

Abstract
Histone modifications play a critical role in the pathological processes of dilated cardiomyopathy (DCM), while the role and expression pattern of histone methyltransferases (HMTs), especially mixed lineage leukemia (MLL) families, in DCM are unclear. To this end, 12 normal and 15 DCM heart samples were included in the present study. A murine cardiac remodeling model was induced by transverse aortic constriction (TAC). Real-time polymerase chain reaction was performed to detect the expression levels of MLL families in the mouse and human left ventricles. The mRNA level of MLL3 was significantly increased in the mouse hearts treated with TAC surgery. Compared with normal hearts, higher mRNA and protein level of MLL3 was detected in the DCM hearts, and its expression level was closely associated with left ventricular end diastolic diameter and left ventricular ejection fraction. However, there was no obvious change in the expression levels of other MLL families (MLL, MLL2, MLL4, MLL5, SETD1A and SETD1B) between control and DCM hearts or remodeled mouse hearts. Furthermore, the dimethylated histone H3 lysine 4 (H3K4me2) but not H3K4me3 was significantly increased in the DCM hearts. The protein levels of Smad3, GATA4 and EGR1, which might be regulated by MLL3, were remarkably elevated in the DCM hearts. Our hitherto unrecognized findings indicate that MLL3 has a potential role in the pathological processes of DCM by regulating H3K4me2 and the expression of Smad3, GATA4 and EGR1.
INTRODUCTION
Dilated cardiomyopathy (DCM) is characterized by left ventricular enlargement or dilatation and systolic dysfunction, and remarkable progress had been made in understanding its genetic basis (1). However, the effect of post-translational modifications of proteins, common and typical regulatory mechanisms that control key cellular processes (eg, cell cycle, survival, proliferation and differentiation), especially histone modification, on DCM was largely unknown. Recently, pioneering work by Nguyen et al. demonstrated that cardiac-specific DOT1L (an H3K79-specific histone methyltransferase) deficiency led to DCM-like pathological changes in mice, including heart chamber dilation, cardiomyocyte death, systolic dysfunction and conduction abnormalities (2). Loss of H3K27 methyltransferase EZH2 in cardiac precursors results in cardiac hypertrophy and fibrosis (3). These studies indicate that histone modifications have critical roles in the pathologic processes of DCM and cardiac remodeling.
Histone modifications, including methylation, acetylation, phosphorylation and ubiquitination, play a critical role in gene transcription regulation (4). Among them, histone methylation was newly found; it is balanced by methyltransferases and demethylases, which add or remove methyl groups from specific lysine or arginine. Methylation at different sites has diverse biological functions. For example, methylation at histone H3 lysine 27 (H3K27) is usually associated with gene repression, while di- and trimethylation at histone H3 lysine 4 (H3K4) often occur in coding regions of active genes (4,5). In mammals, there are at least nine H3K4-specific histone methyltransferases (HMTs), including mixed lineage leukemia (MLL) families consisting of MLL, MLL2, MLL3, MLL4, MLL5, SETD1A and SETD1B (6–8). The MLL family members, each possessing the highly conserved suppressor of variegation, enhancer of zeste, trithorax (SET) domain responsible for HMT activity, exist as distinct multiprotein complexes (namely complex of proteins associated with Set1, or COMPASS) with several common subunits, including Ash2, Wdr5, Rbbp5 and Dpy30 (9,10). MLLs have been credited as key H3K4 mono-, di- and trimethyltransferases at enhancers or promoters to facilitate gene expression (9,10). Previous studies demonstrated that MLLs contribute to several types of cancers, acute myeloid leukemia, embryo development, circadian rhythm and brown adipocyte differentiation (11–17). However, the role of MLLs in clinical DCM remains unknown.
In the present study, we aimed to investigate the expression pattern of MLLs in human normal and DCM hearts, and in mouse hypertrophic hearts. Our results show that there was no significant change in mRNA expression levels of all MLLs, except MLL3, in mouse hypertrophic hearts and in human DCM hearts compared with the control group. MLL3 mRNA level was positively correlated with left ventricular end diastolic diameter (LVEDD), but negatively correlated with left ventricular ejection fraction (LVEF). Furthermore, H3K4me2 and protein expression levels of Smad3, GATA4 and EGR1, which are downstream of MLL3, were significantly elevated in human DCM hearts compared with normal hearts.
MATERIALS AND METHODS
Human Heart Samples
The human sample study was approved by Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, China. The left ventricle samples of DCM were collected from patients undergoing heart transplantation (18,19). The normal left ventricle controls were obtained from donor hearts that were not suitable for transplantation for noncardiac reasons (18,19). Informed consent was obtained from the families of all subjects. All the procedures related to human samples complied with the principles outlined in the Declaration of Helsinki.
