

ABSTRACT
Diabetic retinopathy (DR) is the most common complication of diabetes and remains one of the major causes of blindness in the world; infants born to diabetic mothers have higher risk of developing retinopathy of prematurity (ROP). While hyperglycemia is a major risk factor, the molecular and cellular mechanisms underlying DR and diabetic ROP are poorly understood. To explore the consequences of retinal cells under high glucose, we cultured wild type or E2f1−/− mouse retinal explants from postnatal day 8 with normal glucose, high osmotic or high glucose media. Explants were also incubated with cobalt chloride (CoCl2) to mimic the hypoxic condition. We showed that, at 7 days post exposure to high glucose, retinal explants displayed elevated cell death, ectopic cell division and intact retinal vascular plexus. Cell death mainly occurred in excitatory neurons, such as ganglion and bipolar cells, which were also ectopically dividing. Many Müller glial cells reentered the cell cycle; some had irregular morphology or migrated to other layers. High glucose inhibited the hyperoxia-induced blood vessel regression of retinal explants. Moreover, inactivation of E2f1 rescued high glucose-induced ectopic division and cell death of retinal neurons, but not ectopic cell division of Müller glial cells and vascular phenotypes. This suggests that high glucose has direct but distinct effects on retinal neurons, glial cells and blood vessels, and that E2f1 mediates its effects on retinal neurons. These findings shed new light onto mechanisms of DR and the fetal retinal abnormalities associated with maternal diabetes, and suggest possible new therapeutic strategies.
KEYWORDS: Diabetic retinppathy, E2f1, retinal explant, ectopic cell division, apoptosis, angiogenesis, Müller glia
Introduction
Diabetic retinopathy (DR) is the most common complication of diabetes, and remains one of the leading causes of preventable blindness in working-age adults. The overall prevalence of DR among the diabetic population is about one third.1 About one-tenth have proliferative DR or diabetic macular edema, which are the most frequent causes of blindness in diabetic patients.2 Also infants born to diabetic mothers have higher risk of developing retinopathy of prematurity (ROP), which is a potentially blinding eye disorder that primarily affects premature infants.3 Although DR is considered a microvascular disease, there is mounting evidence to show that retinal degeneration is an early event,4 many studies revealed that diabetes increases apoptosis in the inner retina, especially the retinal ganglion cells (RGC).5 However, the response of other retinal cell types exposed to hyperglycemia prior to the vascular changes, and the underlying mechanisms have not yet been thoroughly studied. For DR in vivo research, rodents are the most common models and play a critical role in understanding the disease.6,7 However due to the short lifespan of the animals, rodent DR models cannot faithfully reproduce most important hallmark lesions typical of DR in spite of prolonged disease progression.7,8 Cell culture-based approaches have proved invaluable for examining retinal cell types and how they respond to factors associated with diabetes, however since the retina is a complex, multi-cellular tissue, isolated cell cultures cannot reproduce the complex interplay of different retinal cell types in diabetes.9 Ex vivo retinal explant cultures offer a system that mimics the in vivo physiological situation to directly study the pathogenesis of retinal diseases.10,11
E2f transcription factors are downstream effectors of the tumor suppressor pRB and have a pivotal role in controlling cell-cycle progression. E2fs also participate in cellular processes beyond the cell cycle, including apoptosis, differentiation and development.12,13 Of particular interest is the observation that E2f1 could be involved in the regulation of angiogenesis14 and oxidative metabolism,15 which are known features of diabetes and its complications. However the role of E2f1 in diabetic complications has never been investigated.
In this study, we used a modified method of retinal explant culture16-18 to directly investigate the effects of high glucose on different mouse retinal cell types. Here we show that high glucose induces ectopic division and cell death of excitatory neurons, ectopic glial cell division and sustained blood vessel growth in the retina, which are similar to phenotypes of Rb1 deficiency mouse retina;12,19 and demonstrate that E2f1 is an important mediator of diabetic retinal neuronal defects.
Results
High glucose induced excitatory neuronal cell death in retinal explants
To study the effects of high glucose on mouse retinas, we compared retinal explants cultured in normal glucose, osmotic control and high glucose media. The glucose concentration in the normal control group (NG) was 7.5 mM, which is similar to the blood glucose concentration of wild type mice20 and rats;21 and was 35 mM (with 5 μg/ml insulin) in the high glucose (HG) group, which mimics type 2 diabetes and was used in previous retinal explants studies.16,22 The osmotic control group (GM) had 7.5 mM glucose and 27.5 mM mannitol to a final concentration of 35 mM. Retinal explants were harvested from postnatal day 8 (P8) C57 BL/6 mice and cultured for 7 days (P8-P15). The reason to use P8 retina is that, in general at P8, most mouse retinal progenitors exit the cell cycle and begin to differentiate into all seven retinal cell types,23 and retinal superficial vascular plexus develops and reaches the peripheral retina.24 Even though P8 retinal explant is not fully differentiated, it can be successfully cultured to study the response to high glucose of many types of retinal cells, including neurons, glial cells and vascular endothelium cells, a situation similar to infant retinas born to diabetic mothers.3
We first assessed the survival of retinal cells in our culture system by TUNEL and active caspase 3 staining, as cell death is the earliest feature of diabetic retinopathy.25 In our groups, the number of TUNEL+ cells in retinas cultured in HG medium was significantly higher than that in NG and GM groups (Fig. 1A and D). Most TUNEL+ cells were in the ganglion cell layer (GCL) and inner nuclear layer (INL), some of them also in the outer nuclear layer (ONL) (Fig. 1A). The number of cleaved caspase-3+ cells of HG retinal explants, mainly located in the GCL and INL, were also significantly higher than that in NG and GM groups (Fig. 1B and D).
Figure 1.

High glucose induced excitatory neuronal death in retinal explants. (A) Sections from P8 retinal explants cultured for 7 days under indicated conditions were stained for nuclei (DAPI, blue), cell death (TUNEL, green), rod photoreceptors (Rhodopsin, green), cone photoreceptors (Cone arrestin, red), horizontal cells (Calbindin, green) and amacrine cells (Calretinin, green). (B) Sections from P8 retinal explants cultured for 7 days under indicated conditions were stained for nuclei (DAPI, blue), apoptosis (active caspase 3, red) and bipolar cells (Chx10, green). (C) Wholemont retinas from P8 retinal explants cultured for 7 days under indicated conditions, and P15 wild type (WT) retina were stained for nuclei (DAPI, blue) and ganglion cells (Brn3, green). (D) Quantification of TUNEL+ cells, active caspase 3 (cas3)+ cells, Chx10+ bipolar cells, Chx10+/Cas3+ dying bipolar cells, Cone arrestin+ Cone cells, Calretinin+ amacrine cells and Calbindin+ horizontal cells per mm2 retinal section area. (E) Quantification of Brn3+ ganglion cells per mm2 of wholemont of retinal explants and P15 WT retinas. (F) Quantification of the thickness of ONL of retinal explants (μm). (G) The relative expression level of indicated genes in retinal explants by RT-PCR. Error bars represent SD of measurements from at least three animals and asterisks indicate significant differences between retinas of NG group (D, E, F) or WT NG group (G) and the indicated groups (*, p < 0.05, **, p < 0.01, t test). Scale bar = 50 μm. NG: normal glucose control (7.5 mM); GM: osmotic control (7.5 mM glucose+27.5 mM Mannitol); HG: high glucose (35 mM). ONL: outer nuclear layer; INL: inner nuclear layer; GCL: ganglion cell layer.
