

Abstract
Advancements in research have added several new therapies for castration-resistant prostate cancer (CRPC), greatly augmenting our ability to treat patients. However, CRPC remains an incurable disease due to the development of therapeutic resistance and the existence of cross-resistance between available therapies. Understanding the interplay between different treatments will lead to improved sequencing and the creation of combinations which overcome resistance and prolong survival. Whether there exists cross-resistance between docetaxel and the next-generation taxane cabazitaxel is poorly understood. In this study, we use C4-2B and DU145 derived docetaxel-resistant cell lines to test response to cabazitaxel. Our results demonstrate that docetaxel resistance confers cross-resistance to cabazitaxel. We show that increased ABCB1 expression is responsible for cross-resistance to cabazitaxel and that inhibition of ABCB1 function through the small molecule inhibitor elacridar re-sensitizes taxane resistant cells to treatment. Additionally, the anti-androgens bicalutamide and enzalutamide, previously demonstrated to be able to re-sensitize taxane resistant cells to docetaxel through inhibition of ABCB1 ATPase activity, are also able to re-sensitize resistant cells to cabazitaxel treatment. Lastly, we show that re-sensitization using an anti-androgen is far more effective in combination with cabazitaxel than docetaxel. Collectively, these results address key concerns in the field including that of cross-resistance between taxanes and highlighting a mechanism of cabazitaxel resistance involving ABCB1. Furthermore, these preclinical studies suggest the potential in utilizing combinations of anti-androgens with cabazitaxel for increased effect in treating advanced CRPC.
Keywords: prostate cancer, docetaxel, cabazitaxel, resistance, ABCB1
Introduction
Mortality from advanced or metastatic prostate cancer remains as a significant clinical challenge. Patients initially respond to androgen deprivation therapy (ADT), but inevitably progress to incurable castration resistant disease (1). The standard of care for castration-resistant prostate cancer (CRPC) has been docetaxel based chemotherapy regimens. Despite a demonstrated survival benefit, about 50% of patients do not respond initially and those that do eventually fail treatment due to the development of resistance (2).
Recent efforts have added novel therapies which have augmented our ability to treat advanced CRPC. Enzalutamide, abiraterone, sipuleucel-T, and radium-223 have been shown to provide a survival benefit and may be administered before or after treatment with docetaxel (3–8). Cabazitaxel, a next-generation taxane, was also shown to provide a survival benefit but is approved only to treat docetaxel pre-treated patients (9). Despite these improvements in care, patients eventually progress and still succumb to the disease highlighting three key concerns. First, it is not known what sequence to use these treatments in for optimized care, nor is it known which combinations will produce the most benefit. Second, presentation with or the development of resistance is inevitable which ultimately renders these new agents ineffective. Lastly, the issue of potential cross-resistance between available therapies further complicates treatment decisions. It is imperative that these questions be addressed.
Elucidating and understanding mechanisms of acquired drug resistance and subsequent cross-resistance to other available therapies will greatly improve our ability to treat advanced CRPC patients. The mechanisms of resistance to docetaxel have been investigated and shown to include loss of p53 functioning, increased expression of β-tubulin isoforms, decreased expression of BRCA1, and increased expression of the drug efflux pump ABCB1 (10–12). Mechanisms of cabazitaxel resistance are lesser known but are thought to include the presence of the retinoblastoma protein and loss of the membrane transporter SLCO1B3 (13, 14). Whether there exists cross-resistance between docetaxel and cabazitaxel is poorly understood. Although it has been shown that cabazitaxel is beneficial post-docetaxel, the survival benefit is modest (9). It may be that responses to cabazitaxel are blunted by common mechanisms of resistance with docetaxel. Ascertaining this information is critical for the rational design of treatment regimens leading to improved outcomes and prolonged survival.
Our previous work investigated mechanisms of resistance to docetaxel in prostate cancer and found that ABCB1 overexpression mediates robust resistance to this drug (12). In this study, we explore whether cross-resistance between docetaxel and cabazitaxel exists and its potential underlying mechanisms in advanced prostate cancer. Our data demonstrates that docetaxel-resistant prostate cancer cells exhibit cross-resistance to cabazitaxel and that this cross-resistance is mediated through ABCB1. Strategies to inhibit ABCB1 function are capable of re-sensitizing cells to cabazitaxel treatment thus highlighting new combinations that may be efficacious for prostate cancer treatment. Lastly, we show that combination strategies are more effective with cabazitaxel than with docetaxel thus providing a potential rationale to favor cabazitaxel over docetaxel re-challenge in the clinic.
Materials and Methods
Cell culture and reagents
DU145 cells were obtained from the American Type Culture Collection (ATCC) in 2008. C4-2B cells were kindly provided by Dr. Leland Chung (Cedars-Sinai Medical Center, Los Angeles, CA) in 2005. Short tandem repeat (STR) analysis was used for testing and authentication of the cell lines. All experiments with these cell lines and their derivatives were conducted within 6 months of receipt or resuscitation after cryopreservation. Cells were maintained in RPMI 1640 media supplemented with 10% fetal bovine serum, 100 units/ml penicillin and 0.1 mg/ml streptomycin. TaxR and DU145-DTXR cells were described previously and maintained in complete RPMI 1640 supplemented with 5nM docetaxel (12, 15). C4-2B and DU145 cells were cultured alongside TaxR and DU145-DTXR cells as respective appropriate controls. TaxR-control and TaxR-shABCB1 cells were described previously and maintained in complete RPMI 1640 media supplemented with 2ug/ml puromycin (12). All cells were maintained at 37ºC in a humidified incubator with 5% carbon dioxide. Puromycin (Cat#: BP2956-100) was purchased from Fisher Scientific. Docetaxel (Ca#: RS019) was purchased from TSZ CHEM. Cabazitaxel (Cat#: S3022) was purchased from Selleckchem. Elacridar (Cat#: 143664-11-3) was purchased from Sigma-Aldrich. Bicalutamide (Cat#: B3209) was purchased from LKT Laboratories. Enzalutamide (Cat#: S1250) was purchased from Selleckchem.
Cell growth assay
For all cell growth assays described, cells were plated at a density of 30,000 cells/well in 24-well plates in complete RPMI 1640 media without any selection agent. After 24 hours, cells were subjected to indicated single or combination treatments. Unless otherwise noted in the text, docetaxel (DTX) = 1nM, cabazitaxel (CTX) = 1nM, elacridar (Elac) = 0.5μM, bicalutamide (Bic) = 10μM, and enzalutamide (Enz) = 10μM. 72hrs post-treatment, total cells were counted via coulter counter. Cell counts were normalized to control cell growth treated only with vehicle (DMSO). Data is displayed as cell survival rate as percent of control cell growth. Cell survival rate (%) = (Treatment group cell number/Control group cell number) × 100. All conditions were performed in triplicate. All experiments were performed at least twice.
