and ZF12 (orange). K, Arg841 of the RD dipeptide of ZF12 makes a DNA phosphate interaction. L, Arg869 at the −8 position of the RN dipeptide of ZF13 interaction with a DNA phosphate group.
The adaptive interaction by Arg–Asp dipeptide at positions −8 and −7 to sequence variation
The Arg–Asp (RD) dipeptides at positions −8 and −7 of ZF9, ZF10, and ZF12 (shaded green in Fig. 1B) provide additional examples of the adaptability of PRDM9c to sequence variations. In ZF9, Arg757 conforms to C4:G4 as the first base pair of its triplet (CCA) and forms bidentate H-bonds with the guanine in the bottom strand (Fig. 3C), whereas Asp758 hydrogen-bonds with the arginine as well as a water-mediated network with the paired cytosine of the opposite strand (Fig. 3C). In ZF10 and ZF12, these same amino acids adopt different conformations and partners. Specifically, Asp786 matches to the first base pair of its triplet (GTG) and makes an H-bond with the cytosine in the bottom strand (Fig. 2H); by doing so, the adjacent Arg785 instead interacts with a backbone phosphate group (Fig. 3, H and I). The same conformational change of an RD dipeptide is also evident in ZF12 (Fig. 3, J and K). Thus, the RD dipeptide can accommodate changes from C:G (with G–Arg interaction) to G:C (with C–Asp interaction). We note that Asp can bind unmodified cytosine (21, 26, 31). The same adaptability could apply to ZF13, which contains the Arg–Asn (RN) dipeptide at positions −8 and −7 (shaded green in Fig. 1B), allowing recognition of the first base pair of a triplet when it is either C:G (G–Arg) or T:A (A–Asn) (Fig. 3L). The latter interaction would be analogous to A8–Asn789 of ZF10 and A14-Asn845 of ZF12 (Fig. 2, I and O).
The thymine–tryptophan van der Waals interaction in ZF11
As noted earlier, the expansion of ZF11 of PRDM9a into two fingers resulted in an extra finger in PRDM9c (Fig. 1A). Allele C ZF11 possesses hydrophobic (Val) and aromatic (Trp) residues at positions −4 and −7, whereas ZF12 has all of the usual features of a regular zinc finger unit, with polar or charged residues at the DNA base-interacting positions (Fig. 1B). The indole ring of Trp814 of ZF11 forms a π interaction with the methyl group of thymine T10 (Fig. 2K). The methyl-π interactions are also used by the zinc finger protein ZNF217 in interaction with DNA, where a tyrosine contacts a thymine methyl group (32). In contrast, the hydrophobic residue Val817 of ZF11 allows a variable base at base pair position 11, probably due to its ability to rotate the side chain torsion angle, moving the terminal methyl groups into or out of contact (Fig. 2L). In contrast, the prediction strongly forecasted that the Val817 corresponding nucleotide position 11 was a G:C base pair (supplemental Fig. S2), probably influenced by the within-finger context of amino acids immediately surrounding Val817 (22).
Although Trp/Tyr–thymine methyl interactions have not often been described in classical native ZF-DNA complexes, aromatic (Phe, Tyr) or hydrophobic (Val, Ala) residues have been observed in the bacterial one-hybrid screen against three-finger proteins in response to selection for binding thymine (22). In addition, a negatively charged glutamate was observed interacting with the methyl groups of thymine or 5-methylcytosine in Klf4 (31) and Kaiso (33).
Structural comparison of PRDM9 A and C alleles in complex with allele-specific DNA
To gain insights from the available structural information, we compared PRDM9a-DNA and PRDM9c-DNA complex structures. In the structure of PRDM9a, five-finger fragment ZF8–12 was used for crystallization, but the last finger (ZF12) could not be seen in the structure (18). Superimposition of the four fingers shared between the two structures (ZF8–11) yielded an RMSD of ∼3 Å over 106 pairs of Cα atoms. The largest difference lies at one end of the DNA molecule, where PRDM9a has no DNA contacts and PRDM9c has two C-terminal zinc fingers (ZF12–13) bound (Fig. 4, A and B). DNA flexibility in the absence of paired ZF unit and crystal packing lattice force involving DNA ends might have influenced the DNA conformation observed here.