The major cell types in GCL are RGCs and displaced amacrine cells. To evaluate the effect of high glucose on RGCs, we performed Brn-3 staining of retinal wholemont. As optic nerve axotomy can induce RGC death,26 RGCs tend to degenerate in ex vivo explant cultures.27 So we included a wild type P15 mouse retina (not cultured ex vivo) as control (Fig. 1C). The results revealed that ex vivo explant culture for 7 days obviously reduced the number of Brn-3+ RGCs to about 50% of the un-cultured age-matched retinas, similar to the previous report27 (Fig. 1C and E), and the number of RGCs in retinas treated with high glucose dropped further in comparison with those cultured in NG or GM medium (Fig. 1C and E).
Chx10 is a pan-marker of bipolar cells, a major cell type in INL.28 Immunostaining of retinal sections indicated that there were significantly fewer Chx10+ bipolar cells in HG group compared to the NG and GM group (Fig. 1B and D). In addition, double labeling experiments found that there were remarkably more cleaved caspase 3+/Chx10+ cells in retinas of HG group than NG and GM groups (Fig. 1B and D), confirming that bipolar cells are vulnerable in diabetes.
The two other neurons in the INL/GCL, amacrine (Calretinin+) and horizontal (Calbindin+) cells, survived high glucose as their cell counts were similar between HG, NG and GM groups (Fig. 1A and D). We detected some TUNEL+ cells in the outer nuclear layer (ONL), but the thickness of ONL (an indicator of the number of rod photoreceptors) and cone arrestin+ cones were the same in these three groups (Fig. 1A, D, and F).
We also analyzed the gene expression of cell death regulators by quantitative RT-PCR and confirmed that, the expression of caspase 3, Aspp2, Atm, Chk1, P19arf, Puma and Rad51 genes in HG treated retinas substantially increased compared to control retinas in NG and GM groups, however the expression level of Gadd45 and p53 gene was the same in all three groups (Fig. 1G).
In summary, high glucose induced ganglion and bipolar cell death (both excitatory retinal neurons), but had no major effects on inhibitory neurons (horizontal and amacrine cells). Even through high glucose also induced some cell death in ONL, the numbers of cones and ONL thickness had not reduced in this ex vivo system. It is well known that Atm, Chk1 and Rad51 are DNA-damage related factors, Aspp2, P19arf and Puma are factors related to p53-depedent cell death. So it is likely that high glucose induces DNA-damage related and p53-depedent cell death.
High glucose induced ectopic cell division of Müller glial cells and neurons in retinal explants
In contrast to RGCs and bipolar cells, retinal glial cells (Müller glia) survived high glucose, there were almost no active caspase-3+/glutamine synthetase (GS)+ double labelled Müller cells in all three groups (Fig. 2A). Actually the GS+ Müller cells increased in HG group compared to control groups (Fig. 2A and C). MCM3 is a pan-cell cycle marker; EdU is a nucleoside analog of thymidine and can be incorporated into DNA during active DNA synthesis, thus directly measuring the S-phase of the cell cycle. PH3 is a mitotic marker and specifically labels the M phase of cell cycle. MCM3+/GS+, EdU+/GS+ and PH3+/GS+ cell counts both increased significantly in HG group, indicating ectopic cell division of Müller glia cells (Fig. 2A and C). We also assessed the morphology and location of Müller glia in retinas treated with high glucose. In control retinas, Müller glia had a characteristic radial morphology, with elongated cell bodies located in the middle of INL and processes that extended towards the apical and basal retinal boundaries (Fig. 2A). However some EdU+ and MCM3+ Müller glial cells in HG group had irregular morphology with cell bodies mislocalized to the GCL and other locations in INL (Fig. 2A, arrows). These cells always had weak GS staining, indicating an immature differentiation status.
Figure 2.

High glucose induced ectopic cell division of Müller glia cells and neurons in retinal explants. (A) Sections from P8 retinal explants cultured for 7 days under indicated conditions, and were labeled with 100 μM EdU for 1 hour before fixation, were stained for nuclei (DAPI, blue), Müller glia cells (GS, green), apoptosis (active caspase 3, red), all cell cycle phases (MCM3, red), S phase of cell cycle (EdU, red) and M phase of cell cycle (PH3, red). Arrows in (A) indicate some mislocalised Müller glia cells with irregular shapes. Dash arrows in A indicate some GS− ectopic dividing cells. (B) Sections from P8 retinal explants cultured for 7 days under indicated conditions, and were labeled with 100 μM EdU for 1 hour before fixation, were stained for nuclei (DAPI, blue), bipolar cells (Chx10, green) or bipolar/ganglion/cholinergic amacrine cells (Isl-1, green) and S phase of cell cycle (EdU, red). Arrows in B indicate some ectopic diving bipolar cells. (C) Quantification of total Müller glia cells (GS), ectopic dividing Müller glia cells (including MCM3+/GS+ cells, EdU+/GS+ cells and PH3+/GS+ cells) and ectopic diving bipolar cells (EdU+/Chx10+ cells, EdU+/Isl-1+ cells)per mm2 retinal section area. (D and E) The relative expression level of indicated genes in retinal explants by RT-PCR. Error bars represent SD of measurements from at least three animals and asterisks indicate significant differences between retinas of NG group (C) or WT NG group (D and E), and the indicated groups (*, p < 0.05, t test). Scale bar = 50μm. ONL: outer nuclear layer; INL: inner nuclear layer; GCL: ganglion cell layer.
In the HG treated retinal explants, there were some GS−/MCM3+ or GS−/EdU+ cells, but no GS−/PH3+ cells (Fig. 2A, dash arrows). These GS− dividing cells could be neurons, de-differentiated Müller glial cells, microglial cells or vascular endothelium cells. Some are likely neurons fail to permanently exit the cell cycle, similar to neurons with aberrant cell-cycle activation in some neurodegenerative disorders, such as Alzheimer's disease, Parkinson's disease.29 As EdU also labels cells that are dying or repairing their DNA, these GS−/EdU+ cells may also be dying neurons.30 They were mainly in the GCL and INL (Fig. 2A and B). We found many EdU+/Chx10+ cells in HG-treated retina (Fig. 2B and C). However Chx10 is a marker of bipolar cells and retinal progenitor cells, and Müller glia also up-regulate Chx10 as they de-differentiate after injury. Thus it is possible that some EdU+/Chx10+ cells could be de-differentiated proliferating Müller glia. To confirm these dividing cells were neurons, we used the marker Islet-1(Isl-1). This LIM-homeodomain protein is expressed in retinal ganglion cells, nascent cholinergic amacrine cells and bipolar cells. The majority of Isl-1+ cells locate in the outer INL, which are ON-bipolar cells. A few of Isl-1+ cells locate in the inner INL and GCL, which are ganglion cells and cholinergic amacrine cells.31-33 By this analysis, indeed, many of these dividing cells were bipolar cells, as the EdU+/Islet1+ cells (mainly in the inner INL) increased in HG group (Fig. 2B and C, arrows).
We also analyzed the gene expression of cell cycle regulators by quantitative RT-PCR and confirmed that the expression of Ccna1, Ccnb1, Ccne2, Cdk2, Mcm3, Ki67, Pcna, E2f1, E2f2 and p57kip2 genes in HG treated retinas significantly increased compared to control retinas in NG and GM groups, the expression of P21cip1 and p27 kip1 was reduced in HG group, however the expression level of E2f3 was the same in these three groups (Fig. 2D and E).
In summary, high glucose induced ectopic cell division of Müller glia cells and retinal neurons, such as bipolar cells. Up-regulation of cell cycle genes and down-regulation Cdk inhibitors (such as P21cip1 and p27 kip1) may be the major mechanism.