Clonogenic assay
For all clonogenic assays described, cells were plated at 500 cells/well in 6-well plates in complete RPMI 1640 with no selection agent. Plated cells were subsequently treated 24 hours later as indicated. Unless otherwise noted in the text, docetaxel (DTX) = 1nM, cabazitaxel (CTX) = 1nM, elacridar (Elac) = 0.5μM, bicalutamide (Bic) = 10μM, and enzalutamide (Enz) = 10μM. Colonies were allowed to form for 8–14 days as indicated. At the completion of each assay, cell colonies were washed in PBS and then fixed in 100% methanol for 10 minutes. This was followed by staining using 0.5% crystal violet for 10 minutes. After staining, colonies were counted. Data is displayed as a percent of control cell colony growth (control is vehicle (DMSO) treatment only). All conditions were performed in duplicate. All experiments were performed at least twice.
WST-1 Proliferation Assay
Cells were plated at a density of 30,000 cells/well in 24-well plates in complete RPMI 1640 media without any selection agent. After 24 hours, cells were subjected to indicated single or combination treatments; cabazitaxel (CTX) = 1nM, bicalutamide (Bic) = 10μM, enzalutamide (Enz) = 10μM. 72 hours post-treatment, cell proliferation was assessed via WST-1 assay purchased from Takara Bio (Cat#: MK400) according to manufacturer’s protocol. Cell proliferation was normalized to control cell proliferation treated only with vehicle (DMSO). Data is displayed as proliferation rate as percent of control. Cell proliferation rate (%) = (Treatment group proliferation/Control group proliferation) × 100. All conditions were performed in triplicate. All experiments were performed at least twice.
Preparation of whole-cell extracts
Cells were harvested, washed with PBS, and lysed in high-salt buffer (10mM HEPES pH 7.9, 0.4M NaCl, 1mM EDTA, 0.5mM PMSF, 1mM DTT, 1mM NaV, 20mM NaF, 1mM EGTA, 20% Glycerol) with a freeze thaw procedure. High salt buffer was supplemented with protease inhibitors (Cat#: 11836153001) purchased from Sigma-Aldrich. Protein concentration was determined with Pierce Coomassie Plus (Bradford) Assay Kit (Cat#: 23236) purchased from Thermo Fisher Scientific.
Western Blot
Whole cell protein extracts were resolved by SDS-PAGE and proteins were transferred to nitrocellulose membranes. After blocking for 1hr at room temperature in 5% milk in PBS/0.1% Tween-20, membranes were incubated overnight at 4°C with the indicated primary antibodies. ABCB1 antibody (SC-8313, rabbit-polyclonal, 1:500 dilution) was purchased from Santa Cruz Biotechnology. PARP antibody (9542, rabbit-polyclonal antibody, 1:1000 dilution) was purchased from Cell Signaling Technology. Cleaved-PARP antibody (9541, rabbit-polyclonal antibody, 1:1000 dilution) was purchased from Cell Signaling Technology. Tubulin (T5168, mouse monoclonal antibody, 1:6000 dilution) was purchased from Sigma-Aldrich. Tubulin was used to monitor the amounts of samples applied. Following secondary antibody incubation, proteins were visualized with a chemiluminescence detection system (Cat#: WBLUR0500) purchased from Millipore.
Statistics
All quantitated data is displayed as percent of control mean ± SD. Significance was assessed using a two tailed two sample equal variance students t-test. A p-value of ≤0.05 was accepted as significant.
Results
Docetaxel-resistant prostate cancer cells are cross-resistant to cabazitaxel
Our previous work utilized C4-2B and DU145 docetaxel-resistant derivative cell lines (hence forth referred to as TaxR and DU145-DTXR respectively) to investigate mechanisms of resistance (12, 15). Clinically, cabazitaxel is approved to treat patients who have progressed during or after docetaxel chemotherapy (16). Whether docetaxel resistance confers any cross-resistance to cabazitaxel treatment is unclear. Thus, we tested if docetaxel-resistant cell lines would respond to cabazitaxel. TaxR and DU145-DTXR cells were subjected to cell growth assays utilizing a cabazitaxel dose curve to assess response to treatment versus parental C4-2B and DU145 cells respectively. We found that in both TaxR and DU145-DTXR cells, response to cabazitaxel was blunted compared to their respective parental cells (Fig. 1A). The IC50 of C4-2B cells to cabazitaxel was 0.7 nM, while the IC50 of TaxR cells to cabazitaxel was 7.3 nM, an ~10-fold increase. The IC50 of the DU145 cells was 0.7 nM, while the IC50 for DU145-DTXR cells was 3.3 nM, an ~5-fold increase. These data were confirmed using colony formation assays. We found that parental cell lines were completely unable to form colonies even at the lowest tested dose of 0.5 nM cabazitaxel, while TaxR and DU145-DTXR cells were far less affected (Fig. 1B). Western blots for PARP and cleaved-PARP indicate that cabazitaxel’s ability to induce apoptosis is decreased in TaxR and DU145-DTXR cells further demonstrating resistance to treatment (Fig. 1C). These data demonstrate that docetaxel-resistant cells are cross-resistant to cabazitaxel. This suggests that mechanisms involved in docetaxel resistance may be shared by cabazitaxel. However, resistance to cabazitaxel is not as robust as that to docetaxel, as both TaxR and DU145-DTXR cells are completely resistant to 5nM docetaxel but do exhibit some response to the same dose of cabazitaxel (Fig. 1D). Thus, our model mimics clinical findings in that cabazitaxel retains efficacy post-docetaxel. However, clinical responses may be blunted by mechanisms of cross-resistance.
Figure 1. Docetaxel-resistant prostate cancer cell line derivatives are cross-resistant to cabazitaxel.