Cardiac Remodeling Mouse Model
Transverse aortic constriction (TAC) was performed as previously reported to induce a cardiac remodeling mouse model (20). Briefly, male mice (C57BL/6J) 8–10 wks old with body weight 24–27 g were anesthetized. The mice were connected to a MouseVent Automatic Ventilator (Kent Scientific) after endotracheal intubation. Then, after thoracotomy was performed, the sternum was retracted using a chest retractor. After freeing the aortic arch, TAC was performed between the right innominate and left carotid artery using 6.0 silk suture against a 27-gauge blunt needle, which was removed after constriction was completed. Finally, sternal closure and skin coverage were performed. A similar procedure, but without constricting the aorta, was performed in the sham-operated group. A Vevo-2100 High-Resolution Micro-Imaging System (VisualSonics, Toronto, ON, Canada) was used to measure cardiac function.
Histological Staining
Human and mouse heart sections were stained with hematoxylin and eosin (H&E) for cardiomyocytes cross-sectional area analysis, and picrosirius red to evaluate collagen deposition. These two procedures were performed as previously described (21–24).
Immunohistochemistry Analysis
Standard immunohistochemistry protocol with minor changes was used for MLL3-specific staining. The human heart sections were deparaffinized with xylene, then hydrated in ethanol gradient and eventually rinsed in deionized water. Antigen was unmasked by 10 mM sodium citrate unmasking solution at sub-boiling temperature for 10 min. The slides were blocked with 5% normal goat serum (ThermoFisher Scientific) in tris-buffered saline and Tween 20 for 1 h at room temperature after being treated with 3% hydrogen peroxide for 10 min. Slides were incubated with MLL3 primary antibody (1:25 dilution; Abgent) overnight at 4°C. The next day, the primary antibody was removed, then incubated with peroxidase-conjugated secondary antibody (1:2500 dilution; Jackson ImmunoResearch Laboratories) for 1 h at room temperature. The DAB kit was used to develop color, and finally sections were dehydrated and mounted. The images were acquired by using an Olympus BX53 light microscope.
Western Blot
Western blot was performed as previously described (20). In brief, total protein was extracted from human left ventricles by using radioimmunoprecipitation assay lysis buffer, and protein concentration was quantified by using Pierce™ BCA Protein Assay Kit (ThermoFisher Scientific). After being denatured, 20 μg protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and then transferred to a polyvinylidene fluoride membrane (Millipore). After being blocked by 5% skim milk solution, the membrane was incubated with primary antibody overnight at 4°C, including H3K4me2 (1:1000 dilution; Cell Signaling Technologies), H3K4me3 (1:1000 dilution; Cell Signaling Technologies), GATA4 (1:1000 dilution; Abcam), EGR1 (1:1000 dilution; Cell Signaling Technologies), Smad3 (1:1000 dilution; Cell Signaling Technologies) or GAPDH (1:1000 dilution; Cell Signaling Technologies). The protein was detected by using a ChemiDoc™ XRS+ system (Bio-Rad) after being incubated with peroxidase-conjugated secondary antibody (1:25000 dilution; Jackson ImmunoResearch Laboratories) for 1 h at room temperature.
Real-time Polymerase Chain Reaction
TRI Reagent® Solution (ThermoFisher Scientific) was used to extract total mRNA from human and mouse left ventricles. The selected gene mRNA levels were detected by CFX Connect™ Real-Time PCR Detection System (Bio-Rad) using iQ™SYBR® Green Supermix (Bio-Rad) after reverse transcription of mRNA into cDNA. GAPDH gene expression was used as internal reference. The primers used in the present study are listed in Table 2.
Table 2.
The primers used in the present study.