High glucose inhibited the regression of retinal blood vessels in retinal explants
Microvascular defect is the major characteristic of diabetic retinopathy. So we assessed the blood vessels of retinal explants. In the mouse retina, a superficial primary vascular plexus in the nerve fiber layer (NFL) develops from the optic nerve head between postnatal P0 and P8. Subsequently, vessels sprout vertically into the retina and ramify in the outer plexiform layer (OPL) and inner plexiform layer (IPL) to form the deep plexus and intermediate plexus respectively, resulting in a three-layered vascular architecture.24 Generally speaking, ex vivo retinal explant is not suitable for evaluation of the overall growth of the vascular plexus, as the hyperoxic conditions experienced by the retina once extracted from the eye generally result in regression of the blood vasculature, either by increased cell death or normal vessel pruning.18 Indeed, retinal explants cultured from P8-P15 under normal glucose or osmotic control medium only had scattered IB4 positive endothelial cells at the NFL without any vascular plexus (Fig. 3A, B, and D–F), suggesting hyperoxia or lack blood supply induced regression; in contrast explants under high glucose had intact superficial primary and deep plexi (Fig. 3A, B, and D–F), suggesting high glucose can inhibit hyperoxia-induced blood vessel regression. The cobalt chloride can induce Hif1a expression and hypoxia in cell culture.34 Hypoxia induced by 200 μM CoCl2 treatment can partially rescue the blood vessel regression in the two control groups, but had no major effect on the HG group (Fig. 3C–F), suggesting high glucose can induce hypoxia in retinal explants. The total vessel length in the CoCl2-treated NG or GM retinal explants was still shorter than CoCl2-untreated HG explants (Fig. 3C and E), suggesting high glucose induce more severe hypoxia than 200 μM CoCl2, or has other protective effects on vasculature.
Figure 3.
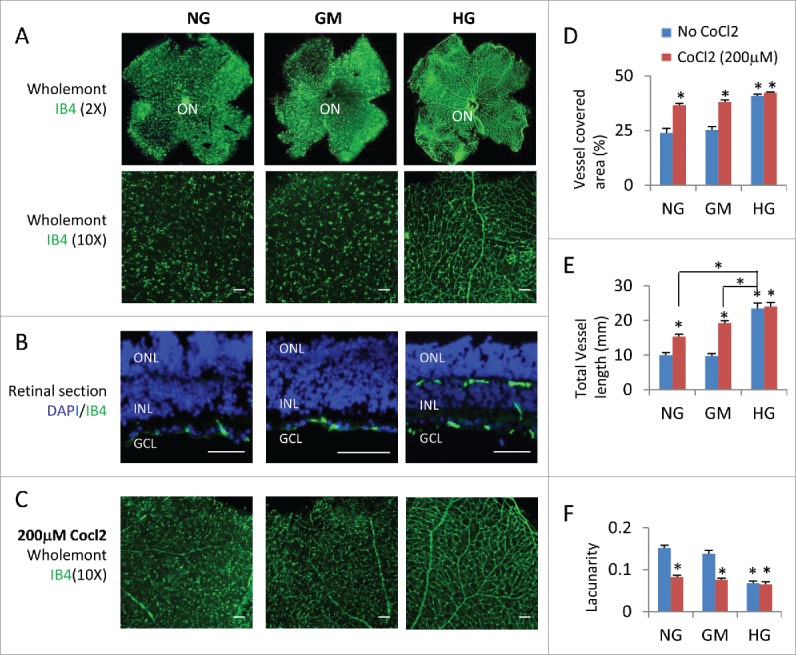
High glucose suppressed hyperoxia-induced regression of retinal vascular vessels. (A) Wholemont retinas (A) and sections (B) from P8 retinal explants cultured for 7 days under indicated conditions were stained for vascular endothelial cells (IB4, green) and nuclei in (B; DAPI, blue). (C) Wholemont retinas from P8 retinal explants cultured for 7 days under indicated conditions and 200 μM CoCl2 were stained for vascular endothelial cells (IB4, green). (D–F) Quantification of vessel coverage (percentage of area covered by IB4+ endothelial cells, D), total vessel length (E) and lacunarity (F, distribution of the gap sizes surrounding the object) by AngioTool software (https://ccrod.cancer.gov/confluence/display/ROB2/Home). Error bars represent SD of measurements from at least three animals and asterisks indicate significant differences between retinas of NG group and the indicated groups and CoCl2 treatments, or between the line-indicated pairs in (E; *p < 0.05, t test). Scale bar = 50 μm. ONL: outer nuclear layer; INL: inner nuclear layer; GCL: ganglion cell layer; ON: optic head.
E2f1 knockout rescued high glucose-induced neuronal cell death
The high glucose induced phenotypes (excitatory neuronal death, ectopic cell division) in retinal explants are similar to the Rb1 gene deficiency (RbKO) retinas in our previous reports.12,19 We reported that ectopic division and cell death of RbKO retinal cells is mediated by E2f112. In addition, E2f1 is also involved in the regulation of angiogenesis14 and oxidative metabolism,15 so we asked if E2f1 also mediated the high glucose induced retinal phenotypes by analyzing E2f1−/− retinal explants. Even though E2f1 RNA level had just slightly increased in HG group (Fig. 2D), inactivation of E2f1 dramatically reduced high glucose-induced cell death, as the TUNEL+ cells and active caspase 3+ cells were the same in the E2f1−/− retinal explants under NG, GM and HG conditions (Fig. 4A and F). There were very few Chx10+/Cas3+ dying bipolar cells (Fig. 4A and F), also Chx10+ bipolar cells (Fig. 4A and F), Brn3+ ganglion cells (Fig. 4D and G) and Isl-1+ bipolar/ganglion/ cholinergic amacrine cells (Fig. 4C and F) were the same in the E2f1−/− retinal explants between the three groups. This result was consistent with our gene expression analysis, the expression of caspase 3, Aspp2, Atm, Chk1, P19arf, Puma and Rad51 genes in HG treated E2f1−/− retinas was the same compared to control E2f1−/− retinas in NG and GM groups (Fig. 1G).
Figure 4.

E2f1 knockout rescued high glucose-induced cell death, but had no effects on high glucose-induced ectopic cell division and hyperoxia-induced regression of retinal vascular vessels. (A) Sections from P8 E2f1−/− mouse retinal explants cultured for 7 days under indicated conditions were stained for nuclei (DAPI, blue), cell death (TUNEL, green), apoptosis (active caspase 3, red) and bipolar cells (Chx10, green). (B) Sections from P8 E2f1−/− mouse retinal explants cultured for 7 days under indicated conditions, and were labeled with 100 μM EdU for 1 hour before fixation, were stained for nuclei (DAPI, blue), Müller glia cells (GS, green), all cell cycle phases (MCM3, red), S phase of cell cycle (EdU, red) and M phase of cell cycle (PH3, red). (C) Sections from P8 E2f1−/− mouse retinal explants cultured for 7 days under indicated conditions, and were labeled with 100 μM EdU for 1 hour before fixation, were stained for nuclei (DAPI, blue), bipolar/ganglion/cholinergic amacrine cells (Isl-1, green) and S phase of cell cycle (EdU, red). (D) Wholemont retinas from P8 E2f1−/− mouse retinal explants cultured for 7 days under indicated conditions, a were stained for nuclei (DAPI, blue) and ganglion cells (Brn3, green). (E) Wholemont retinas from P8 E2f1−/− mouse retinal explants cultured for 7 days under indicated conditions were stained for vascular endothelial cells (IB4, green). (F) Quantification of TUNEL+ cells, active caspase 3 (cas3)+ cells, Chx10+ bipolar cells, Isl-1+ bipolar /ganglion/cholinergic amacrine cells, Chx10+/cas3+ dying bipolar cells, ectopic dividing Müller glia cells (including MCM3+/GS+ cells, EdU+/GS+ cells and PH3+/GS+ cells) and ectopic dividing bipolar cells (Isl-1+/EdU+ cells) per mm2 E2f1−/− retinal section areas. (G) Quantification of Brn3+ ganglion cells per mm2 of wholemont of E2f1−/− retinal explants. (H–J) Quantification of vessel coverage (H), total vessel length (I), and lacunarity (J) of E2f1−/− retinal explant by AngioTool software. Error bars represent SD of measurements from at least three animals and asterisks indicate significant differences between retinas of NG group and the indicated groups (*, p < 0.05, t test). Scale bar = 50 μm. ONL: outer nuclear layer; INL: inner nuclear layer; GCL: ganglion cell layer.
