

Abstract
Oxidized modifications of LDL (oxLDL) play a key role in the development of endothelial dysfunction and atherosclerosis. However, the underlying mechanisms of oxLDL-mediated cellular behavior are not completely understood. Here, we compared the effects of two major types of oxLDL, copper-oxidized LDL (Cu2+-oxLDL) and lipoxygenase-oxidized LDL (LPO-oxLDL), on proliferation of human aortic endothelial cells (HAECs). Cu2+-oxLDL enhanced HAECs’ proliferation in a dose- and degree of oxidation-dependent manner. Similarly, LPO-oxLDL also enhanced HAEC proliferation. Mechanistically, both Cu2+-oxLDL and LPO-oxLDL enhance HAEC proliferation via activation of Rho, Akt phosphorylation, and a decrease in the expression of cyclin-dependent kinase inhibitor 1B (p27kip1). Both Cu2+-oxLDL or LPO-oxLDL significantly increased Akt phosphorylation, whereas an Akt inhibitor, MK2206, blocked oxLDL-induced increase in HAEC proliferation. Blocking Rho with C3 or its downstream target ROCK with Y27632 significantly inhibited oxLDL-induced Akt phosphorylation and proliferation mediated by both Cu2+- and LPO-oxLDL. Activation of RhoA was blocked by Rho-GDI-1, which also abrogated oxLDL-induced Akt phosphorylation and HAEC proliferation. In contrast, blocking Rac1 in these cells had no effect on oxLDL-induced Akt phosphorylation or cell proliferation. Moreover, oxLDL-induced Rho/Akt signaling downregulated cell cycle inhibitor p27kip1. Preloading these cells with cholesterol, however, prevented oxLDL-induced Akt phosphorylation and HAEC proliferation. These findings provide a new understanding of the effects of oxLDL on endothelial proliferation, which is essential for developing new treatments against neovascularization and progression of atherosclerosis.
Keywords: oxidized modifications of low-density lipoproteins, Rho kinase
oxidized modifications of LDL (oxLDL) are known to play a major role in the development of atherosclerosis and endothelial dysfunction (38, 59). Multiple studies have shown that oxLDL has cytotoxic effects on vascular endothelial cells, inhibits bioavailability of nitric oxide (NO), and results in the disruption of the endothelial barrier (41, 42). Our studies demonstrated that oxLDL also has a major impact on endothelial biomechanical properties, resulting in increased endothelial stiffness and contractility (9, 54). Furthermore, we showed that this effect is accompanied by enhanced formation of endothelial networks and capillary lumens in three-dimensional cell cultures (9, 19, 54). Recently, we showed that preexposure to oxLDL facilitates formation of functional capillaries in matrigels in vivo via activation of the RhoA/Rho kinase (ROCK) signaling pathway (45). In this study, we focus on the role of oxLDL in the regulation of endothelial proliferation.
Several studies addressed the role of oxLDL in control of endothelial proliferation, but the results were controversial, with several studies reporting that oxLDL enhances endothelial proliferation, whereas others showed inhibitory effects (12, 20, 52, 63). The emerging trend is that relatively low concentrations of oxLDL (<10–20 µg/ml) have proproliferative effects, whereas higher levels of oxLDL are inhibitory. Because of the lack of a comprehensive study that takes into the account the key parameters, such as the mode and the degree of LDL oxidation, most of these findings have been inconclusive. Multiple studies have shown that there are significant differences in the proinflammatory effects of minimally oxidized, moderately oxidized, and strongly oxidized LDL. These findings indicate that oxLDL particles are highly heterogeneous and that the degree and the mode of oxidation significantly affect the biological activity of oxLDL (35). It is also well known that Cu2+ oxidation, the most common method of LDL oxidation in vitro that was used in the earlier studies of oxLDL effects on endothelial proliferation, does not occur in vivo. Notably, this method yields oxidation products that are significantly different from the products of enzymatic LDL oxidation, such as oxidation by lipoxygenase (LPO) that mimics the oxidation occurring in vivo (35). Thus, the impact of physiologically oxidized LDL on endothelial proliferation remains unclear. Therefore, our first objective was to clarify the impact of oxLDL on endothelial proliferation by testing several key parameters: the degree of oxidation, the mode of oxidation based on the oxidizing agent (Cu2+ vs. lipoxygenase), the concentration range, and the duration of the exposure [short (1 h) vs. prolonged (24 h)], focusing on human aortic endothelial cells (HAECs).
In terms of the mechanism, two pathways have been identified to mediate oxLDL-induced proliferative effects: activation of the RhoA/ROCK pathway linked to proliferation through a decrease in the expression of cyclin-dependent kinase inhibitor 1B (p27kip1) (52) or activation of Akt, a serine/threonine protein kinase that is well known to play a crucial role in the control of cell proliferation and progression of cell cycle (61). However, the effects of oxLDL on AKT1 were also reported to be highly controversial, and whereas some studies showed that oxLDL enhances phosphorylation of Akt in endothelial cells (63), other studies found an inhibitory effect (2, 11). Therefore, our next objective was to provide comparative analysis of Cu2+-oxLDL and LPO-oxLDL on the Rho and Akt signaling pathways and determine whether the two pathways are functionally linked. Furthermore, we also provide the first insights into the mechanism responsible for oxLDL-induced RhoA activation, one of the major unanswered questions in the field. In general, three types of mechanisms were shown to mediate receptor-induced activation of Rho GTPases: 1) GDIs, guanine nucleotide dissociation inhibitors that stabilize Rho GTPases in their inactive GDP-bound form (18, 23); 2) GEFs, guanine nucleotide exchange factors that activate Rho GTPases by facilitating the dissociation of GDP and binding of GTP; and 3) GAPs, GTPase-activating proteins that negatively regulate Rho GTPases by enhancing their GTPase activities (18, 23, 24, 31). Here, we provide the first evidence for the role of Rho-GDI-1 in oxLDL-induced activation of RhoA.
Another major question in oxLDL-induced signaling is the nature of the primary event that links oxLDL to the initiation of the signaling cascades. We proposed earlier that oxLDL-induced activation of the RhoA/ROCK pathway is mediated by the disruption of lipid packing of the plasma membrane, which results from membrane incorporation of oxidized lipids (45). We showed that exposure to oxLDL does not lead to cholesterol loading of endothelial cells, as was previously assumed based on its recognition by scavenger receptors (6, 35), but instead induces effects similar to those of cholesterol depletion on an array of endothelial properties, including lipid packing of the membrane, endothelial stiffness and contractility, sensitivity to flow, and branching behavior (9, 32, 54). Furthermore, enriching endothelial cells with cholesterol prevents oxLDL-induced activation of RhoA and endothelial stiffening (45). These observations led us to conclude that in contrast to the previous belief, normal nonmodified and oxidized forms of LDL may have opposite effects on endothelial function. Clearly, this distinction is expected to have major implications both on the basic understanding of LDL/oxLDL effects on cellular function and on possible therapeutic strategies to alleviate these effects. In this study, we address this hypothesis, focusing on endothelial proliferation.
METHODS
Cell culture and reagents.
Human aortic endothelial cells (HAECs; Lonza, Allendale, NJ) were grown between passages 6 and 10; cells were cultured according to the manufacturer’s instructions at 37°C with 5% CO2 with EGM-2 Bullet Kit media (Lonza). Lipoprotein-deficient serum (Sigma) was used to replace the normal serum in the Bullet Kit for the proliferation study. CuSO4, cholesterol, and soybean lipoxygenase were obtained from Sigma (St. Louis, MO); Y27632 [((+)-(R)-trans-4-(1-aminoethyl)-N-(4-pyridyl))] was obtained from Cayman Chemical (Ann Arbor, MI). MK2206 was from Selleckchem (Houston, TX), and C3 was from Cytoskeleton (Denver, CO). Methyl-β cyclodextrin (MβCD) saturated with cholesterol was prepared as described previously (34). Antibodies for p-Akt no. 4060, Akt no. 4691, p27 Kip1 no. 2552, pMLC2 no. 3674, and MLC2 no. 3672 were purchased from Cell Signaling Technology (Beverly, MA), and for green fluorescent protein (GFP), SC-9996, c-Myc, and SC-40 were purchased from Santa Cruz Biotechnology.