Figure 4.
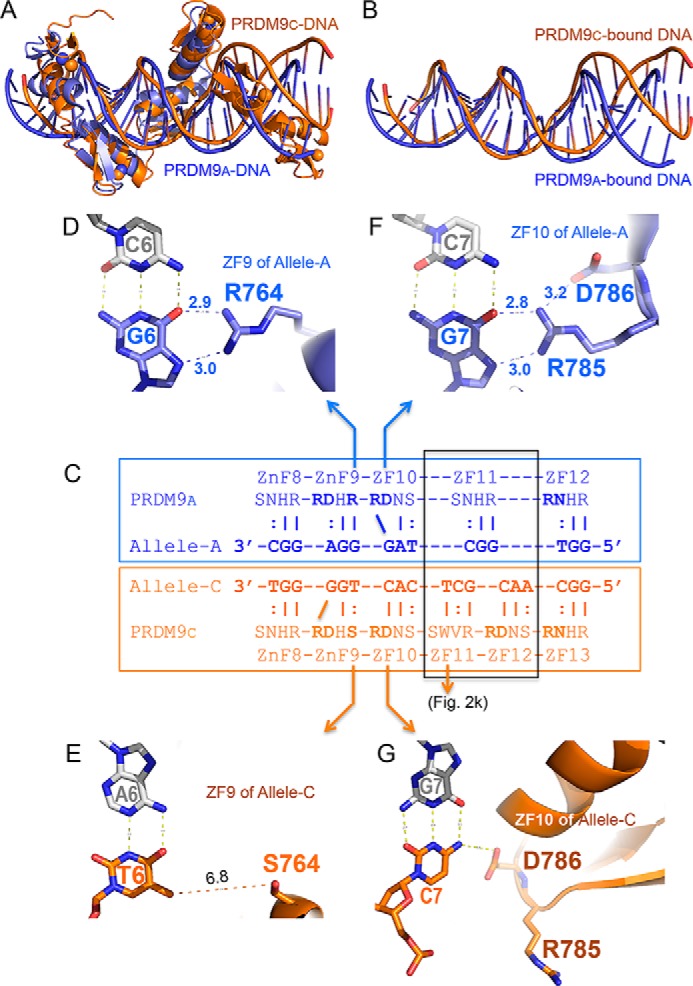
Comparison of PRDM9a and PRDM9c interaction with allele-specific DNA. A, superimposition of PRDM9a-DNA (blue; PDB entry 5EGB) and PRDM9c-DNA (orange) complex structures. B, superimposition of PRDM9a-bound DNA molecule (blue) and PRDM9c-bound DNA molecule (orange). For clarity, the protein components have been removed from the superimposition. C, the allele A–specific sequence (blue) and allele C–specific sequence (orange) are aligned to the corresponding ZF with amino acids at the −8, −7, −4, and −1 positions (see Fig. 1B, top). The expansion of one finger (ZF11) in PRDM9a to two fingers (ZF11–12) in PRDM9c is highlighted in a gray box. D, Arg764 of allele A interacts with G6. E, Ser764 of allele C allows a variable base at base pair position 6. F, RD dipeptide of ZF10 in allele A interacts with G7. G, RD dipeptide of ZF10 in allele C interacts with C7.
From the protein side, two types of differences were observed between the complex structures. One difference results from the protein sequence change. As mentioned, allele C differs from A by an arginine-to-serine change, R764S, at position −1 of ZF9 (Fig. 4C) (12, 13, 17). Arg764 of PRDM9a recognizes the conserved guanine of the C:G base pair in the allele A–specific consensus (18) (Fig. 4D), whereas Ser764 of PRDM9c allows a variable nucleotide at the corresponding position that lacks specific interaction with the base (Fig. 4E). The expansion of ZF11 into two fingers allows PRDM9c to recognize a longer 18-bp consensus sequence, instead of a 15-bp sequence as in PRDM9a. The additional ZF provides an infrequently observed feature of aromatic Trp814 contacting the thymine T10 methyl group (Fig. 2K) (see “Discussion”). These changes allow PRDM9c to bind 3-bp-longer sequences and gain additional interactions with the associated backbone phosphate groups (supplemental Fig. S4). The net result is >10-fold enhanced binding affinity (18).