Inactivation of E2f1 also recued the ectopic division of neurons, as there were almost no EdU+ bipolar cells and ganglion cells (Fig. 4C and F), the EdU+/Isl-1+ cell were dramatically reduced in E2f1 KO retina, comparing to WT retinas (Fig. 2B and C and Fig. 4C and F). However inactivation of E2f1 had no major effect on high glucose-induced ectopic glial cell division, as E2f1−/− retinal explants under HG conditions still had more MCM3+, EdU+ and PH3+ Müller glial cells (Fig. 4B and F). This result was consistent with our gene expression analysis, E2f1−/− only rescued the expression of Ccnb1, p21cip1 and Mcm3 but had no major effect on Ccna1, Ccne2, Cdk2, Ki67, Pcna, E2f2, and p27 kip1 expression in HG treated retinas (Fig. 2D and E). This also suggested that E2f1-induced Ccnb1 activation and p21cip1 suppression may be responsible for diabetic neuronal ectopic division and cell death.
Inactivation of E2f1 also had no effect on the hyperoxia-induced regression of retinal vascular vessels. Similar to WT retinal explants, E2f1−/− retinal explants under NG or GM media still lost most vascular vessels, E2f1−/− retinal explants under HG medium had intact retinal plexus (Fig. 4E and H–J).
Discussions
High glucose is obviously toxic to retinal neurons, inducing many cell deaths in the inner retinal layers (INL and GCL) and some in the ONL. Our study indicated that excitatory neurons (RGCs, bipolar cells and photoreceptors) are most vulnerable to high glucose. During the 7 days culture in 35 mM glucose (about 4–5 times of normal glucose concentration), in comparison with the control groups, about 80% of RGCs and 70% of bipolar cells were lost, which may contribute to decreased contrast sensitivity. Many studies indicated that RGCs are the most vulnerable cells in diabetes, but bipolar cells were seldom reported.25 It was reported that RGCs reduced about 20–25% in in vivo rodent DR models after 4–12 months of diabetes.35-37 So the RGC death in retinal explants under high glucose is much faster and more severe than that in rodent DR models, the possible reason is synthetic lethality between axotomy-induced neurotrophic factor reduction and high glucose. For bipolar cells, a recent study found that the cell numbers of Pkcα+ rod bipolar cells were not different between HG and control groups of retinal explants,22 possibly due to different experimental conditions (they compared 35 mM glucose to 19 mM glucose in the media, but we compared 35 mM glucose to 7.5 mM glucose in the media). In our study, Chx10 was used to label all bipolar cells at P15. We found that Chx10 co-localized with active caspase 3 and Chx10+ cells reduced in HG retinal explants (Fig. 1B).
Photoreceptors are also excitatory neurons, even though we found some TUNEL+ cells in the ONL, we did not detect major changes in ONL thickness or numbers of cones (Cone arrestin+ cells) in HG retinal explants. For cones, a recent study found that Glycogen phosphorylase (GP)+ cone cells reduced in HG (35 mM)-treated retinal explants.22 It is possible that GP is more sensitive than Cone arrestin upon apoptosis, or the expression of GP, an enzyme in the glycogen metabolism pathway, may be suppressed under high glucose, as the GP+ INL cells (mainly amacrine and Müller cells) also reduced in the study.22 Clearance of cellular corpses by phagocytes is a classical feature of apoptosis, but in retinal explant there may be few phagocytes around, thus delayed the clearance of dead photoreceptors during the short period of ex vivo culture.38
Inhibitory neurons (amacrine and horizontal cells) can survive high glucose. During the 7 days of culture, the number of the Calretinin+ amacrine cells and Calbindin+ horizontal cells remained the same. It has been reported that dopaminergic amacrine cells decrease in diabetic rats,39 but dopaminergic amacrine cells account for less than 1% of all amacrine neurons40 and thus it is possible the majority of amacrine cells still survive diabetic conditions.
Müller cells are the major glial cells in the retina, and were involved in DR in many previous studies.41,42 We found that Müller cells reentered the cell cycle (MCM3+, EdU+ and PH3+) and their number increased during the 7 days of culture under high glucose. Many cell cycle regulators were also up-regulated in the HG-cultured retina explants, such as Ccna1, Ccnb1, Ccne2, Cdk2, Mcm3, Ki67, Pcna, E2f1, and E2f2. It is reported that following neuronal injury, Müller cells down-regulate p27kip1 expression, and reenter the cell cycle, but generally only undergo one round of cell division.43 Thus the ectopic cell division of Müller cells may be partially induced by the cell death of excitatory neurons. However as E2f1 KO rescued high glucose-induced neuronal death, but not ectopic cell division of Müller cells, ectopic cell division of Müller cells may has other mechanisms. The RNA level of p27kip1 reduced both in HG-treated WT and E2f1 KO retina, suggesting p27kip1 down regulation is the mechanism of HG-induced Müller glia activation, but not induced by cell death. It is well known that type 2 diabetes is associated with increased cancer risk because of increased IGF-1 signaling and glucose-induced activation of Wnt/β-catenin pathway.44 Indeed, IGF-1 can repress p27kip1 expression in developing rat cerebral cortex.45 Activation of Wnt/β-catenin signaling can mediate proliferation of Müller cells in the injured mammalian retina.46 Oxidative stress also can promote proliferation and de-differentiation of Müller glial cells in vitro.47 Whether these mechanisms trigger the cell-cycle reentry of Müller glia in retinal explant under high glucose needs to be carefully studied.
Interestingly we found that under high glucose, some Müller cells had irregular morphology, abnormal localization and weak expression of glutamate synthetase (GS), indicating immature status of differentiation. This was consistent with reports that diabetes can reduce the GS expression in the retina.48,49 Müller cells normally respond to damage by becoming reactive and undergoing gliosis. However, some of them can de-differentiate and reenter the cell cycle, becoming proliferative multipotent neural progenitors which are then converted into retinal neurons to replace lost retinal cell types.50-52 Future research will determine if diabetic Müller glial cells can gain some properties of retinal progenitors which may have implications for the treatment of DR.
DR has non-proliferative and proliferative stages. The non-proliferative stage produces microvascular injury, leading to retinal ischemia and hypoxia. These changes lead to an increase of proangiogenic factors, disrupting angiogenic homeostasis and facilitating the proliferation of blood vessels, which are characteristics of proliferative DR. However, these hallmarks can never be reproduced in rodent DR models.8 There have been many studies assessing molecular and cellular responses of isolated retinal capillary endothelium cells following exposure to high glucose. However, isolated endothelium is artificial and has limits for uncovering the complexities of the neurovascular unit and how it responds to diabetes.8 Blood vessels of retinal explants generally regress quickly as a result of hyperoxia or lack blood supply,18 making this system not ideal for the study of diabetic vascular defects. However, we found high glucose can inhibit this process, and mimic a hypoxic condition. Thus retinal explants under high glucose have an intact blood vessel plexus, making this system suitable for the study of the diabetic microvascular changes. It was reported that high glucose can protect retinal endothelial cells from lysophosphatidic acid (LPA)-induced regression by elevating ROS (reactive oxygen species), which activated Src-Erk pathway.53 This mechanism needs to be further explored in our system.
