

Abstract
Almost all protein-coding genes are spliced and their majority is alternatively spliced. Alternative splicing is a key element in eukaryotic gene expression that increases the coding capacity of the human genome and an increasing number of examples illustrates that the selection of wrong splice sites causes human disease. A fine-tuned balance of factors regulates splice site selection. Here, we discuss well-studied examples that show how a disturbance of this balance can cause human disease. The rapidly emerging knowledge of splicing regulation now allows the development of treatment options.
1. General principles of alternative splicing
Alternative pre-mRNA splicing regulates the function of the majority of protein-coding genes
An average human protein-coding gene contains a mean of 8.8 exons with a mean size of 145 nt. The mean intron length is 3365 nt and the 5′ and 3′ UTR are 770 and 300 nt, respectively; as a result, this “standard” gene spans about 27 kbp. After pre-mRNA processing the average mRNA exported into the cytosol consists of 1340 nt coding sequence, 1070 nt untranslated regions and a poly (A) tail [1]. This shows that more than 90% of the pre-mRNA is removed as introns, and only about 10% of the average pre-mRNA are joined as exonic sequences by pre-mRNA splicing. Almost all protein-coding genes contain introns that are removed in the nucleus by RNA splicing during pre-mRNA processing. Exon usage is often alternative, i.e. the cell decides whether to remove a part of the pre-mRNA as an intron or include this part in the mature mRNA as an alternative exon [2]; (Figure 1). Alternative pre-mRNA processing is a key regulator of gene expression as it generates numerous transcripts from a single protein-coding gene, which largely increases the use of genetic information. The process is more widely used than previously thought and was recently estimated to affect more than 88% of human protein-coding genes [3]. An estimated 75% of alternative exons encode protein parts [4] and their alternate use allows to generate multiple proteins from a single gene, which increases the coding potential of the genome. Mapping of alternatively spliced regions on known protein structures suggest that most alternative exons are in coiled or loop regions that are located on the surface [5]. Alternative splicing generates protein isoforms with different biological properties that differ in protein:protein interaction, subcellular localization, or catalytic ability [6]. More than a quarter of alternative exons introduce premature stop codons in their mRNAs. This can result either in the formation of truncated proteins or in the degradation of the mRNA in nonsense-mediated decay. Recent array analyses indicate that although frequently found, alternative exons with premature stop codons are present only in low abundance, which question their role as a general shut-off mechanism for protein production [7, 8].
Figure 1. Alternative splicing modes.
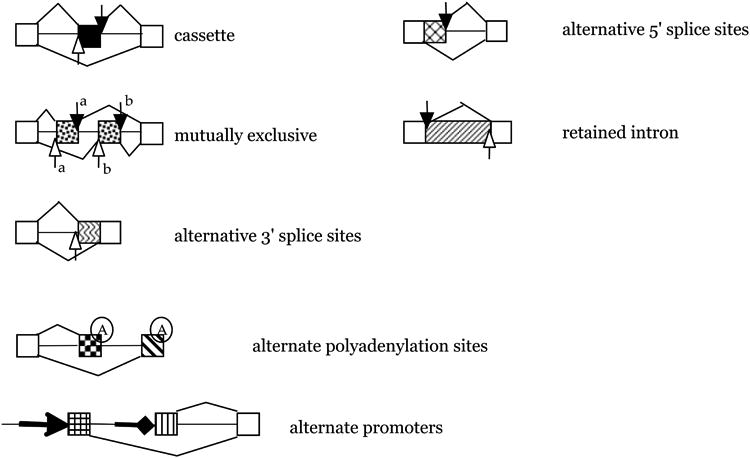
Constitutive exons are shown as white boxes, introns as vertical lines. Open arrows indicate the 3′ splice site, closed arrows the 5′ splice site. A black box indicates a cassette exon, the most frequent form of alternative splicing. Hatched boxes indicate other splicing modes. Alternative polyadenylation sites and alternate promoters are shown as two other mechanisms to increase mRNA diversity. They are mechanistically different from alternative splicing.
Changes in alternative splicing can be the cause or consequence of human diseases
There are only a few reports of mutations in core elements of the splicing machinery that result in human diseases. For example, autosomal dominant forms of retinitis pigmentosa is caused by mutation in the splicing factors PRPF31/U4-61k [9, 10] and PRP8 [11]. It is possible that defects in the general splicing machinery are generally not compatible with life, whereas changes in alternative splicing can be tolerated by an organism, although these changes might manifest in a disease. As alternative splicing affects numerous genes, it is not surprising that changes in alternative splicing are frequently associated with human diseases. It is often not clear whether a change in alternative splicing causes a disease or is an indicator for an underlying defect. A better mechanistical understanding of splice site selection has helped in distinguishing these effects. The first demonstration that exon sequences can have an effect on splice site selection was published 20 years ago [12]. Ten years later, the first review about the impact of exonic mutations on splice site selection postulated that silent mutations can interfere with exon usage and explained how these mutations that do not change the predicted encoded protein can cause a human disease [13]. Since then, a better understanding of alternative splice site selection contributed to a better understanding of human diseases and vice versa. The number of diseases reported to be associated with changes in alterative splicing increased dramatically and has been frequently reviewed [14-17], including in form of a book [18, 19]. To facilitate the access to this fast growing area for colleagues in other fields, we briefly summarize disease-relevant aspects of splice site selection, discuss well-established examples of alternative splicing changes that lead to human disease and point out links between the diseases and aberrant splice site selection.
Alternative exons are regulated by combinatorial control through transient formation of recognition complexes
Since splice sites follow only loose consensus sequences, the key questions in alternative splicing regulation are: How are splice sites recognized in the vast genomic sequence background, and how are they differentially regulated (11)? The mechanism of alternative splice site recognition has been extensively reviewed [16, 20-23]. Exon recognition is regulated by the interaction of proteins and ribonuclear proteins (trans-factors), with sequence elements on the pre-mRNA (cis-factors), which is summarized below.