| Genes | Mouse primers | Human primers | |
|---|---|---|---|
| MLL | forward | AGTGTGTCCGCTGCAAGAG | TGCCAGTAGTGGGCATGTAG |
| reverse | CTGCATCATCTTGCTCTCGT | CCTTCCACAAACGTGACAGA | |
| MLL2 | forward | CAATTGTGCCATTTGGTCAG | TTGTCCTTGGGACTCGAATCA |
| reverse | TGCTGAGACATGAGGACAGG | CCTGTCCAGATCCAACTCTTCT | |
| MLL3 | forward | TCGGGAGAAGATAGCAAGATG | TGTTGTCTCAGGGAGCACAG |
| reverse | AACAGTTGTGGTGCCACTGA | TCAGGAGCTTGGTCAATGTG | |
| MLL4 | forward | TTGGCCTGTTCACAGTGTTC | GCAGAACTGAATCCCAACTCG |
| reverse | GGGGATCCAGGCAGTATGTA | GGAGCGGATAGTCTGACCTC | |
| MLL5 | forward | GACTTGCCTCCTGATGCACT | GCAAGGACTTTTGGGAATGA |
| reverse | TCCTCGCATCAACACACATT | CCATAGTCAAAATCAAAGGCAAT | |
| SETD1A | forward | CGTATCCATGAGTGGGGTCT | CTGACGAGATGGTCATCGAA |
| reverse | AATGATGGTGTCGTGATCCA | TGCAGCAGTGGTTGATGAAT | |
| SETD1B | forward | CCACCACCAGGCTACATACC | AGGGGCATCATAAACTGTACCG |
| reverse | CTTCTTGTCCCACCATTCGT | GGGGATCTTCGACAATTTCCAC | |
| GAPDH | forward | ACTCCACTCACGGCAAATTC | GAGTCAACGGATTTGGTCGT |
| reverse | TCTCCATGGTGGTGAAGACA | TTGATTTTGGAGGGATCTCG |
Statistical Analysis
Data are shown as mean ± standard error of the mean. Differences between two groups were assessed by two-tailed Student t test. Pearson correlation coefficients were used to evaluate the consistency of MLL3 expression level, LVEDD and LVEF. All statistical analyses were performed with SPSS software, version 13.0. P < 0.05 was considered statistically significant.
RESULTS
Expression Level of MLL3, but Not Other MLL Family Members, Was Remarkably Increased in the Hearts of Mice Subjected to TAC Surgery
To investigate the relationship between MLLs and cardiac remodeling, we first established the cardiac remodeling mouse model via TAC surgery, as previously reported (20). After treatment with TAC for 4 or 8 wks, the hearts underwent left ventricular dilatation and impaired cardiac function, as evidenced by increased LVEDD and left ventricular end systolic diameter (Figures 1A and B) and reduced fractional shortening (Figure 1C). In addition, enlarged cardiomyocyte size and obvious collagen deposition indicate cardiac remodeling (Figures 1D and E). Subsequently, we detected the mRNA levels of MLL to MLL5 by real-time polymerase chain reaction. The results show that compared with the sham group, the mRNA levels of MLL, MLL2, MLL4 and MLL5 did not change significantly after TAC stimulation (Figure 1F). On the contrary, the mRNA expression level of MLL3 remarkably increased after TAC treatment for 4 and 8 wks (Figure 1F). These results might indicate that MLL3 participates in TAC-induced cardiac remodeling.
Figure 1.

The expression levels of MLL family members in mouse hearts. (A–C) Parameters of echocardiographic results for mice: (A) left ventricular end diastolic dimension; (B) left ventricular end systolic dimension; (C) fractional shortening (n = 8 per experimental group). (D) H&E staining of left ventricle of sham-treated mouse hearts and hearts treated with transverse aortic constriction for 4 or 8 wks (n = 4 mice per experimental group, scale bar 50 μm). (E) Picrosirius red staining of left ventricles of mouse hearts, red staining indicating collagen deposition (n = 4 mice per experimental group, scale bar 100 μm). (F,G) Real-time PCR detected mRNA levels of MLL families in mouse hearts (n = 4 mice per experimental group).
Except for MLL-MLL5, MLL family members also included SETD1A and SETD1B (10). To investigate whether SETD1A and SETD1B are related to cardiac remodeling, we detected the mRNA levels of these two genes in the left ventricles of mouse hearts. Our results demonstrate that after 4 and 8 wks of TAC treatment, there was no significant difference in SETD1B between groups, while SETD1A transcription level in mice was decreased after TAC treatment for 8 wks (Figure 1G).
Higher MLL3 Expression Was Detected in Human DCM Hearts
Human DCM hearts displayed some similar pathology changes with TAC-induced cardiac remodeling, such as cardiomyocyte hypertrophy and cardiac fibrosis (Figures 2A and B), despite having many different pathologic processes. Therefore, we further studied whether MLLs had similar changes in the hearts of DCM patients as in TAC-induced mouse hearts. Compared with normal hearts, DCM hearts had larger LVEDD but lower LVEF (Table 1). Furthermore, the heart failure biomarkers brain natriuretic peptide, atrial natriuretic peptide and α-myosin heavy chain were significantly increased in the left ventricles of DCM hearts, while α-myosin heavy chain expression level was decreased (Figure 2C). More importantly, we detected mRNA levels of MLL family members in the left ventricles of human hearts. Our results show that MLL, MLL2, MLL4, MLL5, SETD1A and SETD1B had comparable expression levels in donor and DCM left ventricles (Figures 3A and B), while MLL3 was strikingly elevated in the left ventricles of DCM hearts (Figure 3A). Further immunohistochemistry staining also demonstrates that DCM hearts had higher MLL3 expression level in cardiomyocytes (Figure 3C). Notably, the MLL3 expression level was positively correlated with LVEDD, but negatively correlated with LVEF (Figures 4A and B).