Transfection, siRNA, and cDNAs.
Cells were transfected using a Nucleofector Kit for Mammalian Endothelial Cells from Lonza (no. VPI-1001), following the manufacturer’s optimized protocol on the Amaxa nucleofector apparatus. Immediately after pulsing, the cells were transferred to six-well plates and cultured for 48 h before experiments. siRNAs were ordered from IDT (TriFECTa Kit). Eighty to 90 percent confluent cells were transfected in Opti-MEM (Thermo Fisher Scientific) media using Lipofectamine 3000 Reagent (Invitrogen) and a siRNA mix for 4 h. HAEC-enriched cell culture media (Lonza) was then used to replace the Opti-MEM solution, and cells were allowed to grow for 48 h before experiments were performed. GFP-tagged-RhoGDI-1 was generated by PCR amplification (PFU; Stratagene) of the cDNA clone pOTB7-GDI-1 into p-EGFP-C3 vector using specific primer pairs [p-EGFP-FL-GDI-1 (AA1-204)] as described (19): forward primer 5′-AGGAATTCGAATGGCTGAGCAGGAGCCCAC-3′ and reverse primer 5′-CGGGATCCTCATCAGTCCTTCCAGTCCTTC-3′. We used a mutant containing the p115-RGS domain (RGS mutant) to address the role of p115RhoGEF in regulating RhoA activity as described (24)
LDL isolation and oxidation.
Human plasma was obtained from healthy subjects from the local blood bank (Lifesource, Chicago, IL). Briefly, plasma was separated by ultracentrifugation in potassium bromide (KBr) with a final density of 1.063 g/ml. Then pooled human LDL was dialyzed to remove EDTA and KBr. Copper-oxidized oxLDL was prepared by incubating 1 mg/ml native LDL with 25 μM copper sulfate for 16 h at 37°C, and then 1 mM EDTA was added to stop the reaction, as described previously (54). The content of thiobarbituric acid-reactive substances (TBARS) in LDL and oxLDL was determined by using a thiobarbituric acid-reactive substance assay kit (ZeptoMetrix, Buffalo, NY), as expressed with malondialdehyde equivalents. Cu-oxLDL with TBARS at 15 ± 3 was used in the experiments. Lipoxygenase-oxidized LDL (LPO-oxLDL) was made by incubating 1 mg/ml native LDL with 5,000 U/ml soybean lipoxygenase overnight at 37°C, as described previously (54). Increase in absorbance at 234 nm was used to monitor the oxidation of LDL, a method used in previous studies (10). The average increase of OD 234 after overnight incubation is ∼0.4 in our experiments. TBARS value typically increased to ∼3 for LPO-oxLDL, as previously reported (not shown) (24). For cell proliferation assays [5-bromo-2′-deoxyuridine (BrdU) and MTT), HAECs were grown to 50% confluency, serum starved for 2 h, and then exposed to 10–100 µg/ml oxLDL in lipoprotein-deficient serum for 1 or 24 h. For Western blot confluent or subconfluent, HAECs were also serum starved for 2 h and exposed to 50 µg/ml oxLDL for 15 min (Akt) or 24 h (p27kip).
BrdU assay.
Proliferation was measured using the 5-bromo-2′-deoxyuridine (BrdU) labeling detection kit III (Roche), as described (46). After exposure to oxLDL, BrdU labeling reagent was added for 6 h, and then cells were fixed with precooled ethanol fixative for 20 min in −20°C. The fixed cells were incubated with nuclease working solution followed by anti-BrdU-POD Fab fragments working solution and then anti-mouse-Ig-AP solution [1:100 in PBS+]. Cells were then covered in color substrate solution and NBT/BCIP solution 1:50 in substrate buffer [100 mM Tris·HCl buffer, 100 mM NaCl, and 50 mM MgCl2, pH 9.5] at 15–25°C for 15–30 min. Pictures were taken at ×10 with a Zeiss microscope, and at least three pictures per well were captured. After the cells were fixed, all incubations were done for 30 min at 37°C.
MTT assay.
MTT assay kit was purchased from ATCC (Manassas, VA). MTT assay was performed by following the protocol, with minor modifications. Briefly, HAECs were seeded into 48-well plates at ∼50% confluence after cells were attached to the plates and medium was changed to supplement free media with 2% LDS with or without corresponding treatment. Then, oxLDL was added for 24 h. MTT (0.5 mg/ml) dye was added to each well, and the cells were further incubated at 37°C for 2 h. The cells were lysed with DMSO contain ammonia, as described previously (57). Cell lysates were collected, and absorbance at 570 nm was measured using a plate reader (SpectraMax M5; Molecular Devices, Sunnyvale, CA).
Western blot analysis.
HAECs were lysed in lysis buffer (Cell Signaling Technology); 30 μg of total protein samples were resuspended in a reduced sample buffer and then electrophoresed on a 10–15% Tris gel with Tris running buffer, blotted to PVDF membrane, and sequentially probed with primary antibodies against phosphorylated Akt (Ser473) and Akt (Cell Signaling Technology). A horseradish peroxidase-conjugated goat anti-rabbit antibody was then added, and secondary antibodies were detected through autoradiography using enhanced chemiluminescence (ECL Plus; General Electric Healthcare, Milwaukee, WI). To test the effect of oxLDL on p27kip, subconfluent HAECs were used. C3 or MK2206 was introduced into HAECs first, and then cells were incubated with 50 µg/ml oxLDL for 24 h before lysis.
RhoA activation ELISA.
RhoA activation was monitored by RhoA G-LISA quantitative assay (Cytoskeleton) according to the manufacturer’s protocol. Briefly, confluent HAECs were serum starved first, and corresponding treatments were applied. After treatments, the medium was immediately aspirated and the cells washed with ice-cold PBS, lysed in lysis buffer from the kit, and centrifuged at 10,000 g for 1 min at 4°C. The supernatants were collected, snap-frozen in liquid nitrogen, and stored at −80°C until they were used. The protein concentration was determined by using the Precision Red Advanced protein assay supplied with the kit. The same amount of protein was used for ELISA. For all experiments, positive (constitutively active RhoA) and negative (lysis buffer) controls were used. After incubation with the first and second antibody and color development, absorbance was read at 490 nm using a microplate ELISA reader.
RESULTS
Cu-oxLDL and LPO-oxLDL induce endothelial proliferation.
First, we focused on Cu2+-oxidized LDL (Cu2+-oxLDL), the most commonly used oxidation mode of LDL. Effects of Cu2+-oxLDL on proliferation of HAECs were examined for 10 to 100 µg/ml concentration range, a typical range used in previous studies (see, e.g., Refs. 15, 51, and 56) for short (1 h) and more prolonged (24 h) exposures. Our data show that exposure to Cu-oxLDL results in a dose-dependent positive effect on the proliferation of HAECs for both short and prolonged exposures, as assayed by the BrdU or MTT assays (Fig. 1). For the short-term exposure, oxLDL increases HAECs proliferation by ∼40%, with LDL having no effect (Fig. 1, A–C; see typical images of proliferating HAECs identified by the BrdU staining in Fig. 1A). Over 24 h of exposure, we see a significantly stronger, greater than twofold increase in the number of proliferating HAECs treated with 50 µg/ml oxLDL as compared with untreated cells and also a smaller but significant effect of LDL at the same dose (50 µg/ml; Fig. 1D). It is most likely that this effect should be attributed to LDL oxidation by the endogenous endothelial LPO, as described earlier (47). We show here that increasing the degree of LDL oxidation results in a stronger pro-proliferation effect and that this effect is doze-dependent at both levels of oxidation, as assayed by MTT proliferation assay (Fig. 1E). Importantly, enzymatic oxidation of LDL using lipoxygenase (LPO) that generates a more physiological form of oxLDL also results in significant pro-proliferation effect (Fig. 1F).