Five small nuclear RNAs form the core of the spliceosome
During processing, the pre-mRNA has extensive, specific interaction via base pairing with five small nuclear RNAs (U1, U2, U4, U5, and U6). Early in spliceosome assembly, U1 forms a base-pairing interaction with the 5′-splice site and U2 similarly base-pairs with the branch-point. Then a tri-snRNP complex containing U4, U5 and U6 associates with the forming spliceosome and U4 is removed from the complex. This allows U6 to replace U1 at the 5′ splice site and leads to a U6-U2 interaction that brings the 5′-splice site and the branch point close together, allowing for a transesterification step. By forming non-canonical interactions, U5 brings the two exons into close proximity and allows for the second step of splicing, when the two exons are joint. This shows that small RNAs play a crucial role in splicing and there is increasing evidence that some of the RNAs also play a role in catalysis of the splicing reaction [24, 25].
Protein assembling on the pre-mRNA allow exon recognition by interaction with the core spliceosome
Since the information in splice sites is not sufficient for regulation, ribonuclear-protein complexes (RNPs) forming on the pre-mRNA help in the recognition of exons (Figure 2). The majority of splicing regulatory proteins in these complexes belong to two major classes: hnRNPs and SR-proteins. HnRNPs are operationally defined as proteins binding to RNA and SR-proteins are characterized by serine and arginine-rich protein domain. These proteins contain RNA-binding and protein interaction domains [26]. They bind with low specificity to accessible, mostly single-stranded parts of the pre-mRNA. To overcome the low RNA-binding specificity, splicing regulatory proteins use their protein-interaction domains to bind to each other. Another frequent strategy to overcome the low affinity between splicing factors and their recognition sequence is a repetitive arrangement of regulatory sequences. For example, exon 4 of the doublesex pre-mRNA contains six repeats, each 13 nt long, that bind to the SR-proteins tra, tra2 and 9G8 [27]. Once these protein complexes have been formed around an exon, they aid ribonuclear protein components of the core spliceosome in establishing an RNA:RNA interaction at the 5′ splice site and at the branchpoint. A well-studied interaction occurs between SR-proteins and the U1 snRNP, which is part of the spliceosome. The RNA component of the U1 snRNP interacts with the 5′ splice site, which defines one border of an exon. Since SR-proteins that bind to an exon stabilize the interaction with U1 snRNP and the 5′ splice site, they generally promote inclusion of exons to which they bind. The high fidelity of exon recognition is thus achieved by the combination of multiple weak protein:protein, protein:RNA and RNA:RNA interactions, which are schematically shown in Figure 1. Different pre-mRNAs seem to associate with a unique arrangement of proteins, which constitutes the ‘splicing- or mRNP code’[28]. By interacting with the spliceosome, the protein complexes forming on the pre-mRNA, i.e. the splicing code, determine which RNA parts will be removed as introns and which parts will be included in the mature mRNA. The low affinity of each interaction is an intrinsic property of pre-mRNA processing and allows the transient formation of a specific protein:RNA complex from several intrinsically weak interactions. This has several advantages: (i) it allows a high sequence flexibility of exonic regulatory sequences that puts no constrains on coding requirements, (ii) the protein interaction can be influenced by small changes in the concentration of regulatory proteins which allows the alternative usage of exons depending on a tissue and/or developmental- specific concentration of regulatory factors, (iii) phosphorylation of regulatory factors that alter protein:protein-interactions can influence splice site selection, (iv) the regulatory proteins can be exchanged with other proteins after the splicing reaction, allowing a dynamic processing of the RNA. Alternative splice site selection is connected to other processing steps, such as transcription, 5′ end capping, 3′end polyadenylation and nuclear export. Splicing regulatory proteins often function in multiple processing events while they remain bound to ‘their’ pre-mRNA [29].
Figure 2. Elements involved in alternative splicing of pre-mRNA.

Exons are indicated as boxes, introns as thin lines. Splicing regulator elements (enhancers or silencers) are shown as green or red boxes in exons. The 5′ splice-site (CAGguaagu) and 3′ splice-site (y)10ncagG, as well as the branch point (ynyyray), are indicated (y=c or u, n=a, g, c or u). Upper-case letters refer to nucleotides that remain in the mature mRNA. Two major groups of proteins, hnRNPs (green) and SR or SR related proteins (red), bind to splicing regulator elements; the protein:RNA interaction is indicated by shading. The protein complex assembling around the exon stabilizes binding of spliceosomal copmponents, indicated in blue. For example, SR-proteins can interact with U1 snRNA, which helps in the recognition of the 5′ splice site, as it allows hybridization (thick black line) of the U1 snRNA (black line) with the 5′ splice-site. The formation of the multi-protein:RNA complex allows discrimination between proper splice-sites (bold letters) and cryptic splice-sites (small gt ag) that are frequent in pre-mRNA sequences. Factors at the 3′ splice-site include U2AF, which recognizes pyrimidine rich regions of the 3′ splice-sites, and help stabilize the interaction of U2 snRNP with the adenosine of the branch point.
The secondary structure of pre-mRNA influences splice site selection
Secondary structures of the pre-mRNA can influence splice site selection (reviewed in [30]). For example, the stability of a predicted stem-loop structure at the 5′ splice site of tau exon 10 regulates usage of this exon (15) and the alternative exon of the fibronectin gene is influenced by secondary RNA structure [31]. Bioinformatic analysis indicate that the splicing regulatory sequences are preferentially in a single-stranded conformation, which allows these sequences to interact with RNA binding proteins that mostly have a preference for single strand RNA [32].