The second type of difference results from the adaptability of the RD dipeptide to sequence variations. In PRDM9a, the RD dipeptide in ZF10 conforms to C:G as the first base pair of its triplet (CTA), and the Arg forms two H-bonds with the guanine (18) (Fig. 4F). In contrast, the corresponding RD residues in PRDM9c adopt different conformations and partners, due to the substitution of C:G to G:C base pair, and the Asp hydrogen-bonds with the cytosine (Fig. 4G), whereas the Arg interacts with a backbone phosphate group (Fig. 3I). The same adaptability applies to RD in ZF9 of both alleles, RD in ZF12 of PRDM9c and RN in ZF12 of PRDM9a and ZF13 of PRDM9c (Fig. 4C).
We note that the arginine residue of the RD dipeptide is located outside of the canonical positions of the helix. Here, we have demonstrated that an identical ZF unit (ZF10) interacts with two different sequences: TAG with PRDM9a and CAC with PRDM9c. This is not due to the ability of an amino acid in a given position (for instance, Asp at position −7) to specify different bases at the corresponding 3′ nucleotide position. Rather, it is due to the ability of the RD dipeptide to undergo conformation switching. We first observed this unique RD switch in the two-finger structure of Zfp57 (34), a related KRAB-ZF protein. We do not know whether this feature is unique to the family of KRAB-ZF proteins, which are the largest and most rapidly diversifying family of DNA-binding transcriptional regulators in mammals (35). Nevertheless, our study illustrates that (i) residue(s) outside of the canonical positions of the ZF helix contribute to the DNA-binding specificity, and (ii) an identical ZF can recognize different sequences via the RD conformational switch.
Discussion
To summarize, our results from in vitro studies of interaction between human PRDM9 C-terminal C2H2 ZF tandem array and allele-specific DNA provide a structural explanation for the sequence adaptability between the A and C alleles, the two most common alleles found in African populations (13, 14). The importance of the ZF tandem array in hot spot activation is demonstrated by different alleles that activate completely different, allele-specific hot spots yet differ in the DNA-interacting ZFs, whether due to a subtle single amino acid change or expansion/deletion of an additional unit in the ZF array (12–15). A recent experiment humanized the ZF array in mouse Prdm9 (by replacing the mouse ZF array with the orthologous sequence from humans), reversing the hybrid infertility between musculus and domesticus subspecies by entirely reprogramming recombination hot spots (36). Hybrid sterility is a common mechanism for preventing gene exchange between related species throughout the animal and plant kingdoms (37). Altering one Prdm9 allele in mice mimics the consequences of a newly arising allele and thus links the Prdm9 DNA-binding ZF array to its putative role in speciation. In vitro studies indicate that the PRDM9 ZF array forms a stable complex with its specific DNA sequence, with a dissociation halftime of many hours (38). In addition, each allele can bind DNA with high affinity while recognizing sequences with high variability in the consensus sequence motif (18). This property of PRDM9 can be traced to the ability of specific residues in each ZF unit to adopt alternative conformations, allowing it to establish versatile H-bonds with some bases but not with others.
The first seven fingers (ZF1–7) and the last finger (ZF13 in PRDM9a and ZF14 in PRDM9c) appear to be dispensable for binding of specific hot spot sequences. For example, allele B differs from A by a serine-to-threonine change, S680T, at position −1 of ZF6 (12) (the same change occurs in allele C). However, 88% of hot spots in a heterozygous A/B individual overlapped those in two A/A individuals, which themselves overlapped by 89%, suggesting that PRDM9b does not specify a distinct set of hot spots (4). It is evident that the residues at the base-interacting positions for these dispensable fingers are largely small amino acids (Ser/Thr) and hydrophobic or aromatic (Val/Tyr/Trp), whereas the corresponding residues for the functional fingers are large and charged/polar residues (Arg/His/Asp/Asn).