Fig. 1.

Endothelial proliferation is induced by copper-oxidized (Cu-oxLDL) and lipoxygenase (LPO)-oxidized LDL (LPO-oxLDL). A: representative 5-bromo-2′-deoxyuridine (BrdU) images of human aortic endothelial cells (HAECs) treated with Cu-oxLDL, 0–100 µg/ml for 1 h (scale bar, 10 µm). B–D and F: quantification of BrdU-posive cells for the following conditions: HAECs treated with Cu-oxLDL (0–100 µg/ml) for 1 h (*P < 0.05, n ≥ 3; B), HAECs treated with 50 µg/ml Cu-oxidized LDL or native LDL (nLDL) for 1 h (*P < 0.05, n ≥ 3; C), HAECs treated with 50 µg/ml Cu-oxidized LDL or nLDL for 24 h (*P < 0.05, n ≥ 3; D), MTT assay of oxLDL-induced proliferation for Cu-oxLDL dose response and different degrees of oxidation (*P < 0.05, n = 4; E), and HAECs treated with 50 µg/ml LPO-oxLDL for 24 h (*P < 0.05, n ≥ 4; F). All experiments are done with matched controls.
Signaling pathway of oxLDL-induced endothelial proliferation in HAECs.
Akt is a critical regulator involved in endothelial cells proliferation, but as described above, the effects of oxLDL on Akt phosphorylation in endothelial cells have been controversial (2, 11, 63). Here, we show that in HAECs, both strongly oxidized Cu2+-oxLDL and enzymatically oxidized LPO-oxLDL induce significant increases in Akt phosphorylation, an effect observed after a short (15 min) incubation with 50 µg/ml Cu2+-oxLDL or LPO-oxLDL (Fig. 2, A and B). More specifically, LPO-oxLDL showed a more pronounced response with the exposure to 50 µg/ml Cu2+-oxLDL, resulting in a 2.9-fold increase in p-Akt/total Akt ratio, whereas exposure to the same level of LPO-oxLDL resulted in a 4.7-fold increase. To determine the role of Akt phosphorylation in oxLDL-induced proliferative effects, cells were preincubated with 100 nM MK2206, a selective inhibitor of Akt phosphorylation (22). The inhibitor was applied 1 h before the exposure to oxLDL and also for the duration of oxLDL treatment. As expected, oxLDL-induced Akt phosphorylation is abrogated by MK2206. Furthermore, blocking Akt phosphorylation also abrogates oxLDL-induced increase in HAEC proliferation for both types of oxLDL described above (Fig. 2C).
Fig. 2.

Activation of the Akt pathway is required for oxLDL-induced endothelial proliferation. A: representative Western blot image for oxLDL-induced Akt phosphorylation (50 µg/ml) with or without the presence of the Akt-specific inhibitor MK2206 (100n M); markers’ lane is shown at left. B: densitometric analysis of phosphorylated (p)-Akt/total Akt ratio in cells exposed to oxLDL (50 µg/ml) with or without the presence of MK2206. *P < 0.05, n ≥ 5. C: endothelial proliferation assayed by BrdU staining of HAECs exposed to oxLDL with or without Akt inhibitor MK2206 pretreatment. *P < 0.05, n ≥ 3.
RhoA.
Earlier studies, including studies from our laboratory, showed that Cu2+-oxLDL results in the activation of a small Rho-GTPase, RhoA, in different types of endothelial cells (45, 52). However, the link between RhoA activation and Akt phosphorylation in a signaling pathway induced by oxLDL was not clear. Therefore, we tested whether RhoA activation is required for oxLDL-induced activation of Akt. This question was addressed by treating the cells with 1 µg/ml exoenzyme C3 transferase, an inhibitor of Rho, or with 10 µg/ml Y27632, an inhibitor of Rho kinase (ROCK), a downstream target of RhoA, as described in previous studies (21, 28). We show here that preincubation with 1 µg/ml C3 abrogated oxLDL-induced Akt phosphorylation and oxLDL-induced increase in HAEC proliferation for both types of oxLDL (Fig. 3, A–C). Inhibition of Rho also resulted in an ∼30% decrease in the basal rate of HAEC proliferation in the absence of oxLDL, an effect that appears to be independent of Akt. In addition, blocking ROCK, a downstream target of RhoA, with 10 µg/ml Y27632 also resulted in significant decrease in oxLDL-induced Akt phosphorylation and inhibition of cell proliferation (Fig. 3, D–F). Similarly to the effect of C3, exposure to Y27632 also decreased the basal level of HAEC proliferation but not the basal level of Akt phosphorylation. We also tested whether the link between oxLDL-induced RhoA/ROCK activation and Akt phosphorylation may be mediated by the phosphorylation of myosin (MLC2) and increase in cell contractility, a well-known downstream effect of RhoA/ROCK (30, 49). However, we show here that this is not the case, because blocking MLC2 phosphorylation with blebbistatin, a well-known inhibitor of MLC2, has no effect on oxLDL-induced Akt phosphorylation (Fig. 3, G and H). MLC2 phosphorylation was inhibited as expected (Fig. 3I). More studies are needed to elucidate the signaling pathway responsible for the role of RhoA/ROCK in oxLDL-induced activation of Akt.
Fig. 3.

RhoA/Rho kinase (ROCK) is required for oxLDL-induced Akt phosphorylation and proliferation. A and D: representative Western blot images of oxLDL-induced Akt phosphorylation (50 µg/ml) with or without the presence of the Rho inhibitor C3 (1 µg/ml; A) or the ROCK inhibitor Y27632 (10 µM/ml; D). B and E: densitometric analysis of p-Akt/total Akt ratio in cells exposed to oxLDL (50 µg/ml) with or without the presence of C3 or Y27632. *P < 0.05, n ≥ 4. C and F: MTT assay of oxLDL-induced endothelial proliferation with or without C3 or Y27632 pretreatment. *P < 0.05, n ≥ 3. G: representative Western blot images of oxLDL-induced Akt phosphorylation (50 µg/ml) with or without the presence of blebbistatin. H and I: densitometric analysis of pMLC2/total MLC2 and p-Akt/total Akt ratios in cells exposed to oxLDL (50 µg/ml) with or without the presence of blebbistatin. *P < 0.05, n ≥ 4; markers’ lanes shown in A, D, and G, left.