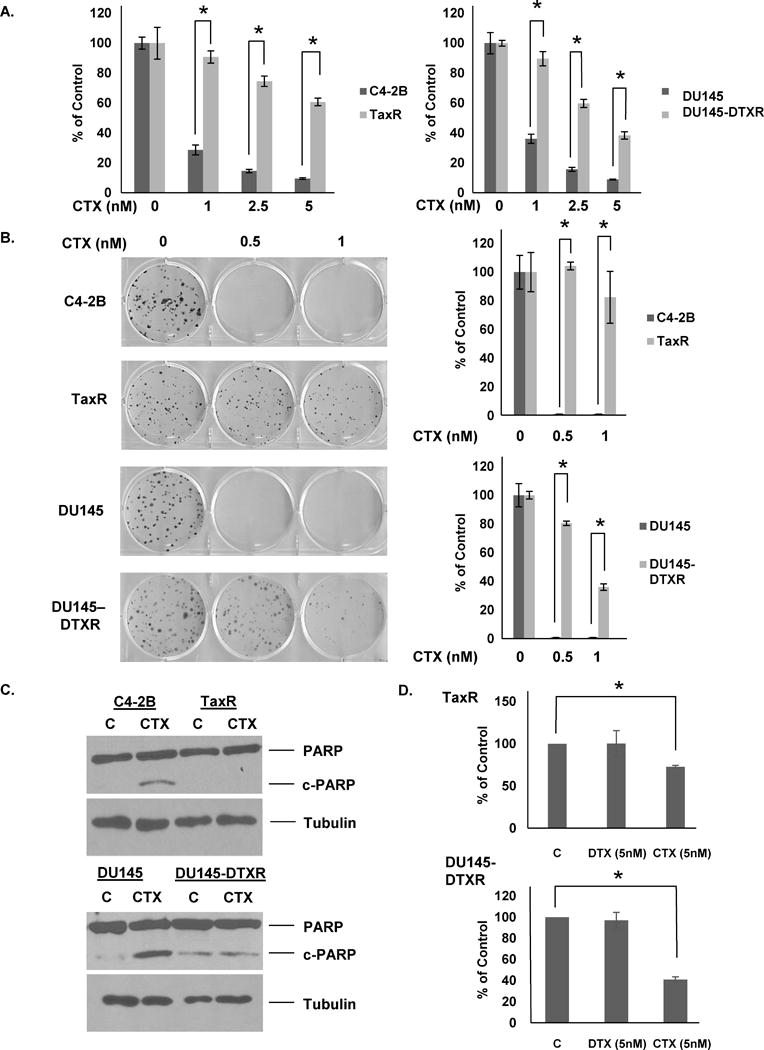
A. C4-2B, TaxR, DU145, and DU145-DTXR cells were subjected to cell growth assays using indicated doses of cabazitaxel. Cells were counted 72 hours post-treatment. B. C4-2B, TaxR, DU145, and DU145-DTXR cells were subjected to clonogenic assays using indicated doses of cabazitaxel. Colonies were allowed to form for 8 (C4-2B and TaxR) or 10 (DU145 and DU145-DTXR) days post-treatment. Colonies were then stained with crystal violet and counted. C. C4-2B, TaxR, DU145, and DU145-DTXR cells were treated with either vehicle (DMSO) or cabazitaxel (1nM) for 72 hours. Whole cell lysates were then prepared and subjected to western blot analysis using indicated antibodies. Tubulin served as a loading control. D. TaxR and DU145-DTXR cells were subjected to cell growth assays treated either with vehicle (DMSO), docetaxel (5nM), or cabazitaxel (5nM). Cells were counted 72 hours post-treatment. c-PARP = cleaved-PARP, C = control (vehicle) treatment, CTX = cabazitaxel, DTX = docetaxel. All data is presented as percent of control mean ± standard deviation. * = p-value ≤ 0.05.
Inhibition of ABCB1 re-sensitizes docetaxel-resistant cells to cabazitaxel
Our previous work demonstrated that ABCB1 expression was increased in both TaxR and DU145-DTXR cells versus parental cells and that this increased expression was responsible for mediating resistance to docetaxel (Fig. 2A left) (12). ABCB1 is a cell membrane efflux protein responsible for pumping a wide array of substrates out of cells and is known to be able to confer resistance to several chemotherapeutic drugs including docetaxel, doxorubicin, and paclitaxel (17). Whether ABCB1 mediates resistance to cabazitaxel in prostate cancer is unclear. To determine whether increased expression of ABCB1 is responsible for cross-resistance to cabazitaxel, we utilized TaxR cells stably expressing a shRNA against ABCB1 (TaxR-shABCB1) and subjected them to cabazitaxel treatment. Cells expressing a shRNA against GFP (TaxR-control) served as a control. We first demonstrate that ABCB1 protein levels are decreased in TaxR-shABCB1 versus TaxR-control (Fig. 2A right). Growth assays clearly demonstrate that TaxR-shABCB1 cells are re-sensitized to 1nM cabazitaxel treatment versus TaxR-control cells (Fig. 2B). These data were further supported with colony formation assays which showed that downregulation of ABCB1 expression using a shRNA specific to ABCB1 re-sensitized TaxR cells to cabazitaxel treatment (Fig. 2C). Western blots for cleaved-PARP demonstrate that TaxR-shABCB1 cells undergo significant apoptosis compared to control cells when exposed to cabazitaxel (Fig. 2D). These data suggest that ABCB1 can mediate resistance to cabazitaxel and that ABCB1 is a critical mechanism of cross-resistance between cabazitaxel and docetaxel.
Figure 2. Reducing ABCB1 expression re-sensitizes docetaxel-resistant cells to cabazitaxel treatment.
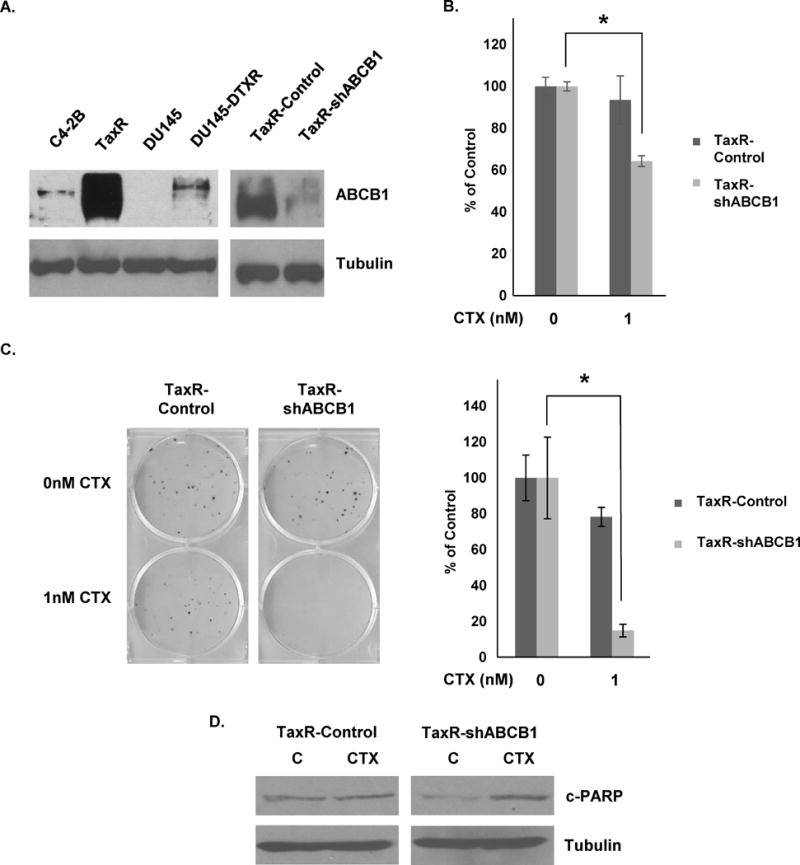
A. C4-2B, TaxR, DU145, DU145-DTXR, TaxR-Control and TaxR-shABCB1 whole cell lysates were subjected to western blot analysis using indicated antibodies. Tubulin served as a loading control. B. TaxR-Control and TaxR-shABCB1 cells were subjected to cell growth assays using either vehicle (DMSO) or cabazitaxel (1nM) treatment. Cells were counted 72 hours post-treatment. C. TaxR-Control and TaxR-shABCB1 cells were subjected to clonogenic assays using indicated doses of cabazitaxel. Colonies were allowed to form for 8 days post-treatment. Colonies were then stained with crystal violet and counted. D. TaxR-Control and TaxR-shABCB1 cells were treated with either vehicle (DMSO) or cabazitaxel (1nM) for 72 hours. Whole cell lysates were then prepared and subjected to western blot analysis using indicated antibodies. Tubulin served as a loading control. c-PARP = cleaved-PARP, C = control (DMSO) treatment, CTX = cabazitaxel. All data is presented as percent of control mean ± standard deviation. * = p-value ≤ 0.05.