Recent results indicate that DNA damage and cell cycle re-entry may constitute a common pathway in apoptosis in neurological diseases. DNA damage and repair after oxidative stress may activate the enzyme ATM (ataxia telangiectasia mutated), which is a cell-cycle regulator. Once the cell cycle is activated, E2f1 could induce cell cycle reentry and neuronal apoptosis.29,54 Surprisingly, the high glucose induced phenotypes (excitatory neuronal death, ectopic cell division) are similar to that of Rb1 gene deficiency retinas as we reported, which depend on E2f112,19. Indeed, we found that there were many ectopic dividing bipolar cells (Fig. 2A and B), and inactivation of E2f1 can rescue the diabetic neuronal cell cycle reentry and cell death (Fig. 4A and C). This result suggests E2f1 is an important mediator of high glucose induced neuronal cell cycle reentry and cell death, maybe downstream of diabetic oxidative stress, and E2f1 pathway is potentially a therapeutic target for DR treatment.25 Our analysis suggested that cyclin B1 activation and p21 suppression may relate with diabetic ectopic division of retinal neurons (Fig. 2D and E), and diabetic neuronal death may be p53-depedent (Fig. 1G).
However, E2f1 KO had no effects on ectopic cell division of Müller glial cells and vascular phenotypes, suggesting different mechanisms (such as Wnt/β-catenin pathway44 or Src-Erk pathway53) underlying these diabetic events. Brownlee suggested that oxidative stress is the unifying factor to active the five pathways involved in the pathogenesis of diabetic complications.55 This notion may be true in our ex vivo system, as all three high glucose-induced phenotypes can be downstream of oxidative stress,29,47,53 but their detailed mechanism needs to be further explored respectively.
In summary, this study introduces a comprehensive analysis of retinal explants under high glucose. We show that high glucose had direct but different effects on neurons, glial cells and blood vessels of retinal explants. E2f1 is an important mediator of diabetic neuronal defects, but not ectopic cell division of Müller glial cells and vascular phenotypes. Future study will focus on the interplay between retinal neurons, glial cells and blood vessel cells in diabetic conditions using this system, to investigate the pathogenesis mechanisms and intervention means of DR and maternal diabetes-related fetal abnormalities.
Materials and methods
Animals
C57 BL/6 WT mice and E2f1−/− (M. Greenberg) mice were housed with a normal 12-h/12-h light/dark schedule with free access to food and water. Genotyping of E2f1−/− mice was performed as before.12 All procedures were reviewed and approved by the Ethics Committee of Animal Research of West China Hospital, Sichuan University (2015001B), and performed in compliance with the Association for Research in Vision and Ophthalmology (ARVO) statement for the use of animals in ophthalmic and visual research.
Ex vivo retinal explant cultures
Retinal explants obtained from postnatal day 8 (P8) mice were cultured using a modified protocol from previously described methods.16-18 Briefly, both eyes were enucleated and retinas were carefully peeled away from the retinal pigment epithelium, and radial cuts were made to flatten the retina. The flattened retina was transferred to the membrane of a Millicell insert (Millipore, PICM03050) with the photoreceptors facing down. The insert was placed into the wells of a 6-well plate (Costar 3516, Corning), each contained 1000 μl of retinal explant media, which was replaced every two days, and was maintained in a 37°C incubator with 5% CO2 for 7 days. The retinal explant basal medium was serum-free and made from Neurobasal A, DMEM/F12 medium and N2/B27 supplements (Life Technologies, USA). The final basal medium contains 5μg/ml insulin, 1.4 mg/ml D-glucose (7.5 mM), 10% bovine albumin and 0.1 mM cpt-cAMP (Life Technologies, USA). There were 3 groups of retinal explants: the normal-glucose medium group (NG, in basal medium containing 7.5 mM D-glucose); the high-glucose medium group (HG, in NG medium with 27.5 mM D-glucose added to a final concentration of 35 mM); and the high osmotic control medium group (GM, in NG medium with 27.5 mM D-mannitol added to a final concentration of 35 mM).
Histology, immunostaining, wholemont retinal staining
Retinal explants cultured for 7 days were fixed by 4% paraformaldehyde in 0.1 M phosphate buffer saline (PBS, pH 7.4) at 4°C for 45 minutes, dehydrated in 30% sucrose overnight, and frozen in OCT at −80°C. Cryostat sections were cut at 14 μm and collected on glass slides and stored at −20°C. For immunohistochemistry, the retinal sections were dried at room temperature (RT) and incubated in blocking solution (5% normal donkey serum, 1% BSA and 0.03% Triton X-100 in 1X PBS) for one hour, then were incubated with primary antibodies (Calbindin-D28 K 1:500, Chemicon, AB1778; Calretinin 1:200, Santa Cruz, SC-11644; Chx10 1:2000, Bremner lab, University of Toronto; Cleaved caspase-3 1:200, Cell Signaling 9661; Glutamine synthetase 1:500, Millipore, MAB302; Islet-1, 1:200, Edlund Lab, UCMM, Umeå, Sweden; Isolectin B4 1:200, Sigma; MCM3 1:200, Santa Cruz, SC-9850; Rhodopsin 1:1000, Santa Cruz, SC-57433; p-Histone H3-ser 10 1:200, Santa Cruz, SC-8656) at 4°C overnight. Subsequently, slides were incubated with corresponding secondary antibodies, conjugated to Alexa Fluor 488 or 568 (Invitrogen, USA), for one hour at RT in the dark. Finally, slides were counterstained with 4′6-diamidino-2- phenylindole (DAPI; Sigma Aldrich Corp.) and mounted with Mowiol mounting medium. The negative control was performed by replacing primary antibodies with PBS. No specific signals were detected in negative control sections (data not shown).
For whole-mount staining, retinas or retinal explants were fixed and dehydrated, and were incubated at 4°C with either anti-Brn3 antibodies (1:200, Santa Cruz, SC-6026) for 3–4 days or biotin conjugated isolectin B4 (IB4, 1:200, Sigma) for 1 day, then with secondary antibody (donkey anti-goat Alexa Fluor 488), or Streptavidin Alexa Fluor 488 and DAPI for 2 days (Brn3) or 2 hours (IB4) at 4°C. The antibodies were diluted in 0.1 M PBS containing 0.5% Triton X-100. The negative controls were performed by replacing primary antibody with PBS.
TUNEL assay, EdU labeling and CoCl2 induced-hypoxia assay
To determine whether cell death had occurred, the frozen sections of retinal explants were labeled by TdT-dUTP terminal nick-end labeling (TUNEL) with an apoptosis detection kit (Promega, USA) according to the manufacturer's instructions. Non-specific signals were detected by omission of the enzyme reaction. To explore the cell proliferation of retinal explant, we used EdU (5-ethynyl-2′-deoxyuridine) incorporation to label the S-phase cells. Briefly cultured retinal explants were treated with 100 μM EdU (RiboBio Corp, Guangzhou, China) for one hour before retinal explants were fixed. The EdU was detected by a click-iT EdU detection kit (Cell-Light TM kit, C10314, RiboBio Corp, Guangzhou, China) according to the manufacturer's instructions. To explore the effects of hypoxia on retinal vascular blood vessels, we used cobalt chloride (CoCl2, Sigma 232696) to mimic hypoxia by stabilizing the hypoxia inducing factor 1 (Hif1). Briefly cultured retinal explants were treated with 200 μM CoCl2 for 7 days before retinal explants were fixed. The blood vessels were detected by IB4 staining.