Previously, our group identified a guanine nucleotide exchange factor, p115RhoGEF, and a guanine nucleotide dissociation inhibitor, RhoGDI-1, as major factors in regulating RhoA activity in endothelial cells (24, 31). We showed that guanine nucleotide dissociation inhibitors (GDI-1) bind with the GDP form of RhoGTPases and prevent their dissociation from the complex inhibiting RhoA activity (31). It was also shown previously, including studies from our group, that expression of p115-RGS inhibits endogenous p115RhoGEF function by blocking the interaction of p115RhoGEF with α-subunits of G12/13 and by its GAP activity on Gα12/13 (24, 33). Therefore, using GDI-1 and p115-RGS constructs, we tested whether RhoGDI-1 and/or p115RhoGEF affect oxLDL-induced activation of RhoA and the downstream effects described above in HAECs. To verify the expression of the two constructs, GDI-1 construct was labeled with the GFP tag, and p115-RGS was labeled with the myc-tag. The expression of both GDI and p115-RGS in HAECs was verified using Western blot analysis (Fig. 4A). RhoA activity was measured by ELISA, as described in our recent study (45). Here, we show that transfecting HAECs with RhoGDI-1 abrogated oxLDL-induced activation of RhoA (Fig. 4B) as well as oxLDL-induced activation of Akt1 (Fig. 4, C and D) and an increase in endothelial proliferation (Fig. 4E). In contrast, transfecting HAECs with p115RGS, had no effect on oxLDL-induced activation of RhoA or any of the downstream effects described above (Fig. 4), suggesting that GDI-1, but not p115RhoGEF, plays a significant role in oxLDL-induced activation of RhoA in HAECs.
Fig. 4.

Guanine nucleotide dissociation inhibitors (GDI), but not RGS, are involved in oxLDL-induced Rho activation. A: a representative Western blot verifying the expression of GDI-green fluorescent protein (GFP) and RGS-myc constructs in HAECs, with a markers’ lane shown at left. B: RhoA activity as measured by G-LISA at 15 min when exposed to oxLDL (LPO; 50 μg/ml) with or without overexpression GDI or RGS (*P < 0.05, n ≥ 4). C: representative Western blot images of oxLDL (LPO)-induced Akt phosphorylation (50 µg/ml) with or without overexpression of GDI or RGS (markers’ lane shown at left). D: densiometric analysis of p-Akt/total Akt ratio in cells exposed to oxLDL (LPO; 50 µg/ml) with or without overexpression of GDI or RGS. *P < 0.05, n ≥ 4. E: MTT assay of oxLDL-induced endothelial proliferation with or without GDI or RGS overexpression. *P < 0.05, n ≥ 3.
Also, since it was shown earlier that Cu2+-oxLDL induces activation of another Rho-GTPase, Rac1, we tested whether Rac1 activation contributes to oxLDL-induced Akt phosphorylation or cell proliferation. Rac1 was inhibited by exposing the cells to 100 µg/ml NSC23766, as described previously (5). However, our data show that inhibiting Rac1 affected neither oxLDL-induced Akt phosphorylation in HAECs nor the rate of cell proliferation (Fig. 5). Both types of oxLDL, Cu2+-oxidized, and LPO-oxidized were tested, yielding the same effect.
Fig. 5.

Inhibition of Rac1 has no effect on oxLDL-induced Akt phosphorylation or proliferation. A: representative Western blot image of oxLDL-induced Akt phosphorylation (50 µg/ml) with or without the presence of NSC23766 (markers’ lane shown at left). B: quantification of Akt phosphorylation induced by oxLDL with or without the presence of NSC23766. *P < 0.05, n = 4. C: MTT assay of oxLDL-induced proliferation with or without the presence of NSC23766. *P < 0.05, n = 4.
p27kip1 is a well-known cell cycle inhibitor (13) and shown to be downregulated under conditions that enhance cell proliferation. It was also shown previously that Cu-oxLDL decreased p27kip1 expression in human umbilical vein endothelial cells (HUVECs) (52). We show here that exposures to both Cu-oxLDL and LPO-oxLDL for 24 h result in a significant decrease in p27kip1 expression in HAECs (Fig. 6). Consistent with a previous study showing that VEGF-induced Akt activation results in a decrease in p27kip1 expression in endothelial cells (1), our data show that inhibition of Akt phosphorylation results in a pronounced increase in p27kip1 expression. No significant effect of oxLDL on p27kip1 expression is observed in the presence of the Akt inhibitor, indicating that it is regulated by Akt phosphorylation (Fig. 6, A and B). Furthermore, blocking Rho activation with C3 also results in a pronounced increase in p27kip1 expression and abrogates the effect of both Cu2+-oxLDL and LPO-oxLDL on p27kip1 (Fig. 6, C and D). Consistent with these observations, Akt1 knockdown increased p27kip1 expression and abrogated oxLDL-induced decrease in p27kip1 expression (Fig. 6, E and F).
Fig. 6.

OxLDL-induced downregulation of cell cycle regulatory protein p27kip1 is mediated by Akt and Rho. A: representative Western blot image of p27kip1 after 24 h of oxLDL (50 µg/ml) with or without the presence of the Akt inhibitor MK2206. B: densitometric analysis of p27kip1 under corresponding conditions. *P < 0.05, n ≥ 4. C: representative Western blot image of p27kip1 after 24 h of oxLDL (50 µg/ml) with or without the presence of C3. D: densitometric analysis of p27kip1 under corresponding conditions. *P < 0.05, n ≥ 4. E: representative Western blot image of p27kip1 after 24 h of oxLDL (50 µg/ml) with or without the presence of Akt siRNA. F: densiometric analysis of p27kip1 under corresponding conditions. n ≥ 4; markers’ lanes shown in A and E, left. NS, not significant.
Preloading with cholesterol blocks oxLDL-induced Akt phosphorylation and endothelial proliferation.
Our recent studies showed that Cu2+-oxLDL-induced activation of RhoA is blocked by preloading the cells with cholesterol using methyl-β-cyclodextrin (MβCD) saturated with cholesterol (45). Therefore, we tested here whether cholesterol preloading affects oxLDL-induced Akt phosphorylation and cell proliferation. HAECs were preloaded with cholesterol by exposing them to 5 mM MβCD cholesterol for 60 min, as we described previously (8). As shown above, exposure to 50 µg/ml of Cu2+-oxLDL or LPO-oxLDL induced a significant increase in Akt phosphorylation, with LPO-oxLDL having a stronger effect (Fig. 7, A and B). However, both Cu2+-oxLDL- and LPO-oxLDL-induced Akt phosphorylation were significantly reduced by cholesterol preloading. The effect of Cu2+-oxLDL was totally blocked, and the effect of LPO-oxLDL was reduced by 65% but was still above the basal level. Cholesterol enrichment alone had a small (∼20%), negative effect.
Fig. 7.

Preloading cells with cholesterol blocks oxLDL-induced Akt activation and endothelial proliferation. A: representative Western blot images of phosphorylated and total Akt for both types of oxLDL (50 µg/ml) with or without cholesterol preloading; markers’ lane is shown at left. B: quantification of Akt phosphorylation induced by oxLDL with or without cholesterol preloading. *P < 0.05, n ≥ 4. C: MTT assay of oxLDL-induced proliferation with or without cholesterol preloading. *P < 0.05, n = 4.
We also show here that cholesterol preloading blocks oxLDL-induced increase in cell proliferation of HAECs for both Cu2+-oxLDL and LPO-oxLDL (Fig. 7C). Cholesterol enrichment alone also had a significant, negative effect on the rate of cell proliferation (∼30% decrease). Exposing cells preloaded with cholesterol to either Cu2+-oxLDL or LPO-oxLDL had no effect on cell proliferation, which remained at the same level as in cells treated with cholesterol alone.
DISCUSSION
Endothelial proliferation is a key process in neovascularization, wound healing, and repair of vascular injury. Increased endothelial proliferation may also be associated with the disruption of endothelial barrier and increase in endothelial permeability. Both neovascularization and disruption of the endothelial barrier are known to contribute to the development of atherosclerotic lesions under dyslipidemic conditions. Neovascularization that occurs in the developing plaques is considered today one of the components of the development of complex and unstable plaques leading to serious complications (16). Therefore, it is essential to determine the impact of the key dyslipidemic factors on endothelial proliferation and elucidate the mechanisms underlying these effects.