To further demonstrate that ABCB1 mediates resistance to cabazitaxel, we utilized elacridar, a small molecule inhibitor of ABCB1. Our previous work demonstrated that elacridar could effectively inhibit ABCB1 activity and render docetaxel resistant cells sensitive to treatment (12, 15). Cell growth assays demonstrate that we can re-sensitize both TaxR and DU145-DTXR cells to 1nM cabazitaxel treatment using pharmacologic ABCB1 inhibition via elacridar (Fig. 3A). We additionally demonstrate that elacridar restores the cell death response to cabazitaxel treatment through western blots for cleaved-PARP (Fig. 3B). Taken together, these results support a role for ABCB1 in mediating resistance to cabazitaxel and cross-resistance between taxanes in prostate cancer.
Figure 3. ABCB1 functional inhibition using elacridar re-sensitizes TaxR and DU145-DTXR cells to cabazitaxel treatment.
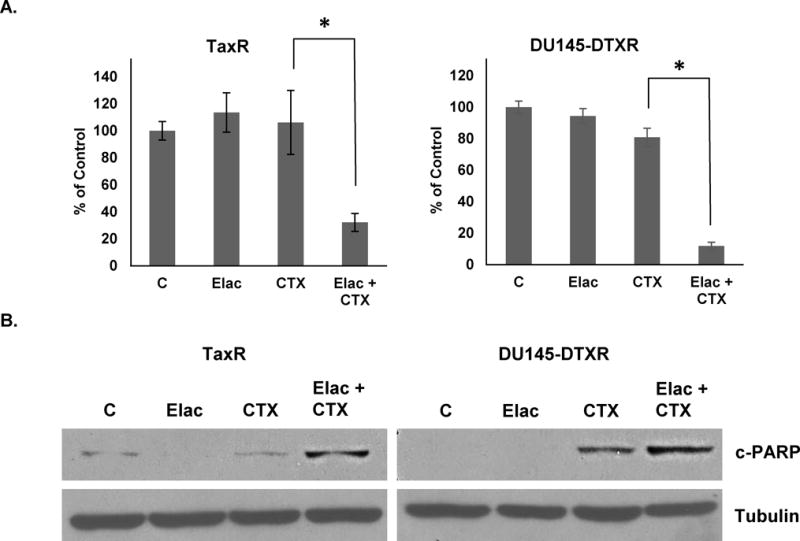
A. TaxR and DU145-DTXR cells were subjected to cell growth assays using either vehicle (DMSO), elacridar (0.5μM), cabazitaxel (1nM), or a combination of elacridar and cabazitaxel. Cells were counted 72 hours post-treatment. B. TaxR and DU145-DTXR cells were treated with either vehicle (DMSO), elacridar (0.5μM), cabazitaxel (1nM), or a combination of both elacridar and cabazitaxel for 72 hours. Whole cell lysates were then prepared and subjected to western blot analysis using indicated antibodies. Tubulin served as a loading control. c-PARP = cleaved-PARP, C = control (DMSO) treatment, Elac = elacridar, CTX = cabazitaxel. All data is presented as percent of control mean ± standard deviation. * = p-value ≤ 0.05.
Anti-androgen mediated inhibition of ABCB1 function sensitizes to cabazitaxel
Previous work demonstrates novel functionality of the anti-androgens bicalutamide and enzalutamide in blocking ABCB1 function through inhibition of its ATPase activity (15). Furthermore, it was shown that inhibition of ABCB1 functioning using either bicalutamide or enzalutamide could re-sensitize docetaxel resistant cells to docetaxel treatment (15). Since we showed that increased ABCB1 expression and activity is a common mechanism of resistance to taxanes, we hypothesized that we could use a similar strategy to re-sensitize these cells to cabazitaxel treatment through anti-androgen mediated inhibition of ABCB1 ATPase activity. Cell growth assays using bicalutamide (10μM), cabazitaxel (1nM), or combinations of bicalutamide with cabazitaxel demonstrate that bicalutamide could indeed re-sensitize both TaxR and DU145-DTXR cells to cabazitaxel treatment (Fig. 4A). These data were confirmed by colony formation assays which robustly show the ability of bicalutamide to increase the efficacy of cabazitaxel (Fig. 4B).
Figure 4. Bicalutamide re-sensitizes TaxR and DU145-DTXR cells to cabazitaxel treatment.