However, the apparently dispensable fingers provide a potential pool for duplication and variation when the need arises. The Trp-containing ZF11 could be considered as a duplication of identical ZF3, ZF6, or ZF7, although with the smaller threonine at position −1 replaced by a larger and charged arginine (Fig. 1B), generating a functional finger. On the other hand, alleles A and C might evolve independently; in PRDM9a, ZF11 is a duplication of ZF8, whereas in PRDM9c, ZF12 is a duplication of ZF10. The amount of variability among ZFs, as indicated by the number of unique residues at a given position comparing all ZFs in a protein, is highest in the first two and last ZFs and at ZF8 and very low in the intervening and DNA-binding ZFs of PRDM9 (unique positions highlighted in yellow in Fig. 5A). This pattern is similar to that seen for the chimpanzee ortholog, with higher variability near the ends (ZF2 and ZF14) (Fig. 5B). In contrast, the orthologs for macaque and gibbon show a different pattern, with the least variability at the two ends and at ZF8 and the highest variability between these three points (Fig. 5, C and D). This may reflect a particularly important role in DNA recognition by ZF8 in those orthologs, perhaps contributing to the presently only partially explained basis for speciation of gibbons and related primates (39). Together, our structures of PRDM9-DNA complexes provide important insights into molecular mechanisms of PRDM9 action for the diversity, flexibility, and conservation of allele-specific DNA binding across the human genomes.
Figure 5.

Sequence alignments of tandem ZF arrays. A, Hsa Hominidae (great apes/humans) NP_001297143.1 PRDM9 allele B (Thr680) (Homo sapiens). (Note that in the NCBI reference sequence, it was named as isoform A; a second sequence, NP_064612.2, was named as isoform B.) For human PRDM9 allele A, B, and C annotation, see Ref. 4. B, Ptr Hominidae (great apes/humans) ACZ82295.1 zinc finger region PR domain-containing 9, partial (Pan troglodytes). C, Mne Cercopithecidae (old world monkeys) XP_011763301.1 PREDICTED: Krueppel-related zinc finger protein 1 isoform X2 (Macaca nemestrina). D, Nle Hylobatidae (gibbons) XP_012365846.1 PREDICTED: Krueppel-related zinc finger protein 1 isoform X7 (Nomascus leucogenys). We note that aside from the fact that the gibbon protein is the best BlastP hit, there is no biochemical proof that it is the PRDM9. The numbers indicate the unique residues at a given position comparing all ZFs in a protein.
Experimental procedures
The gene encompassing the C-terminal ZF array of human PRDM9c (ZF8–14) was synthesized by GENEWIZ and subcloned into pGEX-6p1 vector (pXC1289). The ZF8–13 construct (pXC1505) was generated by PCR from the ZF8–14 plasmid DNA and subcloned into the pGEX-6p1 vector. The protein was expressed and purified using the protocol as described (18, 40) with a few changes. First, after Precission cleavage, the protein was not diluted for salt concentration but was loaded directly at 0.5 m NaCl onto tandem Hitrap Q-SP columns (GE Healthcare). Most of the protein flowed through the Q column onto the SP column from which it was eluted as a single peak at ∼0.8 m NaCl using a linear gradient of NaCl from 0.5 to 1 m. Second, the protein was further purified on a Superdex-200 (16/60) column with the storage buffer containing 20 mm Tris (pH 7.5), 500 mm NaCl, 5% glycerol, 25 μm ZnCl2, and 0.5 mm tris(2-carboxyethyl)phosphine hydrochloride.
Fluorescence polarization was used to measure the dissociation constants (KD), as described (18, 40). KD values were calculated as [mP] = [maximum mP] × [C]/(KD + [C]) + [baseline mP], where [mP] is millipolarization and [C] is protein concentration. Because the last finger (ZF14) did not contribute substantially to the DNA-binding affinity (supplemental Fig. S5), we used ZF8–13 for co-crystallization. Purified ZF8–13 proteins were incubated with the double-stranded DNA (Table 1), synthesized by Integrated DNA Technologies, at an equimolar ratio to a final concentration of 25 μm on ice in the storage buffer. The protein-DNA complexes were formed by dialysis against the same buffer components with 250 mm NaCl but without ZnCl2. The complex was further concentrated up to ∼0.6 mm before crystallization. Crystallization conditions were screened using a crystallization robot (PHOENIX, Art Robbins Instruments) and commercial screens from Hampton Research. This method utilized the sitting drop vapor diffusion method. The PHOENIX instrument mixed an aliquot of protein-DNA complex (0.2 μl) with an equal volume of crystallization mother liquor. The best-diffracting crystal used for data collection was obtained from optimized conditions of 0.1 m BisTris propane, pH 7.3, and 23% (w/v) polyethylene glycol 3350, using manually set-up hanging drop vapor diffusion at 16 °C. The crystals were flash-frozen under liquid nitrogen using 20% glycerol as cryoprotectant.