In this study, we focus on oxidized modifications of LDL, which are believed to be key proatherogenic lipoproteins (38, 59). As described above, a number of earlier studies investigated the effect of oxLDL on endothelial proliferation showing a positive effect at low levels of oxLDL (1–10 µg/ml) and an inhibitory effect at higher levels (12, 20, 52, 63). However, the impacts of the degree and the mode of LDL oxidation, two key parameters in oxLDL biology and function, have not been investigated. In this study, we present the first systematic analysis of these parameters on endothelial proliferation. Briefly, our observations show a positive effect of oxLDL on endothelial proliferation over a large concentration range (10–100 µg/ml), which is also enhanced by an increase in the degree of LDL oxidation. Furthermore, we extend previous observations to test the impact of enzymatically oxidized LDL, which is generated by exposing LDL to lipoxygenase (10), a process that, in contrast to Cu2+ oxidation, occurs in vivo (35). This is critical because, as described above, the products of these two modes of LDL oxidation are very different. More specifically, whereas both Cu2+ and LPO oxidation result in the modifications of PAPC (1-palmitoyl-2-arachidonoyl-sn-glycero-phosphocholine), Cu2 oxidation yields predominantly truncated oxidized phospholipids such as 1-palmitoyl-2-(5′-oxo-valeroyl)-sn-glycero-3-phosphocholine (POVPC) and 1-palmitoyl-2-glutaryl-sn-glycero-3-phosphocholine (PGPC), whereas LPO oxidation yields an array of hydroxyeicosatetraenoic acid (HETE) species (35). Cu2 oxidation also yields an array of other oxidative products, including oxysterols, and modifications of the LDL protein apoB (35). Our study shows that similarly to Cu2+-oxLDL, LPO-oxidized LDL exhibits a significant positive effect on HAEC proliferation, with the effect being comparable or more pronounced than the effect of Cu2+-oxLDL. We propose that the similarities between the effects of Cu2+-oxLDL and LPO-oxLDL on endothelial function are mediated by their common effects on the lipid structure of the membrane.
The question of the physiological range of oxLDL in human plasma has also been a matter of significant controversy and debate. Earlier studies reported 10 µg/ml oxLDL in people with normal cholesterol and 30 µg/ml in hypercholesterolemic patients (25–27). More recent studies estimated significantly lower levels of 0.2–0.4 µg/ml (29). However, the level of oxLDL within the vascular wall is expected to be significantly higher than the levels of circulating oxLDL, with further accumulation in the atherosclerotic plaques (44, 62). The latter was determined using immunohistochemistry, whereas analytical determination of oxLDL in the plaques is still lacking. Based on our observations, we expect that elevation of oxLDL level under dyslipidemia in vivo has a proliferative effect on aortic endothelial cells.
In terms of the signaling pathway responsible for oxLDL-induced endothelial proliferation, we show here that for both Cu2+-oxLDL and LPO-oxidized LDL, it is mediated by RhoA/ROCK signaling via an increase in Akt phosphorylation and downregulation of p27kip1 expression. Furthermore, we also present evidence that oxLDL-induced activation of RhoA is mediated by a guanine nucleotide dissociation inhibitor RhoGDI-1, which was shown previously to be responsible for thrombin-mediated disruption of endothelial permeability (31). Previous studies from several laboratories, including ours, demonstrated a positive effect of Cu2+-oxLDL on activation of Rho-GTPases, including RhoA and Rac1 (45, 51, 52, 56). However, the upstream mechanism leading from oxLDL to RhoA activation has not been identified. Our new data suggest that oxLDL may induce RhoA activation in HAECs by dissociating RhoA from a RhoGDI that stabilizes RhoA in an inactive form. This hypothesis is based on the observations that oxLDL-induced activation of RhoA is abrogated by the overexpression of RhoGDI but is insensitive to the dominant-negative construct of p115RhoGEF, two major factors shown to regulate RhoA activation in endothelial cells (24, 31). It is important to note, however, that the fact that oxLDL-induced activation of RhoA is abrogated by overexpression of GDI does not conclusively prove that oxLDL induces the dissociation between endogenous GDI and RhoA, and further studies will be needed to test this hypothesis directly by analyzing the association between RhoA and GDI using state of the art approaches such as FRET. Notably, it was shown previously that RhoGDI dissociation from RhoA may be mediated by lipids that might compete with the Rho for the hydrophobic pocket of the GDI (17, 40). Therefore, we propose that altering of the lipid composition of the membrane is responsible for oxLDL-induced dissociation of RhoA and rhoGDI leading to RhoA activation and the downstream signaling described above.
The effects of oxLDL on Akt phosphorylation and the link between Rho activation and Akt phosphorylation have been controversial. Specifically, exposure to oxLDL was shown to induce Akt phosphorylation in human coronary ECs (63), vascular smooth muscle cells (36), and monocytes (58) but to result in Akt dephosphorylation in HUVECs (2) and human brain microvascular ECs (39). It was also shown that there is a cross-talk between Rho/ROCK and Akt pathways via phosphatidylinositol kinase (PI3K) or phosphatidylinositol phosphatase (PTEN), two enzymes that are well known to activate or inhibit Akt phosphorylation, respectively (14, 37, 43, 60). In most studies, however, Rho/ROCK was shown to negatively regulate Akt either via inhibition of PI3K (43, 60) or via activation of PTEN (37). It was also reported that RhoA may induce the PI3K/Akt pathway as part of the proliferative response in a model of graft endothelial cells (14). The roles of Rho/ROCK and Akt in Cu2+-oxLDL- and LPO-oxLDL-induced endothelial proliferation are further confirmed by testing the expression of cyclin-dependent kinase inhibitor 1B (p27kip1), a known cell cycle inhibitor that binds to cyclin E-CDK2 or cyclin D-CDK4 complexes and inhibits the cell cycle progression at G1 (50). Indeed, earlier studies showed Cu2+-oxLDL enhances EC proliferation of HUVECs by downregulating of p27kip1 and that this effect is ROCK dependent (52). Clearly, however, we are far from the full understanding of the signaling events involved in the oxLDL-induced endothelial proliferative response. Exposure to oxLDL is known to enhance generation of reactive oxygen species (35) (41), which in turn may promote cell proliferation (7), (55). Also, it was shown that oxLDL-induced endothelial proliferation may be suppressed by the inhibition of endothelial nitric oxide synthase (63), which could also inhibit generation of reactive nitrogen species. Moreover, multiple signaling pathways, including PI3K, MAP kinases, mTOR, and others, may play important roles in oxLDL-induced endothelial proliferation (48). Further studies are needed to fully elucidate the roles of these pathways in oxLDL-induced endothelial proliferation.