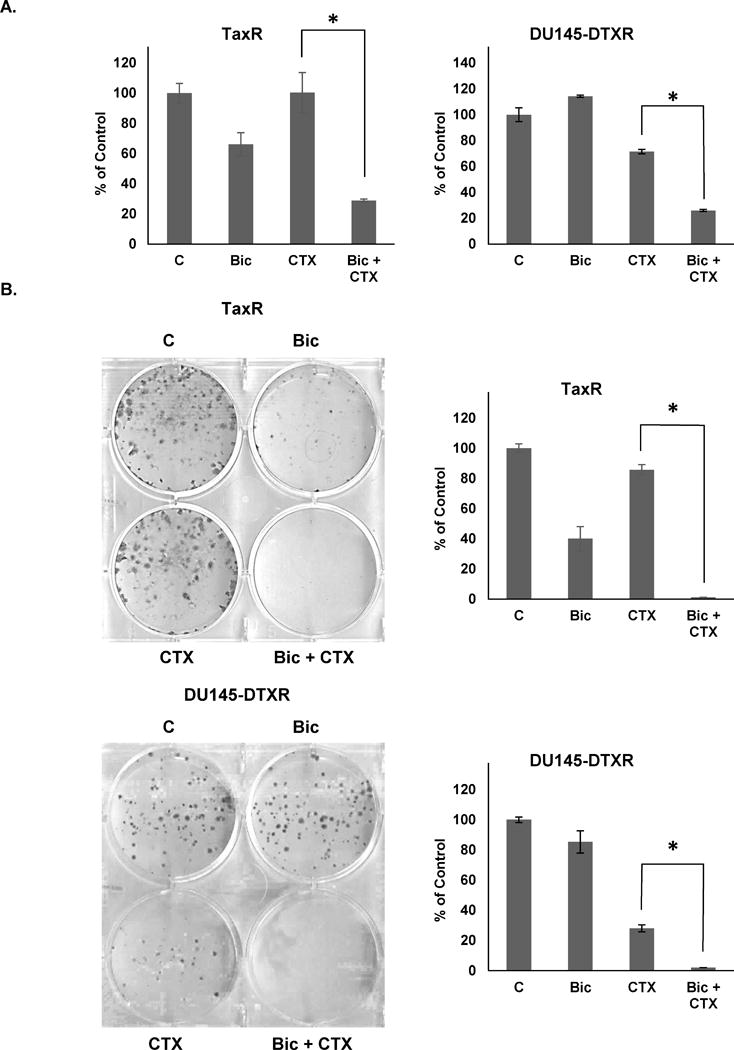
A. TaxR and DU145-DTXR cells were treated with either vehicle (DMSO), bicalutamide (10μM), cabazitaxel (1nM), or a combination of bicalutamide and cabazitaxel. Cells were counted 72 hours post-treatment. B. TaxR and DU145-DTXR cells were subjected to clonogenic assays using either vehicle (DMSO), bicalutamide (10μM), cabazitaxel (1nM), or a combination of bicalutamide and cabazitaxel. Colonies were allowed to form for 14 (TaxR) or 10 (DU145-DTXR) days post-treatment. Colonies were then stained with crystal violet and counted. C = control (DMSO) treatment, Bic = bicalutamide, CTX = cabazitaxel. All data is presented as percent of control mean ± standard deviation. * = p-value ≤ 0.05.
Similar tests were carried out to assess the ability of enzalutamide to affect sensitivity to cabazitaxel in both TaxR and DU145-DTXR cells. Like bicalutamide, cell growth assays demonstrate that enzalutamide (10μM) could re-sensitize both cell lines to cabazitaxel (Fig. 5A). Also, a combination treatment with either bicalutamide or enzalutamide with cabazitaxel could increase the cell death response of TaxR and DU145-DTXR cells as indicated by increased expression of cleaved-PARP (Fig. 5B). WST-1 assays further demonstrate that combinations of either bicalutamide or enzalutamide with cabazitaxel greatly reduce cellular proliferation versus any single agent treatment (Fig. 5C). Taken together, these data demonstrate that resistance to cabazitaxel may be overcome through combinations with anti-androgen drugs.
Figure 5. Anti-androgens re-sensitize TaxR and DU145-DTXR cells to cabazitaxel induced reduction in viability.
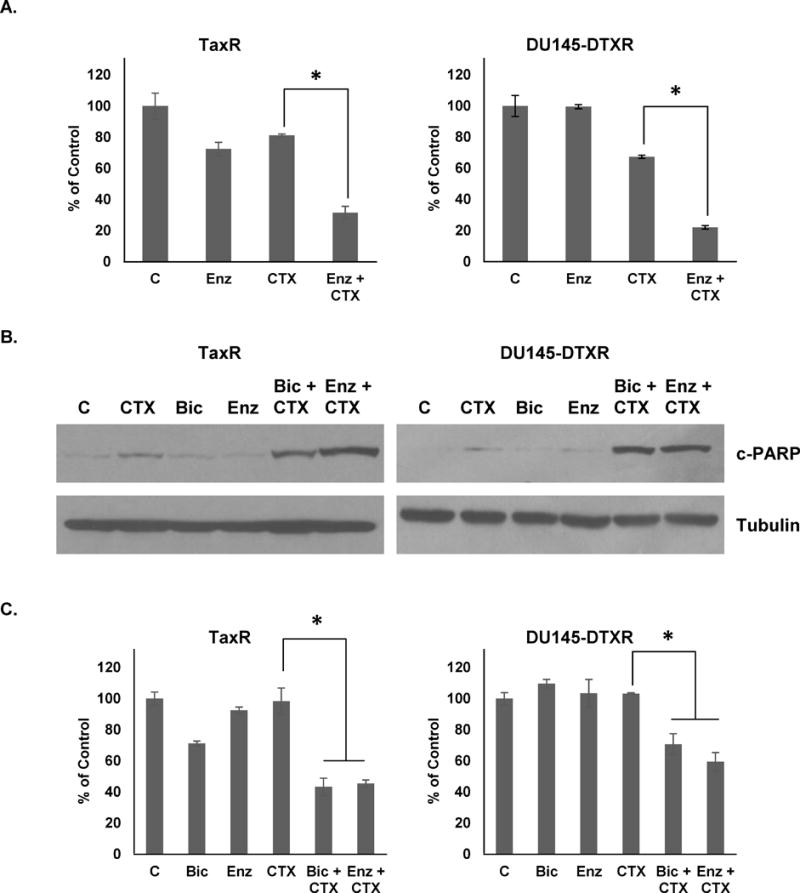
A. TaxR and DU145-DTXR cells were subjected to cell growth assays using either vehicle (DMSO), enzalutamide (10μM), cabazitaxel (1nM), or a combination of enzalutamide and cabazitaxel. Cells were counted 72 hours post-treatment. B. TaxR and DU145-DTXR cells were treated with either vehicle (DMSO), cabazitaxel (1nM), bicalutamide (10μM), enzalutamide (10μM), or a combination of either bicalutamide or enzalutamide and cabazitaxel for 72 hours. Whole cell lysates were then prepared and subjected to western blot analysis using indicated antibodies. Tubulin served as a loading control. C. TaxR and DU145-DTXR cells were treated with either vehicle (DMSO), cabazitaxel (1nM), bicalutamide (10μM), enzalutamide (10μM), or a combination of either bicalutamide or enzalutamide and cabazitaxel for 72 hours. Cells were then subjected to WST-1 proliferation assay 72 hours post-treatment. c-PARP = cleaved-PARP, C = control (DMSO) treatment, CTX = cabazitaxel, Bic = bicalutamide, Enz = enzalutamide. All data is presented as percent of control mean ± standard deviation. * = p-value ≤ 0.05.