Figure 2.
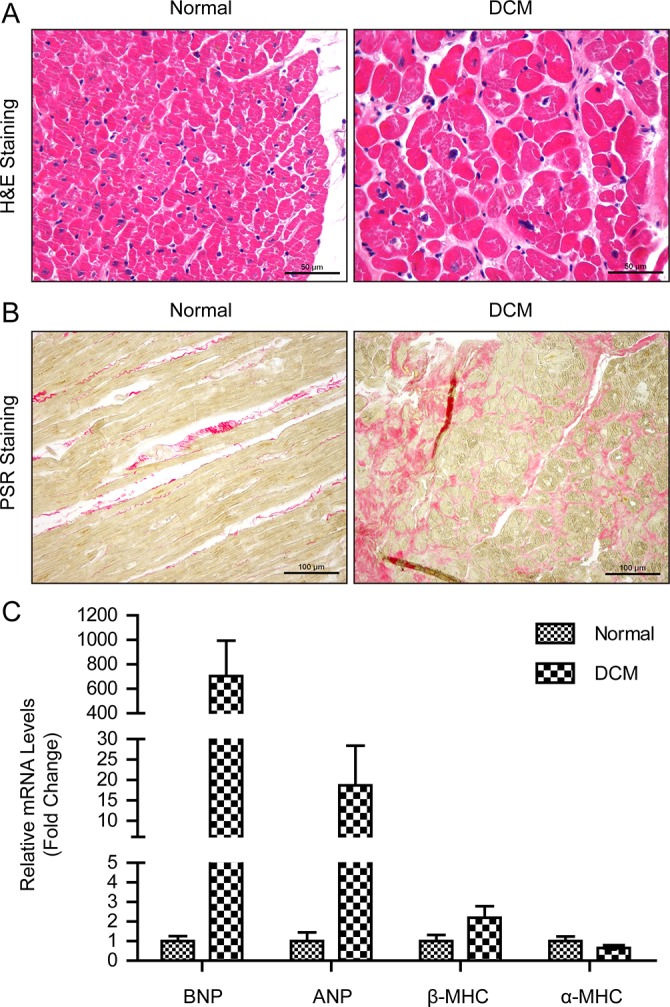
Pathologic and molecular properties of human DCM hearts. (A) H&E staining of left ventricles of donor hearts (n = 12) and DCM hearts (n = 15) (scale bar 50 μm). (B) Picrosirius red staining of left ventricles of human hearts (n = 12–15, scale bar 100 μm). (C) Real-time PCR detected the mRNA levels of heart failure biomarkers brain natriuretic peptide, atrial natriuretic peptide, β-myosin heavy chain and α-myosin heavy chain in human hearts (n = 4 per experimental group).
Table 1.
Clinical information of donors with normal hearts and patients with dilated cardiomyopathy (DCM).
| Characteristics | Normal | DCM |
|---|---|---|
| No. | 12 | 15 |
| Age, y | 28.58 ± 1.60 | 47.5 ± 2.54 |
| Sex, male (%) | 9 (75%) | 10 (66.67%) |
| BMI (kg/m2) | 24.05 ± 0.51 | 22.78 ± 1.06 |
| Medical history | ||
| Hypertension | 0 (0%) | 3 (20%) |
| Diabetes mellitus | 0 (0%) | 0 (0%) |
| Drinking history | 1 (8.3%) | 1 (6.67%) |
| Cigarette smoker | 2 (16.67%) | 2 (13.33%) |
| LVEDD (mm) | 42.17 ± 0.45 | 69.67 ± 3.53 |
| LVEF (%) | 61.67 ± 0.42 | 22.8 ± 1.41 |
| Blood pressure, mmHg | ||
| Systolic | N/A | 105.8 ± 4.4 |
| Diastolic | N/A | 69.9 ± 2.3 |
| Blood glucose, mmol/l | N/A | 6.05 ± 0.46 |
BMI: body mass index; LVEDD: left ventricular end diastolic dimension; LVEF: left ventricular ejection fraction
Figure 3.

MLL3 expression level was increased in human DCM hearts. (A,B) The mRNA levels of MLLs were detected by real-time PCR (n = 8 per experimental group). (C) Specific immunohistochemistry staining of MLL3 and α-actinin in the left ventricles of human hearts. Black arrows indicate MLL3-positive cardiomyocytes (n = 4 per experimental group, scale bar 50 μm).