This study also provides further evidence for the dichotomy between oxLDL and cholesterol loading in their impact on endothelial function. As described briefly in the introduction, this is a novel concept that challenges one of the major dogmas about the role of oxLDL as “a cholesterol loader.” We described this dichotomy previously, first for endothelial stiffness (54) and then for oxLDL-induced activation of RhoA- and oxLDL-induced proangiogenic responses (45). However, since these observations suggest a paradigm shift in our understanding of how oxLDL alters endothelial function, it is important to test whether this is also the case for a variety of endothelial functions. We show here that this is also the case for oxLDL-induced Akt phosphorylation and for the proliferative response and that these effects are consistent for both Cu2+- and LPO-oxidized LDLs. In terms of the mechanism of this dichotomy, based on a series of studies from our laboratory, we suggest that the impact of oxLDL on endothelial function is mediated by altering and disrupting the lipid packing of the plasma membrane via incorporation of oxidized lipids (3, 45, 53, 54). More specifically, we showed that oxLDL is internalized in HAECs via the scavenger receptor CD36 (45), which in turn leads to the incorporation of oxidized lipids into the plasma membrane and disrupts lipid packing. Indeed, we have demonstrated that incorporation of both oxysterols and POVPC/PGPC phospholipids, major oxidation products of Cu2+ oxidation of LDL, results in disrupting lipid order due to their orientations in the membrane (3, 4). The same mechanism is predicted for products of LPO oxidation, HETE, because depending upon the position of its hydroxyl groups in relation to the terminal carboxylic acid, HETE molecules might assume a horizontal orientation in the membrane to limit the exposure of the hydroxyl group to the hydrophobic interior of the membrane. This orientation would cause the phospholipid headgroups to be pushed further away from each other, reducing the membrane packing, resulting in structural disruptions to the membrane similar to those caused by the presence of POVPC and PGPC (3). As we have shown previously, loading the cells with cholesterol “repairs” the disruptive effects of oxidized lipids. Furthermore, based on our new findings reported in this study, we propose the following model for oxLDL-induced endothelial proliferation (Fig. 8); exposure to oxLDL results in the incorporation of oxidized lipids into the endothelial membranes, resulting in the disruption of lipid packing, and these changes in lipid composition and/or physical properties of the membrane bilayer interfere with the association of RhoGDI with the Rho molecule, which favors the activation of RhoA signaling and, consequently, increases endothelial proliferation. Furthermore, based on our findings, EC proliferation should depend on the relative levels of cholesterol and oxLDL in the plaque, but currently there is no direct evidence. Further studies are needed to investigate this in the natural environment of the atherosclerotic plaque in vivo.
Fig. 8.
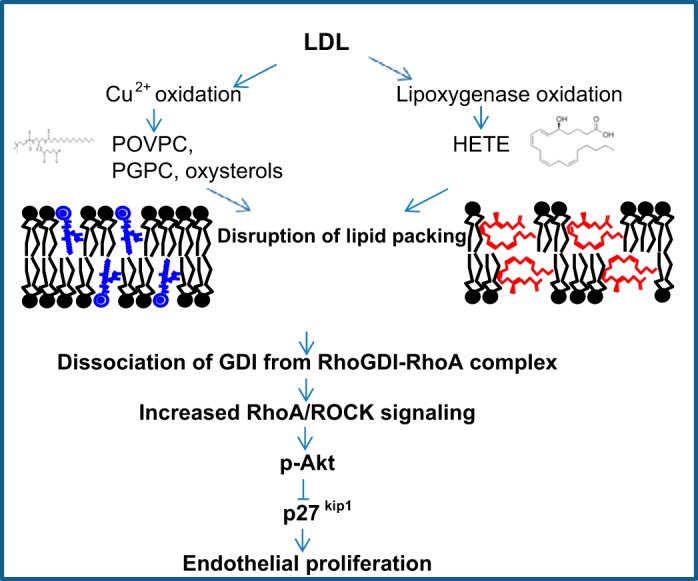
Summary of the proposed signaling events for oxLDL-induced endothelial proliferative response. Briefly, we propose that the products of LDL oxidation, such as oxidized phospholipids and oxysterols, disrupt the lipid bilayer of the membrane, which results in the activation of RhoA/ROCK cascade, possibly via dissociation of the GDI-RhoA complex, and leads to the activation of Akt1, which is known to induce cell proliferation via a decrease in the expression of cyclin-dependent kinase inhibitor 1B (p27kip1). HETE, hydroxyeicosatetraenoic acid; POVPC, 1-palmitoyl-2-(5′-oxo-valeroyl)-sn-glycero-3-phosphocholine; PGPC, 1-palmitoyl-2-glutaryl-sn-glycero-3-phosphocholine.
GRANTS
The work was supported by National Heart, Lung, and Blood Institute Grants 083298 and 073965 to I. Levitan and HL-079356 to K. K. Wary.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.Z., C.A., M.-J.O., and J.B. performed experiments; C.Z., C.A., M.-J.O., J.B., and I.L. analyzed data; C.Z., C.A., M.-J.O., and I.L. interpreted results of experiments; C.Z., C.A., M.-J.O., and M.A.A.A. prepared figures; C.Z. and I.L. drafted manuscript; C.Z., M.-J.O., M.A.A.A., D.M., K.K.W., and I.L. edited and revised manuscript; C.Z., C.A., M.-J.O., D.M., K.K.W., and I.L. approved final version of manuscript; I.L. conceived and designed research.
ACKNOWLEDGMENTS
We thank Yedida Bogachkov for help in preparing LDL. We also thank Gregory Kowalsky for the design of the summary figure.
REFERENCES
- 1.Abid MR, Guo S, Minami T, Spokes KC, Ueki K, Skurk C, Walsh K, Aird WC. Vascular endothelial growth factor activates PI3K/Akt/forkhead signaling in endothelial cells. Arterioscler Thromb Vasc Biol 24: 294–300, 2004. doi: 10.1161/01.ATV.0000110502.10593.06. [DOI] [PubMed] [Google Scholar]
- 2.Ahsan A, Han G, Pan J, Liu S, Padhiar AA, Chu P, Sun Z, Zhang Z, Sun B, Wu J, Irshad A, Lin Y, Peng J, Tang Z. Phosphocreatine protects endothelial cells from oxidized low-density lipoprotein-induced apoptosis by modulating the PI3K/Akt/eNOS pathway. Apoptosis 20: 1563–1576, 2015. doi: 10.1007/s10495-015-1175-4. [DOI] [PubMed] [Google Scholar]
- 3.Ayee MA, LeMaster E, Shentu TP, Singh DK, Barbera N, Soni D, Tiruppathi C, Subbaiah PV, Berdyshev E, Bronova I, Cho M, Akpa BS, Levitan I. Molecular-scale biophysical modulation of an endothelial membrane by oxidized phospholipids. Biophys J 112: 325–338, 2017. doi: 10.1016/j.bpj.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ayee MA, Levitan I. Paradoxical impact of cholesterol on lipid packing and cell stiffness. Front Biosci (Landmark Ed) 21: 1245–1259, 2016. doi: 10.2741/4454. [DOI] [PubMed] [Google Scholar]
- 5.Baumer Y, Spindler V, Werthmann RC, Bünemann M, Waschke J. Role of Rac 1 and cAMP in endothelial barrier stabilization and thrombin-induced barrier breakdown. J Cell Physiol 220: 716–726, 2009. doi: 10.1002/jcp.21819. [DOI] [PubMed] [Google Scholar]
- 6.Boullier A, Bird DA, Chang MK, Dennis EA, Friedman P, Gillotte-Taylor K, Hörkkö S, Palinski W, Quehenberger O, Shaw P, Steinberg D, Terpstra V, Witztum JL. Scavenger receptors, oxidized LDL, and atherosclerosis. Ann NY Acad Sci 947: 214–223, 2001. doi: 10.1111/j.1749-6632.2001.tb03943.x. [DOI] [PubMed] [Google Scholar]
- 7.Bryk D, Olejarz W, Zapolska-Downar D. The role of oxidative stress and NADPH oxidase in the pathogenesis of atherosclerosis. Postepy Hig Med Dosw (Online) 71: 57–68, 2017. doi: 10.5604/17322693.1229823. [DOI] [PubMed] [Google Scholar]