Re-sensitization to cabazitaxel is more effective than that to docetaxel re-challenge
Since inhibition of ABCB1 re-sensitizes taxane resistant cells to both docetaxel and cabazitaxel, we sought to understand whether re-sensitization to one drug is more effective than the other. Docetaxel re-challenge has been demonstrated clinically effective in certain instances and thus, it is of interest to ask which is a better strategy; a re-sensitizing agent in combination with 1) a docetaxel re-challenge or 2) a switch to cabazitaxel (18). Our previous data demonstrated that anti-androgens could re-sensitize docetaxel resistant cells to 10nM docetaxel (15). Here, using additional cell growth assays, we tested lower doses (1nM) of either docetaxel or cabazitaxel in combination with10μM bicalutamide (Fig. 6A). We found that combination with cabazitaxel was superior to docetaxel at this dose. In fact, we found little to no effect with 1nM docetaxel plus bicalutamide. However, a higher dose of 5 nM or 10 nM docetaxel in combination with bicalutamide is effective as shown, consistent with results from a previous study (Fig. 6B) (15). Thus, while bicalutamide can re-sensitize docetaxel resistant cells to docetaxel treatment, combinations with cabazitaxel are more effective. Colony formation assays further demonstrate that combination with cabazitaxel far outperforms a similar combination with docetaxel (Fig. 6C). We also tested combinations of 1 nM docetaxel or cabazitaxel with 10 μM enzalutamide and found that again, combination with cabazitaxel outperformed combination with docetaxel (Fig. 6D). These data suggest that cabazitaxel combinations with either bicalutamide or enzalutamide are superior to similar combinations utilizing docetaxel.
Figure 6. Anti-androgen combinations with cabazitaxel are more effective than similar combinations utilizing docetaxel.
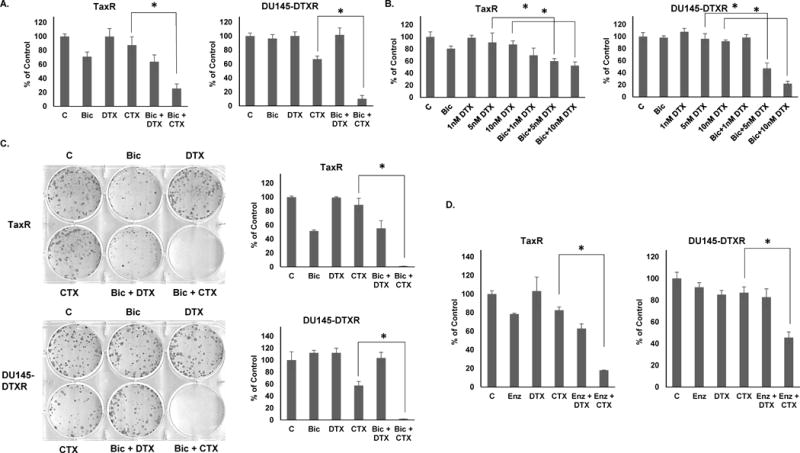
A. TaxR and DU145-DTXR cells were subjected to cell growth assays using either vehicle (DMSO), bicalutamide (10μM), docetaxel (1nM), cabazitaxel (1nM), or a combination of bicalutamide with either docetaxel or cabazitaxel. Cells were counted 72 hours post-treatment. B. TaxR and DU145-DTXR cells were subjected to cell growth assays using either vehicle (DMSO), bicalutamide (10μM), docetaxel (1, 5, or 10nM), or a combination of bicalutamide with either dose of docetaxel. Cells were counted 72 hours post-treatment. C. TaxR and DU145-DTXR cells were subjected to clonogenic assays using either vehicle (DMSO), bicalutamide (10μM), docetaxel (1nM), cabazitaxel (1nM), or a combination of bicalutamide with either docetaxel or cabazitaxel. Colonies were allowed to form for 14 days post-treatment. Colonies were then stained with crystal violet and counted. D. TaxR and DU145-DTXR cells were subjected to cell growth assays using either vehicle (DMSO), enzalutamide (10μM), docetaxel (1nM), cabazitaxel (1nM), or a combination of enzalutamide with either docetaxel or cabazitaxel. Cells were counted 72 hours post-treatment. C = control (DMSO) treatment, Bic = bicalutamide, Enz = enzalutamide, DTX = docetaxel, CTX = cabazitaxel. All data is presented as percent of control mean ± standard deviation. * = p-value ≤ 0.05.
Discussion
In the present study, we have demonstrated that the docetaxel resistant C4-2B and DU145 cell line derivatives TaxR and DU145-DTXR are cross-resistant to the next-generation taxane cabazitaxel. We show that resistance to cabazitaxel is mediated through increased expression of ABCB1 and that inhibition of ABCB1 functioning either with a small molecule inhibitor (elacridar) or anti-androgens (bicalutamide and enzalutamide) can re-sensitize resistant cells to cabazitaxel treatment. Our work addresses key concerns regarding cross-resistance between docetaxel and cabazitaxel for advanced prostate cancer and highlights a common resistance mechanism involving ABCB1 that may be employed by prostate tumors to circumvent the activity of cabazitaxel. We additionally demonstrate the potential in utilizing combinations of anti-androgens with cabazitaxel for increased effect.
Clinically, cabazitaxel is approved only for the treatment of patients who have previously undergone docetaxel treatment (9, 16). The TROPIC clinical trial demonstrated an improved overall survival using cabazitaxel in patients progressing during or after docetaxel treatment. While these results are encouraging, the survival benefit is modest (2.4 months) (9). Our data supports these findings, showing that docetaxel resistant prostate cancer cells are completely resistant to higher doses of docetaxel while still responding to some doses of cabazitaxel (Fig. 1D). However, we show a clear decrease in response to cabazitaxel in docetaxel resistant cells versus parental cells, demonstrating the existence of cross-resistance between docetaxel and cabazitaxel (Fig. 1). Thus, although clinical data demonstrates a clear benefit to using cabazitaxel post docetaxel, it is important to understand potential mechanisms of cross-resistance and design strategies to mitigate or overcome potential treatment impediments.
ABCB1 is an ATP-dependent membrane efflux pump that is known to act on many substrates (19). Furthermore, its upregulated expression is known to correlate with a poor prognosis and disease progression in varying types of cancer including ovarian and bladder (20, 21). In prostate cancer, ABCB1 has also been shown to be upregulated versus non-cancerous tissue and to positively correlate with disease stage and grade (22, 23). Our previous work demonstrated that ABCB1 is dramatically upregulated in docetaxel resistant prostate cancer cells, that ABCB1 mediates resistance to treatment, and that inhibition of ABCB1 re-sensitizes these cells to docetaxel treatment (12, 15). Here, using both shRNA and small molecule (elacridar) inhibition strategies, we show that this increased ABCB1 expression can mediate cross-resistance to cabazitaxel. While it is possible that shRNA off-target effects may have influenced our data, our use of proper controls and multiple methods for inhibiting ABCB1 make us confident that in fact, ABCB1 is a significant factor in resistance to cabazitaxel (24). Interestingly, TaxR cells are more resistant to cabazitaxel than are DU145-DTXR cells and express much higher levels of ABCB1 (Fig. 2A left). While additional, unknown mechanisms of resistance may account for this finding, our data suggests the possibility that cabazitaxel resistance may be a function of ABCB1 expression. Further study is needed to fully understand the clinical significance of ABCB1 expression on response to taxane treatment. Fully understanding to what degree ABCB1 expression levels mediate resistance to each drug may allow for stratification of patients between the two or lead to decisions to try alternative therapies, thus preventing decisions to treat when success is unlikely. Proof of principle studies detecting higher ABCB1 levels in blood or serum samples from docetaxel resistant prostate cancer patients have been performed thus making it feasible considering our novel findings to continue studying ABCB1 expression as a prognostic marker of taxane sensitivity (25, 26).