Small nucleolar RNAs regulate splice site selection
Tiling array data and detailed expression analysis of parts of the human genome in the ENCODE project showed that the expression data and gene structures collected in current databases are largely incomplete [33]. Numerous noncoding regions are transcribed into polyadenylated, stable RNAs, which are named TUF (transcript of unkown function). Within most previously well-characterized protein coding genes, there are large numbers of transcribed fragments (transfrags) that could represent new exons or short RNAs of unknown sequence. The most recent study of human gene expression using tiling arrays demonstrates that about 57% of the transfrags are not annotated in Genbank or EnsEMBL databases [34]. Recently, it was discovered that a snoRNA, HBII-52, regulates alternative splicing by binding to an alternative exon of the serotonin receptor. This snoRNA is not expressed in people with the Prader-Willi syndrome and it is likely that the lack of HBII-52 snoRNA expression contributes to the disease [35]. In support of this theory, a child with a 174,584 bp long microdeletion was reported. The deletion encompasses only snoRNAs: HBII-438A, all snoRNAs of the HBII-85 cluster and 23 of the 47 HBII-52 snoRNAs. Since this child shows most clinical features of Prader-Willi syndrome, it is now very clear that the loss of snoRNAs cause the disease [36], which is in agreement with genetic studies and reflected by two recent mouse models [37-39]. A possible link between snoRNPs and splice site selection was mechanistically hard to understand, as snoRNPs reside in the nucleolus and splicing takes place in the nucleoplasma. Recently, it was shown that nuclear export and import factors are directly involved in U8 C/D box snoRNA biogenesis, suggesting that C/D box snoRNPs are transported through the nucleoplasma while being assembled [40]. Furthermore, the 15.5K protein that was originally identified as part of a the C/D box snoRNP complex where it binds to a conserved kink turn, binds also to a similar structure in the U4 snRNA where it interacts with the splicing factor hPrp31 [41]. These findings raise the possibility that 15.5K protein bound to snoRNPs interferes with the U4/U6 rearrangement during splicing by interacting with hPrp31. It is therefore possible that other small RNAs are also involved in splice site selection.
Exon-recognition complexes are targeted by cellular signal transduction pathways
Cells frequently regulate alternative splicing events, often in response to external stiumuli (reviewed in [42-44]). In most cases studied, rapid changes observed in splice site selection are influenced by phosphorylation events that are regulated by known signal transduction pathways. Reversible phosphorylation is a key regulatory step in the formation of protein complexes on pre-mRNA (reviewed in [45]). SR-proteins are phosphorylated by several kinases, including the Clk/Sty kinase family (cdc2 like kinases, Clk1-4), the SRPK family (SRPK1,2), mammalian PRP4, topoisomerase I, CDC2 kinase 2 [46], and GSK3 [47]. A change in phosphorylation of the RS-domain changes its ability to interact with other proteins. For example, phosphorylation of SRp38 decreases its binding to U1 70K snRNP, but increases its binding to tra2-alpha [48]. Phosphorylation of the SR-protein SF2/ASF increased its binding to U1 70K snRNP [49] and decreased its binding to the RNA export factor TAP/NXF1 [50]. Finally, the ability of the RS-domain to bind to RNA depends on its phosphorylation [49, 51]
The phosphorylation is reversed by phosphatases; only about 25 serine/threonine protein phosphatases are known [52]. This is in contrast to the estimated 474-518 protein kinases representing 1.7% of human proteins [53]. The low number of protein phosphatases is the result of a combinatorial control. The protein phosphatase represents only the catalytically active subunit that forms numerous regulatory complexes with other proteins. Splicing regulatory proteins are dephosphorylated by protein phosphatase 1 (PP1), PP2A and protein phosphatase 2Cgamma [54, 55]. Complete blocking of phosphatases inhibits splicing as dephosphorylation is necessary for the transesterification step [54, 56]. However, modulation of phosphatase activity in vitro [57] and in vivo [58, 59] influences alternative exon usage, demonstrating that adjusting phosphatase activity can be used by cells to control alternative splicing.
Alternative exons are generated during evolution and their usage can be changed by point mutations located outside the splice sites
Alternative exons can be generated by three mechanisms (reviewed by [60]): (i) exon shuffling, where an existing exon is duplicated within the same gene and is then alternatively spliced, (ii) exonization of mobile genetic elements, such as Alu elements [61] and (iii) a transformation of formally constitutive exons into alternative ones [62]. Since the approximately one million human Alu elements are primate-specific elements that account for 10% of the human genome [63], their exonisation provides a large reservoir to generate new alternative exons. Numerous studies showed that synonymous mutations in coding regions can influence splice site selection. There is now also emerging evidence that intronic mutations and single-nucleotide polymorphism can alter exon usage [64]. It is thus likely that alternative exon usage is an evolutionary ‘substrate’ that is subject to a large number of mutations. Due to the complexity of the splicing regulation, the effects of mutations are difficult to predict, but become obvious when they lead to human diseases.
Each of the regulatory principles listed here can be altered to cause a human disease, which is schematically summarized in Figure 3.
Figure 3. Disease causing mutations.

Exons and introns are indicated as in Figure 2. Arrows indicate repetitive elements. A: Mutation in silencer or enhancer sequences change the interaction between regulatory proteins and pre-mRNA, leading to changes in exon recognition. B: Mutations in splice sites change the interaction with core spliceosomal proteins. C: Due to formation of secondary structures, intronic sequences can be brought close to splice sites, accounting for the effect of some intronic mutations. D: Secondary pre-mRNA structures contribute to exon recognition and their mutations can cause aberrant splicing. E. Expanded repeat elements can sequester splicing regulatory proteins, changing splice site selection in other messages. F. Some snoRNAs regulate splice site selection and their loss can cause disease.