Figure 4.

MLL3 expression level closely correlates with severity of DCM. Correlation analysis of MLL3 expression level with (A) left ventricular end-diastolic dimension or (B) left ventricular ejection fraction; r indicates correlation coefficient.
Increased MLL3 Expression Was Accompanied by Elevated H3K4me2, Smad3, GATA4 and EGR1 Protein Levels in Human DCM Hearts
To detect the potential downstream regulated by MLL3, we first evaluated the methylation of H3K4, which is the canonical substrate. The results demonstrate that H3K4me2 level was significantly increased in the DCM hearts compared with normal hearts, while comparable H3K4me3 levels were detected in these two groups Figures 5A and B. As we know, H3K4 methylation indicates gene activation (5). Next, we were curious about what genes were activated by MLL3 and H3K4 methylation. We first searched the ENCODE database (www.encodeproject.org/) using the key words “H3K4me1 heart” or “H3K4me2 heart” or “H3K4me3 heart” or “MLL3 heart” to find the ChIP-sequence data. The results demonstrate that there are multiple H3K4me1 and H3K4me3 binding sites in the genomes of cardiac remodeling regulators Smad3, GATA4 and EGR1 in human hearts (data not shown). In addition, both Valekunja et al. and Kim et al. demonstrated that MLL3 knockout or haploinsufficiency could decrease Smad3, GATA4 and EGR1 expression (14,25). As we have verified that MLL3 expression level was upregulated in the DCM hearts compared with donor hearts, we hypothesized that the expression levels of Smad3, GATA4 and EGR1 would increase in the hearts of DCM patients. As expected, the protein levels of Smad3, GATA4 and EGR1 were significantly elevated in the left ventricles of DCM hearts (Figures 5C F).
Figure 5.

Protein levels of H3K4me2/3, Smad3, GATA4 and EGR1 in human hearts. (A) Di- and trimethylation of H3K4 was detected by Western blot (n = 4–9 per experimental group). (B) The quantitative results of panel A. (C) Protein levels of Smad3, GATA4 and EGR1 were detected via Western blot (n = 4–10 per experimental group). (D–F) The quantitative results of panel C.
DISCUSSION
Several studies have demonstrated that some HMTs are profoundly involved in the development of heart and cardiac remodeling (2,3,15). However, the role of HMTs on human DCM remains largely unknown. In the present study, we identified that MLL3, a H3K4-specific HMT, was upregulated in human DCM hearts and remodeled mouse hearts, suggesting that MLL3 has a potential role in the pathological processes of DCM and heart failure. Intriguingly, Smad3, GATA4 and EGR1, which are regulated by MLL3, were elevated in the human DCM hearts. Therefore, our study suggests, for the first time, that inhibition of MLL3 might be a novel remedy for the treatment of human DCM.
There is accumulating evidence demonstrating that histone methylation and HMTs contribute to heart development and to congenital and adult heart diseases (26,27). Lysine methyltransferase G9a and G9a-like protein play a critical role in the normal morphogenesis of atrioventricular septum via H3-K9 dimethylation, which controls expression levels in multiple genes related to heart development (28). Except for this, G9a has a cardioprotective effect of ischemic preconditioning by regulating Mtor-dependent cardiac autophagy (29), while H3K4- and K36-specific methyltransferase Smyd2 act as endogenous cardioprotective proteins by suppressing p53-dependent cardiomyocyte apoptosis (30). In addition to HMTs, histone demethylase JMJD2A, upregulated in human hypertrophic cardiomyopathy, promotes pressure overload–induced cardiac hypertrophy (31). Unsurprisingly, MLL, a kind of H3K4 HMT, also has an important role in heart development and disease. MLL4 (also known as KMT2D) affects heart development by regulating genes related to ion transport, hypoxia-reoxygenation and cell cycle regulation (15). Cardiac lineage differentiation of mouse embryonic stem cells is controlled by MLL2 via promoting H3K4me3 deposition at cardiac-specific genes (32). In addition, UTX, a component of the COMPASS complex, demethylates H3K27 residue in cardiac genes to affect cardiac development (33). However, its role in human DCM is largely unexplored.