Our previous study highlighted the ability of the anti-androgen dugs bicalutamide and enzalutamide to inhibit ABCB1 function through inhibition of its ATPase activity (15). Furthermore, we demonstrated that use of anti-androgens re-sensitizes docetaxel resistant cells to docetaxel. We show in this study that these same anti-androgens are also able to re-sensitize docetaxel resistant cells to cabazitaxel treatment, suggesting that combinations of drugs currently used for CRPC may be more beneficial than single agents. Our study further shows that cabazitaxel is more efficacious than docetaxel in combination with an anti-androgen in the setting of docetaxel resistance. Thus, our findings suggest that a combination of cabazitaxel with an ABCB1 inhibiting agent, such as an anti-androgen, should be preferred over a similar strategy with docetaxel re-challenge.
Due to the possibility of clinical cross-resistance between docetaxel and cabazitaxel, it is of interest to investigate which taxane would be better when given in the chemotherapy naïve setting. Cabazitaxel is currently approved only to treat patients who have undergone docetaxel treatment. Recent analysis of data from the FIRSTANA clinical trial, which tests cabazitaxel directly against docetaxel in chemotherapy naïve patients, demonstrated that there was no difference in outcome between use of the two taxanes (27). Since cabazitaxel has been shown effective post-docetaxel but is not superior in the chemo-naïve setting, it is unlikely that cabazitaxel would become a first-line therapy. These data in conjunction with our findings suggest important clinical implications in which combination of cabazitaxel with anti-androgens is warranted. Although cabazitaxel alone is a second-line treatment for post-docetaxel CRPC, our work suggests that combination of cabazitaxel with an anti-androgen may improve its efficacy in this setting.
Another implication from our studies is whether anti-androgens should be routinely included in chemohormonal treatment regimens in the hormone sensitive setting preceding the development of CRPC. Our data suggests this may be a strategy for inhibiting the onset of docetaxel resistance through inhibition of ABCB1 function, thus potentially further augmenting the utility of this therapeutic regimen. Results from the CHAARTED and STAMPEDE clinical trials demonstrate a clear and robust increase in overall survival when docetaxel is given upfront with androgen deprivation therapy (ADT) for patients with 4 or more metastases or visceral disease (28, 29). Whether adding an anti-androgen to this treatment is beneficial is unknown. Clinical evidence shows a modest at best improvement in outcomes when adding anti-androgens to ADT (30). As there are additional factors including cost and toxicity to adding anti-androgens at this stage, it remains to be seen whether this addition to chemohormonal therapy is more beneficial (31). Alternatively, anti-androgens could be combined with cabazitaxel post chemohormonal therapy.
Whether taxanes induce cross-resistance to hormonal therapies and vice versa is also a key area of intense study and debate with major implications regarding treatment choices. Understanding therapeutic sequencing and the creation of effective combinations is paramount for future clinical success. In the present study, we see responses to both bicalutamide and enzalutamide in the AR-expressing docetaxel resistant TaxR cells. Several preclinical and clinical studies have suggested the existence of cross-resistance between taxanes and next-generation hormonal therapies in CRPC (32, 33). It is hypothesized that inhibition of AR trafficking to the nucleus is a secondary mechanism of action for taxanes in prostate cancer, thus providing a rationale for this phenomenon (33, 34). Despite these data, others have presented findings in direct opposition, arguing against both cross-resistance between these drug classes and inhibition of AR signaling as a taxane mechanism of action (13, 35). Since it is currently unknown whether cross-resistance between drug classes is a significant clinical factor, it may be better to simply create efficacious combinations rather than wait for potential cross-resistance to develop. Our work suggests that anti-androgens can work synergistically with and may be able to prolong responses to taxanes. Thus, combination therapy may provide better outcomes than sequential use of these drugs.
In summary, we demonstrate that resistance to docetaxel confers cross-resistance to cabazitaxel and that this is mediated by increased expression of ABCB1. We also demonstrate the potential in combining anti-androgen drugs with cabazitaxel for improved efficacy.
Acknowledgments
This work was supported in part by grants NIH/NCI CA168601, CA179970, DOD PC150229, and US Department of Veterans Affairs, ORD VA Merits I01BX0002653.
Financial information: This work was supported in part by grants NIH/NCI CA140468, CA168601, CA179970 (A.C. Gao), DOD PC150229 (A.C. Gao), and US Department of Veterans Affairs, ORD VA Merits I01BX0002653 (A.C. Gao).