RNA extraction, reverse transcription and quantitative real-time PCR
Total RNA was isolated from retinal explants using the TriPure isolation reagent (Roche, USA). First-strand cDNA was synthesized from 0.2–0.5 µg of total RNA using theRT reagent Kit with gDNA Eraser (TaKaRa, China). PCR primers are listed in Table 1. Real-time quantitative PCR was performed using the qTOWER 2.2 PCR machine (Analytik Jena, Germany). Tests were run in duplicate on three separate biological samples with EvaGreen PCR Supermix (SsoFast™, Bio-Rad laboratories, Singapore). PCR consisted of 40 cycles of denaturation at 95°C for 15 sec, annealing and extension at 55°C for 30 sec. An additional cycle (95°C, 15 sec) generated a dissociation curve to confirm a single product. Values obtained for test RNAs were normalized to β-actin mRNA levels.
Table 1.
RT-PCR primer sequences.
| Genes | Forward primer (5′ to 3′) | Reverse primer (5′ to 3′) |
|---|---|---|
| Actb | ACCACCACAGCTGAGAGGGA | GCCATCTCCTGCTCGAAGTC |
| Aspp2 | AGCCATCTGGCTTTGTCAAG | TGCCAAAATCACCCTACACA |
| Atm | GGTGGACAGGTGAACTTGCT | GTCACACCCAAGCTTTCCAT |
| Caspase 3 | CTGTACGCGCACAAGCTAGA | CTTTGCGTGGAAAGTGGAGT |
| Ccna2 | CCTTTTCCCTTGGCCTTTAC | CAGAGTCCCAGTGACCCAAT |
| Ccnb1 | GTAGCTCTTCCAGGGGTGTG | CCAGGTGGCATTACAAGACA |
| Ccne2 | TGTTCACAGGAGGTTTGGCA | GGTTCCAGGTCAGAATGCAGA |
| Cdk2 | TCCTCTGAGAGCAGTGATGCA | TTCCCCCAATGACCTAACCAG |
| Cdkn1 a (p21) | GTGGCCTTGTCGCTGTCTT | GCGCTTGGAGTGATAGAAATCTG |
| Cdkn1b(p27) | TCTCTTCGGCCCGGTCAAT | GGGGCTTATGATTCTGAAAGTCG |
| Cdkn1 c(p57) | CGAGGAGCAGGACGAGAATC | GAAGAAGTCGTTCGCATTGGC |
| Chk1 | GAAGCTCAGCGATGTTGTGA | TTCACCAGGAATGTGCAGAG |
| E2f1 | CTGCAGCAACTGCAGGAGAG | CTCCGAAAGCAGTTGCAGC |
| E2f2 | ACGGCGCAACCTACAAAGAG | GTCTGCGTGTAAAGCGAAGT |
| E2f3 | GGTCCTGGATCTGAACAAGGC | CCTTCCAGCACGTTGGTGAT |
| Gadd45 | GGAAGCTGCGAGAAAAGAGA | TGAAAGTAACCTGGCCATCC |
| Ki67 | GAGTGAGAGGGCAGTTCTGG | GCTGCAGTAACTGTGGGTCA |
| Mcm3 | GCTGTGTCCTGCGTTTGTTA | CAACCTTGTCATCTGCCTGA |
| p19arf | CGCAGGTTCTTGGTCACTGT | TGTTCACGAAAGCCAGAGCG |
| Pcna | GGCTCCCAAGATTGAAGATG | CAGAAAAGACCTCAGGACACG |
| Puma | GGGGGTCTGTGAAGAGCATA | CTGGGCACTGGGTTAAGAAG |
| Rad51 | TGGGGCAGTCTTTTGTCTTC | ACCACCCATTCCCACTTACA |
| p53 | TTGGACCCTGGCACCTACAAT | GGCTTTGCAGAATGGAAGGAA |
Microscopy, quantification and statistics
Stained sections and slides were analyzed using a Zeiss Axio Imager Z2 fluorescence microscope, and Nikon A1RMP confocal microscope. Image J 1.50b with cell counter plugin (https://imagej.nih.gov/ij/) was used for cell counting following the online guide. The positive cells of TUNEL, active caspase 3, EdU, PH3 and cell type markers (including Brn3b, Chx10, Calretinin, GS, Calbindin, Cone arrestin) were counted manually. At least 2 images per section, 2 sections per retina and 3 retinas from each group were counted. For Brn3+ cell counting on wholemont retina, three equivalent areas (each measuring 150 μm × 150 μm) from each quadrant of each retinal wholemont (a total of 12 areas per retina) were selected.56 All images for cell-counting were captured under fluorescence microscope using a 20 × objective lens. For vascular blood vessel analysis, representative images were analyzed using the AngioTool software (https://ccrod.cancer.gov/confluence/display/ROB2/Home).
All experiments were carried out with n = 3 to 6. The data were compared using the Student's t-test and All p-values were two-sided and were considered statistically significant when the values were <0.05.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank M. Greenberg for E2f1−/− mice, R. Bremner for Chx10 antibody, H. Edlund for Islet-1 antibody.
Funding
This study was supported by grants to DC from the National Natural Science Foundation of China (81371022, 81570860).
References
- [1].Yau JW, Rogers SL, Kawasaki R, Lamoureux EL, Kowalski JW, Bek T, Chen SJ, Dekker JM, Fletcher A, Grauslund J, et al.. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care. 2012;35:556-564. doi: 10.2337/dc11-1909. PMID:22301125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Barber AJ, Gardner TW, Abcouwer SF. The significance of vascular and neural apoptosis to the pathology of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52:1156-63. doi: 10.1167/iovs.10-6293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tunay ZO, Ozdemir O, Acar DE, Oztuna D, Uras N. Maternal diabetes as an independent risk factor for retinopathy of prematurity in infants with birth weight of 1500 g or more. Am J Ophthalmol. 2016;168:201-206. doi: 10.1016/j.ajo.2016.05.022. PMID:27287819 [DOI] [PubMed] [Google Scholar]
- [4].Simo R, Hernandez C, European Consortium for the Early Treatment of Diabetic, R . Neurodegeneration is an early event in diabetic retinopathy: Therapeutic implications. Br J Ophthalmol. 2012;96:1285-90. doi: 10.1136/bjophthalmol-2012-302005. PMID:22887976 [DOI] [PubMed] [Google Scholar]
- [5].Barber AJ, Lieth E, Khin SA. Neural apoptosis in the retina during experimental and human diabetes early onset and effect of insulin. J. Clin. Invest. 1998;102:783-791. doi: 10.1172/JCI2425. PMID:9710447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen M, Stitt A. Animal Models of Diabetic Retinopathy. In: Chi-chao Chan. Animal models of ophthalmic diseases. New York: Springer; 2016. p. 67-83. [Google Scholar]
- [7].Robinson R, Barathi VA, Chaurasia SS, Wong TY, Kern TS. Update on animal models of diabetic retinopathy: From molecular approaches to mice and higher mammals. Dis Model Mech. 2012;5:444-456. doi: 10.1242/dmm.009597. PMID:22730475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Stitt AW, Curtis TM, Chen M, Medina RJ, McKay GJ, Jenkins A, Gardiner TA, Lyons TJ, Hammes HP, Simó R, et al.. The progress in understanding and treatment of diabetic retinopathy. Prog Retin Eye Res. 2016;51:156-186. doi: 10.1016/j.preteyeres.2015.08.001. PMID:26297071 [DOI] [PubMed] [Google Scholar]
- [9].Matteucci A, Varano M, Mallozzi C, Gaddini L, Villa M, Gabrielli S, Formisano G, Pricci F, Malchiodi-Albedi F. Primary retinal cultures as a tool for modeling diabetic retinopathy: An overview. Biomed Res Int. 2015;2015:364924. doi: 10.1155/2015/364924. PMID:25688355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hatakeyama J, Kageyama R. Retrovirus-mediated gene transfer to retinal explants. Methods. 2002;28:387-395. doi: 10.1016/S1046-2023(02)00257-8. PMID:12507456 [DOI] [PubMed] [Google Scholar]