- 8.Byfield FJ, Aranda-Espinoza H, Romanenko VG, Rothblat GH, Levitan I. Cholesterol depletion increases membrane stiffness of aortic endothelial cells. Biophys J 87: 3336–3343, 2004. doi: 10.1529/biophysj.104.040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byfield FJ, Tikku S, Rothblat GH, Gooch KJ, Levitan I. OxLDL increases endothelial stiffness, force generation, and network formation. J Lipid Res 47: 715–723, 2006. doi: 10.1194/jlr.M500439-JLR200. [DOI] [PubMed] [Google Scholar]
- 10.Cathcart MK, McNally AK, Chisolm GM. Lipoxygenase-mediated transformation of human low density lipoprotein to an oxidized and cytotoxic complex. J Lipid Res 32: 63–70, 1991. [PubMed] [Google Scholar]
- 11.Chavakis E, Dernbach E, Hermann C, Mondorf UF, Zeiher AM, Dimmeler S. Oxidized LDL inhibits vascular endothelial growth factor-induced endothelial cell migration by an inhibitory effect on the Akt/endothelial nitric oxide synthase pathway. Circulation 103: 2102–2107, 2001. doi: 10.1161/01.CIR.103.16.2102. [DOI] [PubMed] [Google Scholar]
- 12.Chen CH, Jiang W, Via DP, Luo S, Li TR, Lee YT, Henry PD. Oxidized low-density lipoproteins inhibit endothelial cell proliferation by suppressing basic fibroblast growth factor expression. Circulation 101: 171–177, 2000. doi: 10.1161/01.CIR.101.2.171. [DOI] [PubMed] [Google Scholar]
- 13.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer 8: 253–267, 2008. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 14.Coupel S, Leboeuf F, Boulday G, Soulillou JP, Charreau B. RhoA activation mediates phosphatidylinositol 3-kinase-dependent proliferation of human vascular endothelial cells: an alloimmune mechanism of chronic allograft nephropathy. J Am Soc Nephrol 15: 2429–2439, 2004. doi: 10.1097/01.ASN.0000138237.42675.45. [DOI] [PubMed] [Google Scholar]
- 15.Dandapat A, Hu C, Sun L, Mehta JL. Small concentrations of oxLDL induce capillary tube formation from endothelial cells via LOX-1-dependent redox-sensitive pathway. Arterioscler Thromb Vasc Biol 27: 2435–2442, 2007. doi: 10.1161/ATVBAHA.107.152272. [DOI] [PubMed] [Google Scholar]
- 16.Di Stefano R, Felice F, Balbarini A. Angiogenesis as risk factor for plaque vulnerability. Curr Pharm Des 15: 1095–1106, 2009. doi: 10.2174/138161209787846892. [DOI] [PubMed] [Google Scholar]
- 17.Dovas A, Couchman JR. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem J 390: 1–9, 2005. doi: 10.1042/BJ20050104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukuhara S, Chikumi H, Gutkind JS. RGS-containing RhoGEFs: the missing link between transforming G proteins and Rho? Oncogene 20: 1661–1668, 2001. doi: 10.1038/sj.onc.1204182. [DOI] [PubMed] [Google Scholar]
- 19.Gundavaram MS, Shentu TP, Kowalsky GB, Volkov S, Schraufnagel DE, Levitan I. Oxidized low-density lipoprotein alters the effect of matrix stiffness on the formation of endothelial networks and capillary lumens. Pulm Circ 3: 622–631, 2013. doi: 10.1086/674309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinloth A, Heermeier K, Raff U, Wanner C, Galle J. Stimulation of NADPH oxidase by oxidized low-density lipoprotein induces proliferation of human vascular endothelial cells. J Am Soc Nephrol 11: 1819–1825, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Herr MJ, Mabry SE, Jennings LK. Tetraspanin CD9 regulates cell contraction and actin arrangement via RhoA in human vascular smooth muscle cells. PLoS One 9: e106999, 2014. doi: 10.1371/journal.pone.0106999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, Kotani H. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 9: 1956–1967, 2010. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 23.Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol 17: 496–510, 2016. doi: 10.1038/nrm.2016.67. [DOI] [PubMed] [Google Scholar]
- 24.Holinstat M, Mehta D, Kozasa T, Minshall RD, Malik AB. Protein kinase Calpha-induced p115RhoGEF phosphorylation signals endothelial cytoskeletal rearrangement. J Biol Chem 278: 28793–28798, 2003. doi: 10.1074/jbc.M303900200. [DOI] [PubMed] [Google Scholar]
- 25.Holvoet P, Harris TB, Tracy RP, Verhamme P, Newman AB, Rubin SM, Simonsick EM, Colbert LH, Kritchevsky SB. Association of high coronary heart disease risk status with circulating oxidized LDL in the well-functioning elderly: findings from the Health, Aging, and Body Composition study. Arterioscler Thromb Vasc Biol 23: 1444–1448, 2003. doi: 10.1161/01.ATV.0000080379.05071.22. [DOI] [PubMed] [Google Scholar]
- 26.Holvoet P, Mertens A, Verhamme P, Bogaerts K, Beyens G, Verhaeghe R, Collen D, Muls E, Van de Werf F. Circulating oxidized LDL is a useful marker for identifying patients with coronary artery disease. Arterioscler Thromb Vasc Biol 21: 844–848, 2001. doi: 10.1161/01.ATV.21.5.844. [DOI] [PubMed] [Google Scholar]
- 27.Holvoet P, Vanhaecke J, Janssens S, Van de Werf F, Collen D. Oxidized LDL and malondialdehyde-modified LDL in patients with acute coronary syndromes and stable coronary artery disease. Circulation 98: 1487–1494, 1998. doi: 10.1161/01.CIR.98.15.1487. [DOI] [PubMed] [Google Scholar]
- 28.Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, Narumiya S. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol Pharmacol 57: 976–983, 2000. [PubMed] [Google Scholar]
- 29.Itabe H, Ueda M. Measurement of plasma oxidized low-density lipoprotein and its clinical implications. J Atheroscler Thromb 14: 1–11, 2007. doi: 10.5551/jat.14.1. [DOI] [PubMed] [Google Scholar]
- 30.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 273: 245–248, 1996. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 31.Knezevic N, Roy A, Timblin B, Konstantoulaki M, Sharma T, Malik AB, Mehta D. GDI-1 phosphorylation switch at serine 96 induces RhoA activation and increased endothelial permeability. Mol Cell Biol 27: 6323–6333, 2007. doi: 10.1128/MCB.00523-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kowalsky GB, Byfield FJ, Levitan I. oxLDL facilitates flow-induced realignment of aortic endothelial cells. Am J Physiol Cell Physiol 295: C332–C340, 2008. doi: 10.1152/ajpcell.00335.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC. p115 RhoGEF, a GTPase activating protein for Ga12 and Ga13. Science 280: 2109–2111, 1998. doi: 10.1126/science.280.5372.2109. [DOI] [PubMed] [Google Scholar]
- 34.Levitan I, Christian AE, Tulenko TN, Rothblat GH. Membrane cholesterol content modulates activation of volume-regulated anion current in bovine endothelial cells. J Gen Physiol 115: 405–416, 2000. doi: 10.1085/jgp.115.4.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levitan I, Volkov S, Subbaiah PV. Oxidized LDL: diversity, patterns of recognition, and pathophysiology. Antioxid Redox Signal 13: 39–75, 2010. doi: 10.1089/ars.2009.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li HX, Kong FJ, Bai SZ, He W, Xing WJ, Xi YH, Li GW, Guo J, Li HZ, Wu LY, Wang R, Yang GD, Tian Y, Xu CQ. Involvement of calcium-sensing receptor in oxLDL-induced MMP-2 production in vascular smooth muscle cells via PI3K/Akt pathway. Mol Cell Biochem 362: 115–122, 2012. doi: 10.1007/s11010-011-1133-6. [DOI] [PubMed] [Google Scholar]