X-ray diffraction data were remotely collected at the SER-CAT 22-ID beamline of the Advanced Photon Source at the Argonne National Laboratory and processed by HKL2000 (41). The autosolve module of PHENIX (42) was used for initial crystallographic phasing calculation by single-wavelength anomalous dispersion of zinc signals. The initial electron density revealed clearly visible DNA molecules, and a B-DNA model made by the “make-na server” (http://structure.usc.edu/make-na/server.html)3 was placed into the density manually and refined using PHENIX REFINE (43). This partial model was then used to search for the ZF proteins and place them into the density by molecular replacement using PHASER-MR (44). The structure was further refined using PHENIX, and the model was manually adjusted by COOT (45). Structure quality was analyzed and validated by the PDB validation server (46). Molecular graphics were generated using PyMOL (Schroedinger, LLC, New York).
Author contributions
A. P. performed all experimental work. R. M. B. performed data analysis and assisted in preparing the manuscript; X. Z. and X. C. organized and designed the scope of the study. All authors were involved in analyzing data and preparing the manuscript.
Supplementary Material
Acknowledgments
We thank John R. Horton for discussion. The Department of Biochemistry of Emory University School of Medicine supported the use of the SER-CAT beamlines.
This work was supported in part by National Institutes of Health Grant GM049245-23 to (X. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S5, and Table S1.
The atomic coordinates and structure factors (code 5V3G) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- KRAB
- Krüppel-associated box
- ZF
- zinc finger
- RMSD
- root mean square deviation
- PDB
- Protein Data Bank
- H-bond
- hydrogen bond
- BisTris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane.
References
- 1. Baker Z., Schumer M., Haba Y., Bashkirova L., Holland C., Rosenthal G. G., and Przeworski M. (2017) Repeated losses of PRDM9-directed recombination despite the conservation of PRDM9 across vertebrates. eLife 10.7554/eLife.24133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hayashi K., Yoshida K., and Matsui Y. (2005) A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature 438, 374–378 [DOI] [PubMed] [Google Scholar]
- 3. Mihola O., Trachtulec Z., Vlcek C., Schimenti J. C., and Forejt J. (2009) A mouse speciation gene encodes a meiotic histone H3 methyltransferase. Science 323, 373–375 [DOI] [PubMed] [Google Scholar]
- 4. Pratto F., Brick K., Khil P., Smagulova F., Petukhova G. V., and Camerini-Otero R. D. (2014) DNA recombination: recombination initiation maps of individual human genomes. Science 346, 1256442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu H., Mathioudakis N., Diagouraga B., Dong A., Dombrovski L., Baudat F., Cusack S., de Massy B., and Kadlec J. (2013) Molecular basis for the regulation of the H3K4 methyltransferase activity of PRDM9. Cell Rep. 5, 13–20 [DOI] [PubMed] [Google Scholar]
- 6. Eram M. S., Bustos S. P., Lima-Fernandes E., Siarheyeva A., Senisterra G., Hajian T., Chau I., Duan S., Wu H., Dombrovski L., Schapira M., Arrowsmith C. H., and Vedadi M. (2014) Trimethylation of histone H3 lysine 36 by human methyltransferase PRDM9 protein. J. Biol. Chem. 289, 12177–12188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Koh-Stenta X., Joy J., Poulsen A., Li R., Tan Y., Shim Y., Min J. H., Wu L., Ngo A., Peng J., Seetoh W. G., Cao J., Wee J. L., Kwek P. Z., Hung A., et al. (2014) Characterization of the histone methyltransferase PRDM9 using biochemical, biophysical and chemical biology techniques. Biochem. J. 461, 323–334 [DOI] [PubMed] [Google Scholar]