- [11].Gustmann S, Dunker N. In vivo-like organotypic murine retinal wholemount culture. J Vis Exp. 2010. Jan 11;(35). pii: 1634. doi: 10.3791/1634. PMID:20065940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chen D, Opavsky R, Pacal M, Tanimoto N, Wenzel P, Seeliger MW, Leone G, Bremner R. Rb-mediated neuronal differentiation through cell-cycle-independent regulation of E2f3 a. PLoS Biol. 2007;5:e179. doi: 10.1371/journal.pbio.0050179. PMID:17608565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chen D, Pacal M, Wenzel P, Knoepfler PS, Leone G, Bremner R. Division and apoptosis of E2f-deficient retinal progenitors. Nature. 2009;462:925-929. doi: 10.1038/nature08544. PMID:20016601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Qin G, Kishore R, Dolan CM, Silver M, Wecker A, Luedemann CN, Thorne T, Hanley A, Curry C, Heyd L, et al.. Cell cycle regulator E2F1 modulates angiogenesis via p53-dependent transcriptional control of VEGF. Proc Natl Acad Sci U S A. 2006;103:11015-020. doi: 10.1073/pnas.0509533103. PMID:16835303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Blanchet E, Annicotte JS, Lagarrigue S, Aguilar V, Clapé C, Chavey C, Fritz V, Casas F, Apparailly F, Auwerx J. E2 F transcription factor-1 regulates oxidative metabolism. Nat Cell Biol. 2011;13:1146-52. doi: 10.1038/ncb2309. PMID:21841792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Oshitari T, Yoshida-Hata N, Yamamoto S. Effect of neurotrophic factors on neuronal apoptosis and neurite regeneration in cultured rat retinas exposed to high glucose. Brain Res. 2010;1346:43-51. doi: 10.1016/j.brainres.2010.05.073. PMID:20573599 [DOI] [PubMed] [Google Scholar]
- [17].Johnson TV, Martin KR. Development and characterization of an adult retinal explant organotypic tissue culture system as an in vitro intraocular stem cell transplantation model. Invest Ophthalmol Vis Sci. 2008;49:3503-12. doi: 10.1167/iovs.07-1601. PMID:18408186 [DOI] [PubMed] [Google Scholar]
- [18].Sawamiphak S, Ritter M, Acker-Palmer A. Preparation of retinal explant cultures to study ex vivo tip endothelial cell responses. Nat Protoc. 2010;5:1659-65. doi: 10.1038/nprot.2010.130. PMID:20885378 [DOI] [PubMed] [Google Scholar]
- [19].Chen D, Livne-bar I, Vanderluit JL, Slack RS, Agochiya M, Bremner R. Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death-resistant cell-of-origin in retinoblastoma. Cancer Cell. 2004;5:539-551. doi: 10.1016/j.ccr.2004.05.025. PMID:15193257 [DOI] [PubMed] [Google Scholar]
- [20].Takano M, Sango K, Horie H, Sato M, Iijima Y, Ohno S, Inoue S, Ishikawa Y. Diabetes alters neurite regeneration from mouse retinal explants in culture. Neurosci Lett. 1999;275:175-178. doi: 10.1016/S0304-3940(99)00768-5. PMID:10580703 [DOI] [PubMed] [Google Scholar]
- [21].Casson RJ, Chidlow G, Wood JP, Osborne NN. The effect of hyperglycemia on experimental retinal ischemia. Arch Ophthalmol. 2004;122:361-366. doi: 10.1001/archopht.122.3.361. PMID:15006850 [DOI] [PubMed] [Google Scholar]
- [22].Valdes J, Trachsel-Moncho L, Sahaboglu A, Trifunović D, Miranda M, Ueffing M, Paquet-Durand F, Schmachtenberg O. Organotypic retinal explant cultures as in vitro alternative for diabetic retinopathy studies. Altex. 2016;33:459-464. PMID:27159027 [DOI] [PubMed] [Google Scholar]
- [23].Dyer MA, Cepko CL. Regulating proliferation during retinal development. Nat Rev Neurosci. 2001;2, 333-342. doi: 10.1038/35072555. PMID:11331917 [DOI] [PubMed] [Google Scholar]
- [24].Fruttiger M. Development of the retinal vasculature. Angiogenesis. 2007;10:77-88. doi: 10.1007/s10456-007-9065-1. PMID:17322966 [DOI] [PubMed] [Google Scholar]
- [25].Simo R, Hernandez C. Neurodegeneration in the diabetic eye: New insights and therapeutic perspectives. Trends Endocrinol Metab. 2014;25:23-33. doi: 10.1016/j.tem.2013.09.005. PMID:24183659 [DOI] [PubMed] [Google Scholar]
- [26].Galindo-Romero C, Avilés-Trigueros M, Jiménez-López M, Valiente-Soriano FJ, Salinas-Navarro M, Nadal-Nicolás F, Villegas-Pérez MP, Vidal-Sanz M, Agudo-Barriuso M. Axotomy-induced retinal ganglion cell death in adult mice: Quantitative and topographic time course analyses. Exp Eye Res. 2011;92:377-387. doi: 10.1016/j.exer.2011.02.008. PMID:21354138 [DOI] [PubMed] [Google Scholar]
- [27].Pattamatta U, McPherson Z, White A. A mouse retinal explant model for use in studying neuroprotection in glaucoma. Exp Eye Res. 2016;151:38-44. doi: 10.1016/j.exer.2016.07.010. PMID:27450912 [DOI] [PubMed] [Google Scholar]
- [28].Burmeister M, Novak J, Liang MY, Basu S, Ploder L, Hawes NL, Vidgen D, Hoover F, Goldman D, Kalnins VI, et al.. Ocular retardation mouse caused by Chx10 homeobox null allele: Impaired retinal progenitor proliferation and bipolar cell differentiation. Nat Genet. 1996;12:376-384. doi: 10.1038/ng0496-376. PMID:8630490 [DOI] [PubMed] [Google Scholar]
- [29].Folch J, Junyent F, Verdaguer E, Auladell C, Pizarro JG, Beas-Zarate C, Pallàs M, Camins A. Role of cell cycle re-entry in neurons: A common apoptotic mechanism of neuronal cell death. Neurotox Res. 2012;22:195-207. doi: 10.1007/s12640-011-9277-4. PMID:21965004 [DOI] [PubMed] [Google Scholar]
- [30].Arsenijevic Y. Cell cycle proteins and retinal degeneration: Evidences of new potential therapeutic targets. Adv Exp Med Biol. 2016;854:371-377. doi: 10.1007/978-3-319-17121-0_49. PMID:26427434 [DOI] [PubMed] [Google Scholar]
- [31].Elshatory Y, Deng M, Xie X, Gan L. Expression of the LIM-homeodomain protein Isl1 in the developing and mature mouse retina. J Comp Neurol. 2007;503:182-197. doi: 10.1002/cne.21390. PMID:17480014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Elshatory Y, Everhart D, Deng M, Xie X, Barlow RB, Gan L. Islet-1 controls the differentiation of retinal bipolar and cholinergic amacrine cells. J Neurosci. 2007;27:12707-720. doi: 10.1523/JNEUROSCI.3951-07.2007. PMID:18003851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bejarano-Escobar R, Álvarez-Hernán G, Morona R, González A, Martín-Partido G, Francisco-Morcillo J. Expression and function of the LIM-homeodomain transcription factor Islet-1 in the developing and mature vertebrate retina. Exp Eye Res. 2015;138:22-31. doi: 10.1016/j.exer.2015.06.021. PMID:26122047 [DOI] [PubMed] [Google Scholar]