2. Examples of diseases caused by alternative splicing
The sequences of the exons mutates in the diseases are listed in Table 1, further access to internet information is given in Table 4.
Table 1.
Summary of disease-causing mutations, Exons associated with diseases mentioned in the text are listed. Since there are no unique accession numbers for exons, the NCBI accession number is listed. The mutations having an effect on splicing are listed under features and changed nucleotides are underlined. These nucleotides are underlined in the sequence. Capital letters are exons, small letters are introns.
| disease | sequence | Accession number |
Features | Ref. |
|---|---|---|---|---|
| FD | tttaagATGCCAAGGGGAAACTTAGAAGTTGTTCATCATCGAGCCCTGGTTTTAGCTCAGATTCGGAAGTGGTTGG ACAAgtaagtgccat | AF044195 | Change from agtgc to agcgc causes skipping | [168] |
| FTDP-17 | GTGCAGATAATTAATAAGAAGCTGGATCTTAGCAACGTCCAGTCCAAGTGTGGCTCAAAGGATAATATCAAACACG TCCCGGGAGGCGGCAGT | J03778 | AATAAGA to AAGAAGA causes inclusion | [169] |
| FTDP-17 | GTGCAGATAATTAATAAGAAGCTGGATCTTAGCAACGTCCAGTCCAAGTGTGGCTCAAAGGATAATATCAAACACG TCCCGGGAGGCGGCAGT | J03778 | AATAAGAAGCT to AATAAGCT causes skipping | [170] |
| FTDP-17 | GTGCAGATAATTAATAAGAAGCTGGATCTTAGCAACGT CCAGTCCAAGTGTGGCTCAAAGGATAATATCAAACACG TCCCGGGAGGCGGCAGT | J03778 | ATCTTAGCA to ATCTCAGCA causes inclusion | [171] |
| FTDP-17 | GTGCAGATAATTAATAAGAAGCTGGATCTTAGCAACGTCCAGTCCAAGTGTGGCTCAAAGGATAATATCAAACACG TCCCGGGAGGCGGCAGT | J03778 | GCAGT to GCAAT causes inclusion | [172] |
| HGPS | GGCTCCCACTGCAGCAGCTCGGGGGACCCCGCTGAGTACAACCTGCGCTCGCGCACCGTGCTGTGCGGGACCTGCG GGCAGCCTGCCGACAAGGCATCTGCCAGCGGCTCAGGA GCCCAGGTGGGCGGACCCATCTCCTCTGGCTCTTCTGC CTCCAGTGTCACGGTCACTCGCAGCTACCG | BC014507 | GGGCGGA to GGGTGGA or GTCACTCGCA to GTCATTCGCA Activates cryptic splice site | [93] |
| hyperchole sterolemia | ATCTCCTCAGTGGCCGCCTCTACTGGGTTGACTCCAAACTTCACTCCATCTCAAGCATCGATGTCAACGGGGGCAA CCGGAAGACCATCTTGGAGGATGAAAAGAGGCTGGCCC ACCCCTTCTCCTTGGCCGTCTTTGAG | AY114155 | CAACGGG To CAATGGG Causes skipping | [94, 95] |
| MCAD deficiency | GAGGTCTTGGACTTGGAACTTTTGATGCTTGTTTAATT AGTGAAGAATTGGCTTATGGATGTACAGGGGTTCAGACTGCTATTGAAGGAAATTCTTTGGGG | M16827 | GACTGC To GATTGC causes exon skipping, ACAGGG to ACCGGG promotes inclusion | [103] |
| SMA | GGTTTTAGACAAAATCAAAAAGAAGGAAGGTGCTCACATTCCTTAAATTAAGGA | U18423 | TTTAGA to TTCAGA promotes exon skipping | [173] |
Table 4. Information about diseases on the web.
| General information about alternative splicing | http://www.eurasnet.info/ |
| Familial dysautonomia | http://www.familialdysautonomia.org/ |
| Frontotemporal lobar dementias/amyotrophic lateral sclerosis | http://www.alsa.org/ |
| Hutchinson-Gilford progeria syndrome | http://www.progeriaresearch.org/ |
| Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency | http://www.fodsupport.org/mcad_fam.htm |
| Myotonic dystrophy | http://www.myotonic.com http://www.mda.org |
| Prader-Willi Syndrome | http://www.pwsausa.org/http://www.fpwr.org/ |
| Spinal muscular atrophy | http://www.fsma.org/http://www.mda.org |
| Tauopathies | http://www.molgen.ua.ac.be/ADMutations/ |
| Substances that influence alternative splicing | http://www.stamms-lab.net/cpds.htm |
2.1 Diseases caused by point mutations in regulatory sequences
Spinal muscular atrophy as an example of a recessive disease caused by a point mutation in an exonic regulatory element
Spinal muscular atrophy (SMA) describes several different diseases that are characterized by degeneration of alpha motoneurons in the brainstem and spinal cord. Autosomal recessive SMA associated with chromosome 5 is molecularly the best understood. It is characterized by progressive paralysis caused by the loss of alpha-motor neurons in the spinal cord. The incidence is 1:6,000 to 1:10,000 for live births and the carrier frequency is 1 in 40 [65]. SMA is the second most common autosomal recessive disorder. Since children suffering from cystic fibrosis now largely survive childhood, it is the most frequent genetic cause of infantile death. SMA is caused by the loss of the SMN1 gene that encodes the SMN protein, which regulates snRNP assembly. It is not clear how the loss of SMN protein causes the disease and leads to a specific death of motoneurons. Mouse studies revealed that the loss of SMN protein causes cell-type specific changes in snRNAs and a generally reduced snRNP assembly capacity. Numerous pre-mRNA splicing events are deregulated in all tissues analyzed. Some of the changes observed reflect a shift in known alternative splicing patterns. However, the majority of the deregulated splicing events are aberrant mRNAs, which are normally not produced. These findings suggest that (i) the selective death of motoneurons could be caused by the cumulative effect of aberrantly splicing mRNAs and (ii) that changes in cells surrounding the motoneurons cause their death [66]. Genetic studies identified six families with eight female members that were asymptomatic for SMA, although they inherited the same SMN1 and SMN2 alleles as their affected siblings [67]. Plastin 3, an actin binding protein was identified as a modifier. Overexpression of plastin 3 in SMN knock-out mice partially rescued the short neuronal axon length causes by the absence of SMA protein. These findings argue that that the death of motoneurons could be caused by a mechanism different from a change in splicing. Although it is not understood how the loss of SMA protein causes the disease, it is clear that restoration of SMA protein production would be a therapeutic approach.