- 8. Oliver P. L., Goodstadt L., Bayes J. J., Birtle Z., Roach K. C., Phadnis N., Beatson S. A., Lunter G., Malik H. S., and Ponting C. P. (2009) Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genet. 5, e1000753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thomas J. H., Emerson R. O., and Shendure J. (2009) Extraordinary molecular evolution in the PRDM9 fertility gene. PLoS One 4, e8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ségurel L., Leffler E. M., and Przeworski M. (2011) The case of the fickle fingers: how the PRDM9 zinc finger protein specifies meiotic recombination hotspots in humans. PLoS Biol. 9, e1001211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Groeneveld L. F., Atencia R., Garriga R. M., and Vigilant L. (2012) High diversity at PRDM9 in chimpanzees and bonobos. PLoS One 7, e39064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baudat F., Buard J., Grey C., Fledel-Alon A., Ober C., Przeworski M., Coop G., and de Massy B. (2010) PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327, 836–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berg I. L., Neumann R., Lam K. W., Sarbajna S., Odenthal-Hesse L., May C. A., and Jeffreys A. J. (2010) PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat. Genet. 42, 859–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Berg I. L., Neumann R., Sarbajna S., Odenthal-Hesse L., Butler N. J., and Jeffreys A. J. (2011) Variants of the protein PRDM9 differentially regulate a set of human meiotic recombination hotspots highly active in African populations. Proc. Natl. Acad. Sci. U.S.A. 108, 12378–12383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kong A., Thorleifsson G., Gudbjartsson D. F., Masson G., Sigurdsson A., Jonasdottir A., Walters G. B., Jonasdottir A., Gylfason A., Kristinsson K. T., Gudjonsson S. A., Frigge M. L., Helgason A., Thorsteinsdottir U., and Stefansson K. (2010) Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467, 1099–1103 [DOI] [PubMed] [Google Scholar]
- 16. Hinch A. G., Tandon A., Patterson N., Song Y., Rohland N., Palmer C. D., Chen G. K., Wang K., Buxbaum S. G., Akylbekova E. L., Aldrich M. C., Ambrosone C. B., Amos C., Bandera E. V., Berndt S. I., et al. (2011) The landscape of recombination in African Americans. Nature 476, 170–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jeffreys A. J., Cotton V. E., Neumann R., and Lam K. W. (2013) Recombination regulator PRDM9 influences the instability of its own coding sequence in humans. Proc. Natl. Acad. Sci. U.S.A. 110, 600–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Patel A., Horton J. R., Wilson G. G., Zhang X., and Cheng X. (2016) Structural basis for human PRDM9 action at recombination hot spots. Genes Dev. 30, 257–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pavletich N. P., and Pabo C. O. (1991) Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 Å. Science 252, 809–817 [DOI] [PubMed] [Google Scholar]
- 20. Wolfe S. A., Nekludova L., and Pabo C. O. (2000) DNA recognition by Cys2His2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct. 29, 183–212 [DOI] [PubMed] [Google Scholar]
- 21. Choo Y., and Klug A. (1997) Physical basis of a protein-DNA recognition code. Curr. Opin. Struct. Biol. 7, 117–125 [DOI] [PubMed] [Google Scholar]
- 22. Persikov A. V., Wetzel J. L., Rowland E. F., Oakes B. L., Xu D. J., Singh M., and Noyes M. B. (2015) A systematic survey of the Cys2His2 zinc finger DNA-binding landscape. Nucleic Acids Res. 43, 1965–1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim C. A., and Berg J. M. (1996) A 2.2 A resolution crystal structure of a designed zinc finger protein bound to DNA. Nat. Struct. Biol. 3, 940–945 [DOI] [PubMed] [Google Scholar]
- 24. Segal D. J., Crotty J. W., Bhakta M. S., Barbas C. F. 3rd, Horton N. C. (2006) Structure of Aart, a designed six-finger zinc finger peptide, bound to DNA. J. Mol. Biol. 363, 405–421 [DOI] [PubMed] [Google Scholar]
- 25. Peisach E., and Pabo C. O. (2003) Constraints for zinc finger linker design as inferred from X-ray crystal structure of tandem Zif268-DNA complexes. J. Mol. Biol. 330, 1–7 [DOI] [PubMed] [Google Scholar]