To investigate the contribution of MLL families to human DCM, we first detected their expression levels in normal donor hearts and DCM hearts. Our results show that all the MLL family members, except MLL3, have comparable mRNA levels in donor and DCM hearts. Similarly, MLL3 mRNA level also increased in TAC-treated mouse hearts. However, SETD1A mRNA level was unexpectedly decreased in the mouse hearts treated with TAC for 8 wks. The disparity in SETD1A expression pattern between human and mouse may be primarily because TAC-induced cardiac remodeling in the mouse model cannot completely simulate the pathologic processes of DCM. In addition, most DCM is associated with genetic factors but not pressure overload. The MLL3 (KMT2C) protein consists of 4,911 amino acids and includes eight plant homeodomain, FY-rich N-terminal domain and a catalytic domain (ie, SET domain) (34). It is capable of introducing mono-, di- and trimethylation to H3K4 (8). MLL3 deficiency results in stunted growth, reduced cell proliferation and lower fertility of mice (35). MLL3 insufficiency is often identified in myeloid leukemia patients, and MLL3 inactivation in mice leads to urothelial tumors (11,36). In addition, recent studies reported that reduced MLL3 expression or its mutation was found in several kinds of cancers, including breast tumors, gastric cancer, glioblastoma, melanoma and pancreatic cancer (37–40). However, no research has investigated the function of MLL3 on heart diseases currently. Therefore, the present study provides the first evidence that MLL3 is associated with human DCM.
Previous studies demonstrated that the expression levels of Smad3, GATA4 and EGR1 were downregulated in the livers of MLL3-deficient mice, which was detected by RNA-seq and microarray (14,25). Furthermore, we found that H3K4me1 and H3K4me3 had multiple binding sites at the genomes of these three genes via analysis of the ChIP- sequence data released in the ENCODE database. Importantly, we found that Smad3, GATA4 and EGR1 were remarkably elevated, along with increased MLL3 expression level in the DCM hearts compared with donor hearts. The hallmarks of pathologic changes of DCM include cardiomyocyte hypertrophy and cardiac fibrosis (20). Current evidence suggests that Smad3, GATA4 and EGR1 are the critical factors in regulating cardiomyocyte hypertrophy and cardiac fibrosis (41–46). An array of cardiac genes, including atrial natriuretic peptide, brain natriuretic peptide, α-myosin heavy chain and β-myosin heavy chain, are regulated by GATA4 in response to hypertrophic agonists (42). More importantly, GATA4 overexpression in mice was sufficient to induce cardiac hypertrophy and fibrosis (47). Otherwise, GATA4 is essential for formation of the proepicardium and early cardiac development (41). Compared with the control group, EGR1 was increased in the peripheral blood mononuclear cells of non-ischemic dilated cardiomyopathy (48). Isoproterenol and phenylephrine infusion–induced cardiac hypertrophy and fibrosis were alleviated by EGR1 deficiency in mice (45). Hsu et al. demonstrated that phenylephrine-induced neonatal rat ventricular myocyte and H9c2 cell hypertrophy was prevented by EGR1 knockdown via regulating Cav3.2 expression (43). Similarly, Smad3 knockout mice displayed attenuated cardiac remodeling in response to angiotensin II stimulation (46). In parallel, Smad3 loss also prevented myocardial infarction–induced interstitial fibrosis in the non-infarcted myocardium and attenuated cardiac remodeling (44). Therefore, increased Smad3, GATA4 and EGR1 expression might contribute to cardiac remodeling of human DCM hearts.
CONCLUSION
Our results demonstrate that MLL3 is the only significantly changed MLL family member in DCM hearts compared with normal donor hearts. More importantly, its expression level is positively correlated with severity of DCM. In addition, we identified Smad3, GATA4 and EGR1 as downstream of MLL3 affecting the pathologic processes of human DCM. Based on these data, we believe that inhibition of MLL3 activity or downregulation of MLL3 in the heart would be a good therapeutic strategy for the prevention or treatment of human DCM.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (81370201, 81600188, 81370264), the National Key Scientific Instrument Special Program of China (2013YQ030923-0607), the Natural Science Foundation of Hubei Province (2016CFB162) and the Fundamental Research Funds for the Central Universities (2042016kf0074).
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine or other interests that might be perceived to influence the results and discussion reported in this paper.
Cite this article as: Jiang D-S, et al. (2017) The histone methyltransferase mixed lineage leukemia (MLL) 3 may play a potential role in clinical dilated cardiomyopathy. Mol. Med. 23:196–203.