Humans posses a gene, SMN2, that is almost identical to SMN1. SMN2 was generated through a recent duplication. Although both genes are almost identical in sequence, due to a translationally silent C->T change at position 6 in exon 7, they have different splicing patterns and exon 7 is predominantly excluded in SMN2 (reviewed in [68]). This exon-skipping event generates a truncated, less stable and probably non-functional protein. Therefore, SMN2 cannot compensate the loss of SMN1. At least one copy of SMN2 is retained in humans with SMA, as lack of both SMN2 and SMN1 is embryonically lethal. Mice have only one SMN gene where exon 7 is constitutively spliced. A homozygous knock out of this gene is lethal. To study the splicing regulation of the human gene in mice, transgenic animals that contain the human gene were developed [69]. Although, in SMA patients the SMN protein is almost completely absent from all cells, for unknown reasons, alpha motoneurons are most severely affected and die, which causes the muscular atrophy. The disease can manifest in four phenotypes (type I to IV) that differ in onset and severity. The phenotypes correlate roughly with the number of SMN2 copies in the genome, most likely because more SMN2 copies produce more SMN protein [70]. Since stimulation of SMN2 exon7 usage would increase SMN protein levels and potentially cure the disease, work has concentrated on understanding the regulation of exon 7. Typical for the combinatorial control of exon regulation, multiple factors determine the regulation, including a suboptimal polypyrimidine tract [71], a central tra2-beta1-dependent enhancer [72] and the sequence around the C->T change at position 6. Recent large scale mutagenesis studies indicate that a composite regulatory exonic element termed EXINCT (extended inhibitory context) is responsible for the regulation of exon 7 inclusion [73, 74]. The exon skipping event is caused by the C->T change at position 6 and currently two models are proposed for its mechanism. In one model, the base exchange destroys the exonic enhancer that normally binds to SF2/ASF [75, 76] and in the other model, the mutation creates an hnRNPA1 binding site that acts as a silencer [77, 78]. Both models can explain the predominant skipping of exon 7. Inclusion of exon 7 depends on a central tra2-beta1 enhancer sequence [72]. Tra2-beta1 is an SR-related protein. Its activity is regulated by dephosphorylation mediated by protein phosphatase 1 and, not surprisingly, exon 7 usage depends on cellular PP1 activity [79]. SMN illustrates several common features of diseases caused by missplicing. Evolutionary changes in the genome, here the recent dublication of genes that facilitate their recombination, can manifest in splicing changes. Alternative exons are regulated by numerous factors and sequence elements and a single mutation can disturb the balance necessary for normal exon recognition. Finally, splicing factors are regulated by reversible phosphorylation controlled by cellular signaling pathways.
Tauopathies as an example for a disease caused by a change in the ratio of protein isoforms generated by alternative splicing
Tauopathies describe several diseases of the central nervous system that show abnormal intracellular accumulations of abnormal filaments that contain the microtubule associated protein tau. The tau protein is encoded by a single gene (MAPT, (microtubule associated protein tau) located on chromosome 17. The gene undergoes extensive alternative splicing and eight of the sixteen exons are alternatively spliced. In humans, these splicing events are spatially and temporally regulated. For example, exons 2, 3 and 10 are adult specific and show differences in splicing in various brain regions. The tau protein binds to microtubules via microbuble repeat regions. One of these microtubule binding regions is encoded by the alternatively used exon 10. Exon 10 inclusion creates a protein with four microtubule repeats (4R), whereas exon 10 skipping creates an isoform with three repeats (3R). This splicing event is species-specific in the adult. In humans, exon 10 is alternatively spliced in the adult, whereas in mice the exon is constitutively used. In both species, the exon usage is regulated during development.
Genetic studies identified rare dominant mutations in the tau gene that caused frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), which are collected on the world wide web (http://www.molgen.ua.ac.be/ADMutations/) and currently list 42 mutations. The majority of the mutations affect the splicing regulation of exon 10 that encodes part of one microtubule binding site. The mutations in tau exon 10 helped dissect its regulatory elements. The exon shows an alternating arrangement of four enhancer and three silencer regions. A mutation that falls into a silencer or enhancer regions either promotes or decreases exon usage, respectively (reviewed in [80]). Mutations in exon 10 alter its normal fraction of inclusion and changes of pre-mRNA encoding 3R and 4R repeat tau isoforms were found associated with FTDP-17 (recently reviewed by [81], [82]). These data clearly suggested that the splicing mutations cause the neuropathology by changing the ratio between the 3R and 4R isoforms. One mechanistically well understood mutation is N279K [83]. This mutation is caused by changing a TAAGAAG into GAAGAAG. The GAAGAAG sequence forms the core of a tra2-beta1 binding site. Similar to the situation in exon 7 of SMN2, this mutated version contains two partially overlapping versions of the GAAG binding site. Biochemical studies showed that the mutation increases affinity to tra2-beta1 in vitro [84] and cotransfections experiments showed that tra2-beta1 promoted exon 10 inclusion in reporter gene constructs [85]. In vitro studies showed that the asparagine to lysine exchange in the mutation does not alter the binding between tau and tubulin, the tau aggregation or microtubule assembly [86]. These data suggested that mainly the change in the ratio of expressed isoforms is responsible for the disease. Testing this hypothesis in mouse models was difficult, as the mouse tau gene constitutively expresses the 4R isoform in the adult. Therefore, a minigene of human tau, containing the promoter and all exons flanked by shortend intronic regions was expressed in mice. These constructs show alternative splicing resembling the human situation and express human tau protein containing either 3 or 4 microtubule binding domains. When the mutation that promotes exon 10 inclusion (N279K) was introduced into exon 10 of this construct, pre-mRNA splicing was shifted as expected towards exon 10 inclusion. Interestingly, the mice showed similar pathophysiology as humans with the same mutation and also showed behavioral changes [87]. These data suggest that a change in the ratio between 3R and 4R tau isoforms is an important underlying cause for FTDP-17.