- 26. Hashimoto H., Wang D., Horton J. R., Zhang X., Corces V. G., and Cheng X. (2017) Structural basis for the versatile and methylation-dependent binding of CTCF to DNA. Mol. Cell 66, 711–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chandrasegaran S., and Carroll D. (2016) Origins of programmable nucleases for genome engineering. J. Mol. Biol. 428, 963–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Persikov A. V., and Singh M. (2014) De novo prediction of DNA-binding specificities for Cys2His2 zinc finger proteins. Nucleic Acids Res. 42, 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luscombe N. M., Laskowski R. A., and Thornton J. M. (2001) Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 29, 2860–2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vanamee E. S., Viadiu H., Kucera R., Dorner L., Picone S., Schildkraut I., and Aggarwal A. K. (2005) A view of consecutive binding events from structures of tetrameric endonuclease SfiI bound to DNA. EMBO J. 24, 4198–4208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hashimoto H., Wang D., Steves A. N., Jin P., Blumenthal R. M., Zhang X., and Cheng X. (2016) Distinctive Klf4 mutants determine preference for DNA methylation status. Nucleic Acids Res. 44, 10177–10185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vandevenne M., Jacques D. A., Artuz C., Nguyen C. D., Kwan A. H., Segal D. J., Matthews J. M., Crossley M., Guss J. M., and Mackay J. P. (2013) New insights into DNA recognition by zinc fingers revealed by structural analysis of the oncoprotein ZNF217. J. Biol. Chem. 288, 10616–10627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buck-Koehntop B. A., Stanfield R. L., Ekiert D. C., Martinez-Yamout M. A., Dyson H. J., Wilson I. A., and Wright P. E. (2012) Molecular basis for recognition of methylated and specific DNA sequences by the zinc finger protein Kaiso. Proc. Natl. Acad. Sci. U.S.A. 109, 15229–15234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Y., Toh H., Sasaki H., Zhang X., and Cheng X. (2012) An atomic model of Zfp57 recognition of CpG methylation within a specific DNA sequence. Genes Dev. 26, 2374–2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Imbeault M., Helleboid P. Y., and Trono D. (2017) KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 543, 550–554 [DOI] [PubMed] [Google Scholar]
- 36. Davies B., Hatton E., Altemose N., Hussin J. G., Pratto F., Zhang G., Hinch A. G., Moralli D., Biggs D., Diaz R., Preece C., Li R., Bitoun E., Brick K., Green C. M., et al. (2016) Re-engineering the zinc fingers of PRDM9 reverses hybrid sterility in mice. Nature 530, 171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maheshwari S., and Barbash D. A. (2011) The genetics of hybrid incompatibilities. Annu. Rev. Genet. 45, 331–355 [DOI] [PubMed] [Google Scholar]
- 38. Striedner Y., Schwarz T., Welte T., Futschik A., Rant U., and Tiemann-Boege I. (2017) The long zinc finger domain of PRDM9 forms a highly stable and long-lived complex with its DNA recognition sequence. Chromosome Res. 25, 155–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Carbone L., Harris R. A., Gnerre S., Veeramah K. R., Lorente-Galdos B., Huddleston J., Meyer T. J., Herrero J., Roos C., Aken B., Anaclerio F., Archidiacono N., Baker C., Barrell D., Batzer M. A., et al. (2014) Gibbon genome and the fast karyotype evolution of small apes. Nature 513, 195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Patel A., Hashimoto H., Zhang X., and Cheng X. (2016) Characterization of how DNA modifications affect DNA binding by C2H2 zinc finger proteins. Methods Enzymol. 573, 387–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Otwinowski Z., Borek D., Majewski W., and Minor W. (2003) Multiparametric scaling of diffraction intensities. Acta Crystallogr. A 59, 228–234 [DOI] [PubMed] [Google Scholar]
- 42. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 46. Read R. J., Adams P. D., Arendall W. B. 3rd, Brunger A. T., Emsley P., Joosten R. P., Kleywegt G. J., Krissinel E. B., Lütteke T., Otwinowski Z., Perrakis A., Richardson J. S., Sheffler W. H., Smith J. L., Tickle I. J., et al. (2011) A new generation of crystallographic validation tools for the protein data bank. Structure 19, 1395–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.