REFERENCES
- 1.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–47. doi: 10.1038/nrcardio.2013.105. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen AT, et al. DOT1L regulates dystrophin expression and is critical for cardiac function. Genes Dev. 2011;25:263–74. doi: 10.1101/gad.2018511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delgado-Olguin P, et al. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat Genet. 2012;44:343–47. doi: 10.1038/ng.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho YW, et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem. 2007;282:20395–406. doi: 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernstein BE, et al. Methylation of histone H3 Lys 4 in coding regions of active genes; Proc Natl Acad Sci USA; 2002. pp. 8695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee J, et al. Targeted inactivation of MLL3 histone H3-Lys-4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis; Proc Natl Acad Sci USA; 2008. pp. 19229–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ansari KI, Shrestha B, Hussain I, Kasiri S, Mandal SS. Histone methylases MLL1 and MLL3 coordinate with estrogen receptors in estrogen-mediated HOXB9 expression. Biochemistry. 2011;50:3517–27. doi: 10.1021/bi102037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weirich S, Kudithipudi S, Kycia I, Jeltsch A. Somatic cancer mutations in the MLL3-SET domain alter the catalytic properties of the enzyme. Clin Epigenetics. 2015;7:36. doi: 10.1186/s13148-015-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ansari KI, Mandal SS. Mixed lineage leukemia: roles in gene expression, hormone signaling and mRNA processing; FEBS J; 2010. pp. 1790–1804. [DOI] [PubMed] [Google Scholar]
- 10.Sze CC, Shilatifard A. MLL3/MLL4/COMPASS family on epigenetic regulation of enhancer function and cancer. Cold Spring Harb Perspect Med. 2016;6 doi: 10.1101/cshperspect.a026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee J, et al. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4; Proc Natl Acad Sci USA; 2009. pp. 8513–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brun ME, et al. Characterization and expression analysis during embryo development of the mouse ortholog of MLL3. Gene. 2006;371:25–33. doi: 10.1016/j.gene.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 13.Li WD, et al. Exome sequencing identifies an MLL3 gene germ line mutation in a pedigree of colorectal cancer and acute myeloid leukemia. Blood. 2013;121:1478–79. doi: 10.1182/blood-2012-12-470559. [DOI] [PubMed] [Google Scholar]
- 14.Valekunja UK, et al. Histone methyltransferase MLL3 contributes to genome-scale circadian transcription; Proc Natl Acad Sci USA; 2013. pp. 1554–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ang SY, et al. KMT2D regulates specific programs in heart development via histone H3 lysine 4 di-methylation. Development. 2016;143:810–21. doi: 10.1242/dev.132688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Son MJ, et al. Methyltransferase and demethylase profiling studies during brown adipocyte differentiation. BMB Reports. 2016;49:388–93. doi: 10.5483/BMBRep.2016.49.7.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ford DJ, Dingwall AK. The cancer COMPASS: navigating the functions of MLL complexes in cancer. Cancer Genet. 2015;208:178–91. doi: 10.1016/j.cancergen.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 18.Jiang DS, et al. IRF8 suppresses pathological cardiac remodelling by inhibiting calcineurin signalling. Nat Commun. 2014;5:3303. doi: 10.1038/ncomms4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang DS, et al. Signal regulatory protein-alpha protects against cardiac hypertrophy via the disruption of toll-like receptor 4 signaling. Hypertension. 2014;63:96–104. doi: 10.1161/HYPERTENSIONAHA.113.01506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang DS, et al. The potential role of lysosome-associated membrane protein 3 (LAMP3) on cardiac remodelling. Am J Transl Res. 2016;8:37–48. [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang DS, et al. Role of interferon regulatory factor 4 in the regulation of pathological cardiac hypertrophy. Hypertension. 2013;61:1193–1202. doi: 10.1161/HYPERTENSIONAHA.111.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang DS, et al. Interferon regulatory factor 1 is required for cardiac remodeling in response to pressure overload. Hypertension. 2014;64:77–86. doi: 10.1161/HYPERTENSIONAHA.114.03229. [DOI] [PubMed] [Google Scholar]
- 23.Jiang DS, et al. Interferon regulatory factor 7 functions as a novel negative regulator of pathological cardiac hypertrophy. Hypertension. 2014;63:713–22. doi: 10.1161/HYPERTENSIONAHA.113.02653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang DS, et al. Interferon regulatory factor 9 protects against cardiac hypertrophy by targeting myocardin. Hypertension. 2014;63:119–27. doi: 10.1161/HYPERTENSIONAHA.113.02083. [DOI] [PubMed] [Google Scholar]