Abnormal intracellular tau aggregations are also found in other tauopathies, such as Alzheimer's disease [88]. Several studies were performed to investigate whether the 4R to 3R ratio of tau pre-mRNA is changed in Alzheimer's disease. The studies gave conflicting results, which most likely reflects that only post-mortem human tissue can be analysed. Two studies showed no statistical regional differences in the 4R/3R ratio [89, 90], one study showed regional differences but no clear link to Alzheimer pathology [91] and a forth study showed a statistical significant increase of the 4R to3R ratio in temporal cortex. The later study also investigated possible changes in other splicing factors regulating exon 10 and found changes in clk2 and tra2-beta1 splicing, suggesting that multiple changes in alternative splice site selection are either causing or contributing to Alzeheimer's disease [92]. This example shows that a change in the ratio of protein isoforms generated by alternative splicing can cause human diseases. It further illustrates how analysis of genetic mutants helps to understand splicing regulation, which is often complicated due to species-specific splicing differences. Despite these problems, animal models can be generated that reflect the human pathology.
Hutchinson-Gilford progeria syndrome as an example for a disease caused by an intronic mutation that activates a cryptic splice site
Hutchinson-Gilford progeria syndrome (HGPS) is a rare genetic disorder phenotypically characterized by many features of premature aging. It is clinically characterized by postnatal growth retardation, midface hypoplasia, micrognathia, premature atherosclerosis, absence of subcutaneous fat, alopecia and generalized osteodysplasia. At birth, the appearance of patients is generally normal, but by one year of age patients show severe growth retardation, balding and sclerodermatous skin changes. Patients live a median of 13.4 years and die of heart attacks or congestive heart failure. Mutations causing HGPS have been identified in the nuclear lamin A/C (LMNA) gene. Lamin proteins are distributed throughout the nucleoplasm and are involved in numerous functions, including DNA replication, transcription, chromatin organization, nuclear positioning and shape, as well as the assembly/disassembly of the nucleus during cell division. Out of 14 mutations affecting lamin A/C, three have been reported to specifically alter lamin A splicing. The changes in splicing lead to the production of truncated protein products (p.G608G, p.T623S and IVS11+1G>A). Most of the typical Hutchinson-Gilford progeria cases are due to a recurrent, de novo point mutation in LMNA exon 11: c.1824C>T [93]. This mutation occurs in a probable exon splicing enhancer. As a result, a cryptic splice site is activated in transcripts generated from the mutated allele, which is located 5 nucleotides upstream of the mutation. The use of the cryptic splice site leads to the production of a truncated Lamin A protein lacking the last 150 base pairs of exon 11. The truncated protein is called “progerin” and acts in a dominant fashion to generate the HGPS phenotype. This example shows how a mutation can cause activation of a nearby otherwise ‘hidden’, cryptic splice site.
LDL receptor splicing variants caused by a single nucleotide polymorphism are a sex-specific factor for hypercholesterolemia
Hypercholesterolemia is a major risk factor for arteriosclerosis. Low-density lipoproteins are removed from the bloodstream by the LDL receptor (LDLR). Mutations in the LDLR are a primary cause for hypercholesterolemia. Recently, a single nucleotide polymorphism was identified in exon 12 of the LDLR that promotes skipping of this exon. The SNP was found to promote exon 12 skipping in the liver of pre-menopausal women. However, the SNP had no effect on men and post-menopausal women. The SNP and the splicing pattern are associated with a higher level of cholesterol in pre-menopausal women, but not in men. Exon 12 skipping generates a truncated form of the receptor that lacks the transmembrane domain necessary for membrane binding and internalization. It is possible that the protein generated by exon 12 skipping prevents, in a dominant negative form, the uptake of LDL. This model explains the interesting finding that exon 12 skipping caused by this SNP is associated with cholesterol levels. The reason for the sex-dependency of the SNP is unclear, but is possible that high estrogen levels influence transcription level of the gene or its alternative splicing [94]. Apo lipoprotein E (ApoE) is a ligand for the LDLR receptor and the apoE allel status is a major risk factor for Alzheimers disease. It was therefore investigated whether the SNP in exon 12 of the LDLR associates with Alzheimer's disease. It was found that the SNP associates with an increased chance to develop Alzheimer's disease in males, but not females [95]. This example nicely illustrates that a SNP can influence alternative splicing, which in turn predisposes to a disease. Reflecting the combinatorial control of alternative exon regulation, the result of a mutation depends on other factors, in this case the sex and age.