- 37.Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, Tang L, Hla T, Zeng R, Li L, Wu D. Regulation of PTEN by Rho small GTPases. Nat Cell Biol 7: 399–404, 2005. [Erratum. Nat Cell Biol 7: 531,, 2005, and Nat Cell Biol 8: 1038, 2006.] doi: 10.1038/ncb1236. [DOI] [PubMed] [Google Scholar]
- 38.Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol 32: 2045–2051, 2012. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Pan Q, Zhao Y, He C, Bi K, Chen Y, Zhao B, Chen Y, Ma X. MicroRNA-155 regulates ROS production, NO generation, apoptosis and multiple functions of human brain microvessel endothelial cells under physiological and pathological conditions. J Cell Biochem 116: 2870–2881, 2015. doi: 10.1002/jcb.25234. [DOI] [PubMed] [Google Scholar]
- 40.Longenecker K, Read P, Derewenda U, Dauter Z, Liu X, Garrard S, Walker L, Somlyo AV, Nakamoto RK, Somlyo AP, Derewenda ZS. How RhoGDI binds Rho. Acta Crystallogr D Biol Crystallogr 55: 1503–1515, 1999. doi: 10.1107/S090744499900801X. [DOI] [PubMed] [Google Scholar]
- 41.Lubrano V, Balzan S. LOX-1 and ROS, inseparable factors in the process of endothelial damage. Free Radic Res 48: 841–848, 2014. doi: 10.3109/10715762.2014.929122. [DOI] [PubMed] [Google Scholar]
- 42.Maiolino G, Rossitto G, Caielli P, Bisogni V, Rossi GP, Calò LA. The role of oxidized low-density lipoproteins in atherosclerosis: the myths and the facts. Mediators Inflamm 2013: 714653, 2013. doi: 10.1155/2013/714653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ming XF, Viswambharan H, Barandier C, Ruffieux J, Kaibuchi K, Rusconi S, Yang Z. Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol Cell Biol 22: 8467–8477, 2002. doi: 10.1128/MCB.22.24.8467-8477.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishi K, Itabe H, Uno M, Kitazato KT, Horiguchi H, Shinno K, Nagahiro S. Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arterioscler Thromb Vasc Biol 22: 1649–1654, 2002. doi: 10.1161/01.ATV.0000033829.14012.18. [DOI] [PubMed] [Google Scholar]
- 45.Oh MJ, Zhang C, LeMaster E, Adamos C, Berdyshev E, Bogachkov Y, Kohler EE, Baruah J, Fang Y, Schraufnagel DE, Wary KK, Levitan I. Oxidized LDL signals through Rho-GTPase to induce endothelial cell stiffening and promote capillary formation. J Lipid Res 57: 791–808, 2016. doi: 10.1194/jlr.M062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oviedo PJ, Sobrino A, Laguna-Fernandez A, Novella S, Tarín JJ, García-Pérez MA, Sanchís J, Cano A, Hermenegildo C. Estradiol induces endothelial cell migration and proliferation through estrogen receptor-enhanced RhoA/ROCK pathway. Mol Cell Endocrinol 335: 96–103, 2011. doi: 10.1016/j.mce.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 47.Parthasarathy S, Wieland E, Steinberg D. A role for endothelial cell lipoxygenase in the oxidative modification of low density lipoprotein. Proc Natl Acad Sci USA 86: 1046–1050, 1989. doi: 10.1073/pnas.86.3.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol 4: 64, 2014. doi: 10.3389/fonc.2014.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ridley AJ. Rho GTPase signalling in cell migration. Curr Opin Cell Biol 36: 103–112, 2015. doi: 10.1016/j.ceb.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roy A, Banerjee S. p27 and leukemia: cell cycle and beyond. J Cell Physiol 230: 504–509, 2015. doi: 10.1002/jcp.24819. [DOI] [PubMed] [Google Scholar]
- 51.Ryoo S, Bhunia A, Chang F, Shoukas A, Berkowitz DE, Romer LH. OxLDL-dependent activation of arginase II is dependent on the LOX-1 receptor and downstream RhoA signaling. Atherosclerosis 214: 279–287, 2011. doi: 10.1016/j.atherosclerosis.2010.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seibold S, Schürle D, Heinloth A, Wolf G, Wagner M, Galle J. Oxidized LDL induces proliferation and hypertrophy in human umbilical vein endothelial cells via regulation of p27Kip1 expression: role of RhoA. J Am Soc Nephrol 15: 3026–3034, 2004. doi: 10.1097/01.ASN.0000146425.58046.6A. [DOI] [PubMed] [Google Scholar]
- 53.Shentu TP, Singh DK, Oh MJ, Sun S, Sadaat L, Makino A, Mazzone T, Subbaiah PV, Cho M, Levitan I. The role of oxysterols in control of endothelial stiffness. J Lipid Res 53: 1348–1358, 2012. doi: 10.1194/jlr.M027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shentu TP, Titushkin I, Singh DK, Gooch KJ, Subbaiah PV, Cho M, Levitan I. oxLDL-induced decrease in lipid order of membrane domains is inversely correlated with endothelial stiffness and network formation. Am J Physiol Cell Physiol 299: C218–C229, 2010. doi: 10.1152/ajpcell.00383.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stone JR, Collins T. The role of hydrogen peroxide in endothelial proliferative responses. Endothelium 9: 231–238, 2002. doi: 10.1080/10623320214733. [DOI] [PubMed] [Google Scholar]
- 56.Sugimoto K, Ishibashi T, Sawamura T, Inoue N, Kamioka M, Uekita H, Ohkawara H, Sakamoto T, Sakamoto N, Okamoto Y, Takuwa Y, Kakino A, Fujita Y, Tanaka T, Teramoto T, Maruyama Y, Takeishi Y. LOX-1-MT1-MMP axis is crucial for RhoA and Rac1 activation induced by oxidized low-density lipoprotein in endothelial cells. Cardiovasc Res 84: 127–136, 2009. doi: 10.1093/cvr/cvp177. [DOI] [PubMed] [Google Scholar]
- 57.Wang H, Wang F, Tao X, Cheng H. Ammonia-containing dimethyl sulfoxide: an improved solvent for the dissolution of formazan crystals in the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Anal Biochem 421: 324–326, 2012. doi: 10.1016/j.ab.2011.10.043. [DOI] [PubMed] [Google Scholar]
- 58.Wang WY, Li J, Yang D, Xu W, Zha RP, Wang YP. OxLDL stimulates lipoprotein-associated phospholipase A2 expression in THP-1 monocytes via PI3K and p38 MAPK pathways. Cardiovasc Res 85: 845–852, 2010. doi: 10.1093/cvr/cvp367. [DOI] [PubMed] [Google Scholar]
- 59.Witztum JL, Steinberg D. Role of oxidized low density lipoprotein in atherogenesis. J Clin Invest 88: 1785–1792, 1991. doi: 10.1172/JCI115499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, Dominiak P, Liao JK. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol 24: 1842–1847, 2004. doi: 10.1161/01.ATV.0000142813.33538.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu N, Lao Y, Zhang Y, Gillespie DA. Akt: a double-edged sword in cell proliferation and genome stability. J Oncol 2012: 951724, 2012. doi: 10.1155/2012/951724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ylä-Herttuala S, Palinski W, Rosenfeld ME, Parthasarathy S, Carew TE, Butler S, Witztum JL, Steinberg D. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J Clin Invest 84: 1086–1095, 1989. doi: 10.1172/JCI114271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu S, Wong SL, Lau CW, Huang Y, Yu C-M. Oxidized LDL at low concentration promotes in-vitro angiogenesis and activates nitric oxide synthase through PI3K/Akt/eNOS pathway in human coronary artery endothelial cells. Biochem Biophys Res Commun 407: 44–48, 2011. doi: 10.1016/j.bbrc.2011.02.096. [DOI] [PubMed] [Google Scholar]