- 25.Kim DH, et al. Crucial roles of mixed-lineage leukemia 3 and 4 as epigenetic switches of the hepatic circadian clock controlling bile acid homeostasis in mice. Hepatology. 2015;61:1012–23. doi: 10.1002/hep.27578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Q-J, Liu Z-P. Histone methylations in heart development, congenital and adult heart diseases. Epigenomics. 2015;7:321–30. doi: 10.2217/epi.14.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yi X, Jiang XJ, Li XY, Jiang DS. Histone methyltransferases: novel targets for tumor and developmental defects. Am J Transl Res. 2015;7:2159–75. [PMC free article] [PubMed] [Google Scholar]
- 28.Inagawa M, et al. Histone H3 lysine 9 methyltransferases, G9a and GLP are essential for cardiac morphogenesis. Mech Dev. 2013;130:519–31. doi: 10.1016/j.mod.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 29.Gidlof O, et al. Ischemic preconditioning confers epigenetic repression of Mtor and induction of autophagy through G9a-dependent H3K9 dimethylation. J Am Heart Assoc. 2016;5(12):e004076. doi: 10.1161/JAHA.116.004076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sajjad A, et al. Lysine methyltransferase Smyd2 suppresses p53-dependent cardiomyocyte apoptosis. Biochim Biophys Acta. 2014;1843:2556–62. doi: 10.1016/j.bbamcr.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang QJ, et al. The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice. J Clin Invest. 2011;121:2447–56. doi: 10.1172/JCI46277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wan X, et al. Mll2 controls cardiac lineage differentiation of mouse embryonic stem cells by promoting H3K4me3 deposition at cardiac-specific genes. Stem Cell Rev. 2014;10:643–52. doi: 10.1007/s12015-014-9527-y. [DOI] [PubMed] [Google Scholar]
- 33.Lee S, Lee JW, Lee SK. UTX, a histone H3-lysine 27 demethylase, acts as a critical switch to activate the cardiac developmental program. Dev Cell. 2012;22:25–37. doi: 10.1016/j.devcel.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Starczynowski DT, Arcipowski KM, Bulic M, Gurbuxani S, Licht JD. Loss of Mll3 catalytic function promotes aberrant myelopoiesis. PLoS One. 2016;11:e0162515. doi: 10.1371/journal.pone.0162515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S, et al. Coactivator as a target gene specificity determinant for histone H3 lysine 4 methyltransferases; Proc Natl Acad Sci USA; 2006. pp. 15392–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen C, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell. 2014;25:652–65. doi: 10.1016/j.ccr.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang XX, et al. Somatic mutations of the mixed-lineage leukemia 3 (MLL3) gene in primary breast cancers. Pathol Oncol Res. 2011;17:429–33. doi: 10.1007/s12253-010-9316-0. [DOI] [PubMed] [Google Scholar]
- 38.Rabello Ddo A, de Moura CA, de Andrade RV, Motoyama AB, Silva FP. Altered expression of MLL methyltransferase family genes in breast cancer. Int J Oncol. 2013;43:653–60. doi: 10.3892/ijo.2013.1981. [DOI] [PubMed] [Google Scholar]
- 39.Li B, et al. Association of MLL3 expression with prognosis in gastric cancer. Genet Mol Res. 2014;13:7513–18. doi: 10.4238/2014.September.12.18. [DOI] [PubMed] [Google Scholar]
- 40.Kandoth C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–39. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watt AJ, Battle MA, Li J, Duncan SA. GATA4 is essential for formation of the proepicardium and regulates cardiogenesis; Proc Natl Acad Sci USA; 2004. pp. 12573–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bisping E, et al. Gata4 is required for maintenance of postnatal cardiac function and protection from pressure overload-induced heart failure; Proc Natl Acad Sci USA; 2006. pp. 14471–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsu SC, Chang YT, Chen CC. Early growth response 1 is an early signal inducing Cav3.2 T-type calcium channels during cardiac hypertrophy. Cardiovasc Res. 2013;100:222–30. doi: 10.1093/cvr/cvt190. [DOI] [PubMed] [Google Scholar]
- 44.Bujak M, et al. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116:2127–38. doi: 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- 45.Saadane N, Alpert L, Chalifour LE. Altered molecular response to adrenoreceptor-induced cardiac hypertrophy in Egr-1-deficient mice. Am J Physiol Heart Circ Physiol. 2000;278:H796–805. doi: 10.1152/ajpheart.2000.278.3.H796. [DOI] [PubMed] [Google Scholar]
- 46.Huang XR, et al. Smad3 mediates cardiac inflammation and fibrosis in angiotensin II-induced hypertensive cardiac remodeling. Hypertension. 2010;55:1165–71. doi: 10.1161/HYPERTENSIONAHA.109.147611. [DOI] [PubMed] [Google Scholar]
- 47.Liang Q, et al. The transcription factors GATA4 and GATA6 regulate cardiomyocyte hypertrophy in vitro and in vivo. J Biol Chem. 2001;276:30245–53. doi: 10.1074/jbc.M102174200. [DOI] [PubMed] [Google Scholar]
- 48.Cappuzzello C, et al. Gene expression profiles in peripheral blood mononuclear cells of chronic heart failure patients. Physiol Genomics. 2009;38:233–40. doi: 10.1152/physiolgenomics.90364.2008. [DOI] [PubMed] [Google Scholar]