References
- 1.Chandrasekar T, Yang JC, Gao AC, Evans CP. Mechanisms of resistance in castration-resistant prostate cancer (CRPC) Transl Androl Urol. 2015;4(3):365–80. doi: 10.3978/j.issn.2223-4683.2015.05.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351(15):1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 3.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13):1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 4.Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371(5):424–33. doi: 10.1056/NEJMoa1405095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364(21):1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryan CJ, Smith MR, Fizazi K, Saad F, Mulders PF, Sternberg CN, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015;16(2):152–60. doi: 10.1016/S1470-2045(14)71205-7. [DOI] [PubMed] [Google Scholar]
- 7.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 8.Hoskin P, Sartor O, O’Sullivan JM, Johannessen DC, Helle SI, Logue J, et al. Efficacy and safety of radium-223 dichloride in patients with castration-resistant prostate cancer and symptomatic bone metastases, with or without previous docetaxel use: a prespecified subgroup analysis from the randomised, double-blind, phase 3 ALSYMPCA trial. Lancet Oncol. 2014;15(12):1397–406. doi: 10.1016/S1470-2045(14)70474-7. [DOI] [PubMed] [Google Scholar]
- 9.de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376(9747):1147–54. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 10.Liu C, Zhu Y, Lou W, Nadiminty N, Chen X, Zhou Q, et al. Functional p53 determines docetaxel sensitivity in prostate cancer cells. Prostate. 2013;73(4):418–27. doi: 10.1002/pros.22583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duran GE, Wang YC, Francisco EB, Rose JC, Martinez FJ, Coller J, et al. Mechanisms of resistance to cabazitaxel. Mol Cancer Ther. 2015;14(1):193–201. doi: 10.1158/1535-7163.MCT-14-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Y, Liu C, Nadiminty N, Lou W, Tummala R, Evans CP, et al. Inhibition of ABCB1 expression overcomes acquired docetaxel resistance in prostate cancer. Mol Cancer Ther. 2013;12(9):1829–36. doi: 10.1158/1535-7163.MCT-13-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Leeuw R, Berman-Booty LD, Schiewer MJ, Ciment SJ, Den RB, Dicker AP, et al. Novel actions of next-generation taxanes benefit advanced stages of prostate cancer. Clin Cancer Res. 2015;21(4):795–807. doi: 10.1158/1078-0432.CCR-14-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Morree ES, Bottcher R, van Soest RJ, Aghai A, de Ridder CM, Gibson AA, et al. Loss of SLCO1B3 drives taxane resistance in prostate cancer. Br J Cancer. 2016;115(6):674–81. doi: 10.1038/bjc.2016.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu Y, Liu C, Armstrong C, Lou W, Sandher A, Gao AC. Antiandrogens Inhibit ABCB1 Efflux and ATPase Activity and Reverse Docetaxel Resistance in Advanced Prostate Cancer. Clin Cancer Res. 2015;21(18):4133–42. doi: 10.1158/1078-0432.CCR-15-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paller CJ, Antonarakis ES. Cabazitaxel: a novel second-line treatment for metastatic castration-resistant prostate cancer. Drug Des Devel Ther. 2011;5:117–24. doi: 10.2147/DDDT.S13029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaidyanathan A, Sawers L, Gannon AL, Chakravarty P, Scott AL, Bray SE, et al. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br J Cancer. 2016;115(4):431–41. doi: 10.1038/bjc.2016.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petrioli R, Francini E, Roviello G. Is there still a place for docetaxel rechallenge in prostate cancer? World J Clin Oncol. 2015;6(5):99–103. doi: 10.5306/wjco.v6.i5.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan YY, Wang F, Zhao XQ, Wang XK, Chen YF, Liu H, et al. Degradation of P-glycoprotein by pristimerin contributes to overcoming ABCB1-mediated chemotherapeutic drug resistance in vitro. Oncol Rep. 2017;37(1):31–40. doi: 10.3892/or.2016.5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun S, Cai J, Yang Q, Zhu Y, Zhao S, Wang Z. Prognostic Value and Implication for Chemotherapy Treatment of ABCB1 in Epithelial Ovarian Cancer: A Meta-Analysis. PLoS One. 2016;11(11):e0166058. doi: 10.1371/journal.pone.0166058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Z, Zhang Y, Zhang X, Du G, Yang W, Hu Z, et al. Expression of multidrug-associated protein, P-glycoprotein, P53 and Bcl-2 proteins in bladder cancer and clinical implication. J Tongji Med Univ. 2001;21(1):56–8. doi: 10.1007/BF02888038. [DOI] [PubMed] [Google Scholar]
- 22.Karatas OF, Guzel E, Duz MB, Ittmann M, Ozen M. The role of ATP-binding cassette transporter genes in the progression of prostate cancer. Prostate. 2016;76(5):434–44. doi: 10.1002/pros.23137. [DOI] [PubMed] [Google Scholar]
- 23.Bhangal G, Halford S, Wang J, Roylance R, Shah R, Waxman J. Expression of the multidrug resistance gene in human prostate cancer. Urol Oncol. 2000;5(3):118–21. doi: 10.1016/s1078-1439(99)00055-1. [DOI] [PubMed] [Google Scholar]
- 24.Mockenhaupt S, Grosse S, Rupp D, Bartenschlager R, Grimm D. Alleviation of off-target effects from vector-encoded shRNAs via codelivered RNA decoys. Proc Natl Acad Sci U S A. 2015;112(30):E4007–16. doi: 10.1073/pnas.1510476112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kharaziha P, Chioureas D, Rutishauser D, Baltatzis G, Lennartsson L, Fonseca P, et al. Molecular profiling of prostate cancer derived exosomes may reveal a predictive signature for response to docetaxel. Oncotarget. 2015;6(25):21740–54. doi: 10.18632/oncotarget.3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kato T, Mizutani K, Kameyama K, Kawakami K, Fujita Y, Nakane K, et al. Serum exosomal P-glycoprotein is a potential marker to diagnose docetaxel resistance and select a taxoid for patients with prostate cancer. Urol Oncol. 2015;33(9):385e15–20. doi: 10.1016/j.urolonc.2015.04.019. [DOI] [PubMed] [Google Scholar]
- 27.Handy CE, Antonarakis ES. Sequencing Treatment for Castration-Resistant Prostate Cancer. Curr Treat Options Oncol. 2016;17(12):64. doi: 10.1007/s11864-016-0438-9. [DOI] [PubMed] [Google Scholar]
- 28.Sweeney CJ, Chen YH, Carducci M, Liu G, Jarrard DF, Eisenberger M, et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N Engl J Med. 2015;373(8):737–46. doi: 10.1056/NEJMoa1503747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.James ND, Sydes MR, Clarke NW, Mason MD, Dearnaley DP, Spears MR, et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet. 2016;387(10024):1163–77. doi: 10.1016/S0140-6736(15)01037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maximum androgen blockade in advanced prostate cancer: an overview of the randomised trials. Prostate Cancer Trialists‘ Collaborative Group. Lancet. 2000;355(9214):1491–8. [PubMed] [Google Scholar]
- 31.Klotz L, Schellhammer P. Combined androgen blockade: the case for bicalutamide. Clin Prostate Cancer. 2005;3(4):215–9. doi: 10.3816/cgc.2005.n.002. [DOI] [PubMed] [Google Scholar]
- 32.Chandrasekar T, Yang JC, Gao AC, Evans CP. Targeting molecular resistance in castration-resistant prostate cancer. BMC Med. 2015;13:206. doi: 10.1186/s12916-015-0457-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Soest RJ, van Royen ME, de Morree ES, Moll JM, Teubel W, Wiemer EA, et al. Cross-resistance between taxanes and new hormonal agents abiraterone and enzalutamide may affect drug sequence choices in metastatic castration-resistant prostate cancer. Eur J Cancer. 2013;49(18):3821–30. doi: 10.1016/j.ejca.2013.09.026. [DOI] [PubMed] [Google Scholar]
- 34.Zhu ML, Horbinski CM, Garzotto M, Qian DZ, Beer TM, Kyprianou N. Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer Res. 2010;70(20):7992–8002. doi: 10.1158/0008-5472.CAN-10-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al Nakouzi N, Le Moulec S, Albiges L, Wang C, Beuzeboc P, Gross-Goupil M, et al. Cabazitaxel Remains Active in Patients Progressing After Docetaxel Followed by Novel Androgen Receptor Pathway Targeted Therapies. Eur Urol. 2015;68(2):228–35. doi: 10.1016/j.eururo.2014.04.015. [DOI] [PubMed] [Google Scholar]