Familial dysautonomia as an example for a disease caused by a mutation in the 5′ splice site
About 10% of roughly 80,000 mutations reported in the human gene mutation database affect splice sites [96]. Well-studied diseases caused by changes in splice site selection include thalassemias [97] and Familial dysautonomia (FD). FD, (also Riley-Day syndrome, hereditary sensory and autonomic neuropathy type III) is a recessive disease that is caused by loss of function of the i-kappa-B kinase complex associated protein (IKBKAP). In the Ashkenazi jewish population, the incidence is 1/3600 in live birth (carrier frequency 1:30) [98]. Affected children show abnormal development of the nervous system that is associated with demyelination in various regions. This leads to a large clinical spectrum that includes vomiting crises, unsteady gait, and decreased perception of pain. In more than 99.5% of FD patients the 5′ splice site of exon 20 is mutated T->C in position 6 of intron 20. This point mutation interrupts base pairing with U1snRNA. U1 snRNA interacts both with the last three nucleotides of the exon and the first six nucleotides of the downstream intron. The majority of 5′ splice sites show complementarity to seven base pairs of U1 snRNA. This means there are usually three mismatches between the 5′ splice site and U1 snRNA. Bioinformatic analyses indicate that these mismatches are not randomly distributed. They either weaken the exonic portion of the 5′ splice site, which is then compensated by strong binding to the intronic portion, or a weak intronic portion is compensated by a strong exonic portion. In exon 20 of the IKBKAP gene, the exonic part of the splice site is weak, due to an A at position −1. The T->C mutation weakens the intronic part of the 5′ splice site, which causes exon skipping [99]. Exon 20 usage is susceptible to a weak 5′ splice site, as the exon has a weak 3′ splice site that has an A at the –3 position, and contains several exonic silencer elements [100]. Array analysis indicates that IKBKAP promotes expression of genes involved in oligodendrocyte and myelin formation, which could explain the demyelination phenotype caused by the loss of IKBKAP [101].
The example of FD illustrates the complexity of mutations in splice sites that have to be carefully analyzed within the context of other regulatory elements. It further shows that a missplicing event of a key regulatory gene can have profound impact by affecting other genes, and finally indicates that splicing is influenced by small substances.
Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency illustrates how multiple mutations affect exon usage leading to a human disease
Medium-chain acyl-CoA dehydrogenase (MCAD) is a mitochondrial enzyme that participates in the degradation of medium chain length fatty acids. Deficiencies of this enzyme are the most frequently diagnosed defect of mitochondrial beta-oxidation. The patients show metabolic crisis, characterized by hypoglycemia, lethargy and seizures, when first exposed to viral infections or challenged by fasting. About 20% of the infants die. MCAD deficiency results in accumulation of medium-chain acylcarnitines in the urine, which can be analyzed by mass-spectroscopy. A large newborn screen showed an incidence from 1:15,000 in the US population. The major reason for deficiency is a K304E missense mutation leading to a less active protein [102]. The newborn screening project identified a 362C->T missense mutation in exon 5 of the MCAD gene that causes exon skipping and subsequent degradation of the mRNA by nonsense-mediated decay [103]. This mutation disrupts a splicing enhancer that is highly similar to the SF2/ASF enhancer in the SMN2 exon 7. Cotransfection experiments demonstrate that an increase of the SF2/ASF concentration promotes inclusion of the mutated exon, suggesting that the mutation weakens an SF2/ASF-dependent enhancer. Interestingly, a synonymous mutation 351A->C was identified 11 nucleotides upstream in the same exon. This mutation affects an hnRNP A1 dependent silencer. Since the exon is constitutively used in the absence of the 362C->T SF2/ASF enhancer mutation, it has no effect on splicing. However, in the presence of the 362C->T enhancer mutation, it promotes exon inclusion, which antagonized the exon-skipping effect of the 362C->T SF2/ASF enhancer mutation. This example illustrates the fine-tuned balance of positive and negative acting factors that exists in splicing regulation. It also shows that seemingly irrelevant mutations can have an effect on splicing when they are combined with other mutations. Finally, the similarities between the regulation of MCAD exon 5 and SMN2 exon 7 suggest that there are degenerate ‘building blocks’ or ‘regulatory modules’ in the splicing code.
Frontotemporal lobar dementias are caused by the loss of the splicing factor TDP43
TDP43 (TAR DNA binding protein 43 kd) was originally identified as a transcriptional repressor that associates with the transcriptional activator DNA region (TAR) in HIV [104] and was later also found associated with the spermatid-specific gene SP-10 promoter [105], reviewed in [106]. TDP43 is a member of the heterogeneous nuclear ribonucleoproteins (hnRNP) family of proteins. The protein was later identified as a factor that binds to 12 UG repeats that cause aberrant skipping of exon 9 of the CFTR gene, leading to cystic fibrosis [107]. It contains two RNA binding domains. The first RNA binding domain is necessary and sufficient for binding to RNA that contains at least four UG repeats. In addition, TDP43 shows binding to single stranded TG DNA repeats in vitro [108]. TDP43 is a nuclear protein and it was therefore completely surprising when it was detected in ubiquitin-positive, tau and alpha synuclein-negative cytosolic inclusions that are the characteristic of frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS, Lou Gehring's disease) [109]. During the disease, TDP43 is cleaved by caspase-3 and the cleavage fragments accumulate in the cytosol, where they form aggregates. The caspase-3 reaction is inhibited by progranulin, which explains why mutations reducing the progranulin expression cause FTLD [110]. It is not clear whether the disease is caused by a loss of nuclear function of TDP43 or by a possible cytotoxoic accumulation. However, siRNA mediated knock down of TDP43 in HeLa cells followed by RNA microarray analysis demonstrated a strong upregulation of cyclin-dependent kinase 6 (cdk6). The human Cdk6 gene contains numerous intronic TG repeats and interestingly a change in cdk6 expression was not observed for the chicken gene that lacks these repeats. These data strongly suggest that TDP43 represses cdk6 expression by sequestering its RNA by binding to UG repeats. Loss of TDP43 expression leads to abnormal cdk6 activity, resulting in pRB phosphorylation, genomic instability and apoptosis, which could explain the cell death in FTLD [111]. Mutations the gene encoding TDP43 are found in families with amyotrophic lateral sclerosis, as well as in sporadic cases, indicating a direct link between TDP43 and amyotrophic lateral sclerosis [112].