- [34].Wu D, Yotnda P. Induction and testing of hypoxia in cell culture. J Vis Exp. 2011;12:2899. doi: 10.3791/2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998;102:783-791. doi: 10.1172/JCI2425. PMID:9710447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gastinger MJ, Kunselman AR, Conboy EE, Bronson SK, Barber AJ. Dendrite remodeling and other abnormalities in the retinal ganglion cells of Ins2 Akita diabetic mice. Invest Ophthalmol Vis Sci. 2008;49:2635-42. doi: 10.1167/iovs.07-0683. PMID:18515593 [DOI] [PubMed] [Google Scholar]
- [37].Martin PM, Roon P, Van Ells TK, Ganapathy V, Smith SB. Death of retinal neurons in streptozotocin-induced diabetic mice. Invest Ophthalmol Vis Sci. 2004;45:3330-36. doi: 10.1167/iovs.04-0247. PMID:15326158 [DOI] [PubMed] [Google Scholar]
- [38].Murakami Y, Notomi S, Hisatomi T, Nakazawa T, Ishibashi T, Miller JW, Vavvas DG. Photoreceptor cell death and rescue in retinal detachment and degenerations. Prog Retin Eye Res. 2013;37:114-140. doi: 10.1016/j.preteyeres.2013.08.001. PMID:23994436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Seki M, Tanaka T, Nawa H, Usui T, Fukuchi T, Ikeda K, Abe H, Takei N. Involvement of brain-derived neurotrophic factor in early retinal neuropathy of streptozotocin-induced diabetes in rats: Therapeutic potential of brain-derived neurotrophic factor for dopaminergic amacrine cells. Diabetes. 2004;53:2412-19. doi: 10.2337/diabetes.53.9.2412. PMID:15331553 [DOI] [PubMed] [Google Scholar]
- [40].Keeley PW, Reese BE. Morphology of dopaminergic amacrine cells in the mouse retina: Independence from homotypic interactions. J Comp Neurol. 2010;518:1220-31. PMID:20148440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fu S, Dong S, Zhu M, Sherry DM, Wang C, You Z, Haigh JJ, Le YZ. Muller glia are a major cellular source of survival signals for retinal neurons in diabetes. Diabetes. 2015;64:3554-63. doi: 10.2337/db15-0180. PMID:26068541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Matteucci A, Gaddini L, Villa M, Varano M, Parravano M, Monteleone V, Cavallo F, Leo L, Mallozzi C, Malchiodi-Albedi F, et al.. Neuroprotection by rat Muller glia against high glucose-induced neurodegeneration through a mechanism involving ERK1/2 activation. Exp Eye Res. 2014;125:20-29. doi: 10.1016/j.exer.2014.05.011. PMID:24877742 [DOI] [PubMed] [Google Scholar]
- [43].Dyer MA, Cepko CL. Control of Muller glial cell proliferation and activation following retinal injury. Nat Neurosci. 2000;3:873-880. doi: 10.1038/78774. PMID:10966617 [DOI] [PubMed] [Google Scholar]
- [44].Chocarro-Calvo A, Garcia-Martinez JM, Ardila-Gonzalez S, De la Vieja A, Garcia-Jimenez C. Glucose-induced beta-catenin acetylation enhances Wnt signaling in cancer. Mol Cell. 2013;49:474-486. doi: 10.1016/j.molcel.2012.11.022. PMID:23273980 [DOI] [PubMed] [Google Scholar]
- [45].Mairet-Coello G, Tury A, DiCicco-Bloom E. Insulin-like growth factor-1 promotes G(1)/S cell cycle progression through bidirectional regulation of cyclins and cyclin-dependent kinase inhibitors via the phosphatidylinositol 3-kinase/Akt pathway in developing rat cerebral cortex. J Neurosci. 2009;29:775-788. doi: 10.1523/JNEUROSCI.1700-08.2009. PMID:19158303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Osakada F, Ooto S, Akagi T, Mandai M, Akaike A, Takahashi M. Wnt signaling promotes regeneration in the retina of adult mammals. J Neurosci. 2007;27:4210-19. doi: 10.1523/JNEUROSCI.4193-06.2007. PMID:17428999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Abrahan CE, Insua MF, Politi LE, German OL, Rotstein NP. Oxidative stress promotes proliferation and dedifferentiation of retina glial cells in vitro. J Neurosci Res. 2009;87:964-977. doi: 10.1002/jnr.21903. PMID:18855938 [DOI] [PubMed] [Google Scholar]
- [48].Yu XH, Zhang H, Wang YH, Liu LJ, Teng Y, Liu P. Time-dependent reduction of glutamine synthetase in retina of diabetic rats. Exp Eye Res. 2009;89:967-971. doi: 10.1016/j.exer.2009.08.006. PMID:19699197 [DOI] [PubMed] [Google Scholar]
- [49].Lieth E, LaNoue KF, Antonetti DA, Ratz M. Diabetes reduces glutamate oxidation and glutamine synthesis in the retina. The penn state retina research group. Exp Eye Res. 2000;70:723-730. doi: 10.1006/exer.2000.0840. PMID:10843776 [DOI] [PubMed] [Google Scholar]
- [50].Pollak J, Wilken MS, Ueki Y, Cox KE, Sullivan JM, Taylor RJ, Levine EM, Reh TA. ASCL1 reprograms mouse Muller glia into neurogenic retinal progenitors. Development. 2013;140:2619-31. doi: 10.1242/dev.091355. PMID:23637330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Goldman D. Muller glial cell reprogramming and retina regeneration. Nat Rev Neurosci. 2014;15:431-442. doi: 10.1038/nrn3723. PMID:24894585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sanges D, Simonte G, Di Vicino U, Romo N, Pinilla I, Nicolás M, Cosma MP. Reprogramming Muller glia via in vivo cell fusion regenerates murine photoreceptors. J Clin Invest. 2016;126:3104-16. doi: 10.1172/JCI85193. PMID:27427986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Aranda J, Motiejunaite R, Im E, Kazlauskas A. Diabetes disrupts the response of retinal endothelial cells to the angiomodulator lysophosphatidic acid. Diabetes. 2012;61:1225-33. doi: 10.2337/db11-1189. PMID:22415872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zencak D, Schouwey K, Chen D, Ekström P, Tanger E, Bremner R, van Lohuizen M, Arsenijevic Y. Retinal degeneration depends on Bmi1 function and reactivation of cell cycle proteins. Proc Natl Acad Sci U S A. 2013;110:E593-601. doi: 10.1073/pnas.1108297110. PMID:23359713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058-70. doi: 10.1161/CIRCRESAHA.110.223545. PMID:21030723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rojas B, Gallego BI, Ramírez AI, Salazar JJ, de Hoz R, Valiente-Soriano FJ, Avilés-Trigueros M, Villegas-Perez MP, Vidal-Sanz M, Triviño A, et al.. Microglia in mouse retina contralateral to experimental glaucoma exhibit multiple signs of activation in all retinal layers. J Neuroinflammation. 2014;11:133. doi: 10.1186/1742-2094-11-133. PMID:25064005 [DOI] [PMC free article] [PubMed] [Google Scholar]