This example illustrates that splicing factors like TDP43 operate in a regulatory network. A loss of their function can have drastic, but indirect effects. HnRNPs typically perform several different functions, which can obscure possible disease mechanisms. Finally, the interaction between UG repeats of the cdk6 gene and TDP43 illustrate how an hnRNP can be recruited to a new function during evolution.
Diseases associated with repeat elements
Short sequence repeats can be detected in numerous exons, where they serve to increase the recognition by a certain splicing factor [27, 113], reviewed in [114]. A change in length of simple repeat sequences can therefore change the splicing pattern of a gene. For example, the endothelial NO synthase gene contains an intronic polymorphic CA-repeat region and the number of repeats correlate with the risk of coronary artery disease and hyperhomocysteinemie in a sex-specific fashion [115]. SELEX experiments and functional studies showed that CA repeats bind to hnRNP L and the action of hnRNP L depends on the CA repeat length. Microarray studies showed that intronic CA-rich repeats can influence alternative splicing decisions, strongly suggesting that other intronic CA-repeat polymorphisms could cause human disease [116]. Another disease-relevant repeat is the UG repeat that can cause aberrant splicing of exon 9 of the CFTR gene, leading to cystic fibrosis [107]. This repeat binds to TDP43, which is discussed below. Myotonic dystrophy (DM) is the most common form of muscular dystrophy in adults. The disease is caused by extensions of two repeats: a CUG repeat in the 3′ region of the DMPK gene leads to DM1 and extension of a CCUG repeat in an intron of the ZNF9 gene leads to DM2 [117]. The common CUG element binds to at least two groups of RNA binding proteins, muscleblind-like1 and CUG binding protein. Extension of the repeats leads a sequestration of muscleblind protein in nuclear foci. This sequestration of muscleblind-like 1 protein reduces its cellular concentration and changes alternative splicing events that depend on this protein. Surprisingly, the extended CUG repeats lead to an increase of CUG binding protein concentration. This increase is due to a stabilization of CUG-binding protein. CUG RNA repeats induce via an unknown mechanism protein kinase C, which phosphorylates the CUG protein, leading to a more stable protein [118].
Alu elements are the largest group of repetitive elements in the human genome. They represent about 10% of the total genome sequence (reviewed in [63]). Alu elements contain potential splice sites and can evolve into exons [61]. It has been estimated that up to 5% of human alternative exons could be derived from Alu sequences [119]. It is therefore not surprising that mutations in existing Alu elements can cause their abnormal inclusion in mRNA, which leads to human diseases, such as the Alport syndrome [120], congenital cataracts facial dysmorphism neuropathy syndrome [121], and mucopolysaccharidosis type VII [122].
These examples illustrate how repeats can change the balance of alternative splicing regulation, even when they are located in introns or in seemingly unrelated mRNAs. The exonisation of Alu elements shows how new repetitive elements can be used by evolution to aquire new functions, and diseases caused by improper Alu-element exonisations can be viewed as failed evolutionary experiments.
3. Numerous changes in alternative splicing are found in cancer
Numerous reports have shown that alternative splicing patterns are changed in cancer (reviewed in [123]). The expression of alternative or even tumour-specific splice variants significantly affects many cellular events critical for cancer biology such as cell proliferation, motility, and drug response [124]. It is still unclear, however, to what extent alternative splicing functionally contributes to the initiation and progression of cancers. Most altered splicing patterns could be largely symptomatic and attributed to a generalized lack of fidelity of the splicing apparatus in cancer cells. The existence of particular splice variants in cancer could merely be a consequence of the malignant phenotype without contributing to the cancer phenotype. Here we will describe general changes of the splicing machinery in cancer and discuss specific examples illustrating the action on tumor suppressor genes.
3.1 Splicing regulatory factors and cancer
Changes in the concentration, localization, composition, or activity of trans-acting regulatory factors, such as hnRNP and SR proteins, can alter splice site selection. Several studies have demonstrated specific alterations in the expression of splicing factors in cancer. The increased phosphorylation and the elevated mRNA expression levels of Tra2-beta1, YB-1 and classical SR proteins, including SC35 and ASF/SF2, have been observed in human ovarian cancer and in mouse models of breast cancer development [125-128]. One of the best characterized examples of splicing changes observed in cancer tissues is CD44, a type 1 transmembrane glycoprotein involved in cell-cell and cell-matrix interactions. Alternative splicing of 10 of its 20 exons is found in association with several biological processes, such as lymphocyte recruitment, epithelial cell-matrix adhesion, and tumour metastasis. Due to its abundant alternative splicing, it has been correlated with changes in splicing factors, for example the concentration of ASF/SF2 or hnRNP A1 in mouse lung tumours [129] and human colon carcinomas [130].
SF2/ASF is a prototypical splicing factor and has been widely used in studying the connection between cancer and alternative splicing. SF2/ASF controls alternative splicing of the oncogene Ron to modulate cell motility, a general property of metastatic cancer cells [131]. An increased expression of SF2/ASF can transform NIH3T3 and Rat1 cells in vitro, and NIH3T3 cells that express high levels of ASF/SF2 produce tumours in vivo. This increase correlates with a change in splicing of genes involved in the Ras-mitogen-activated protein kinase (MAPK) and the phophatidylinositol-3-kinas (PI3K)-mammalian target of rapamycin (mTOR) pathways, which are often deregulated in cancer. MNK2 and S6K1 kinases involved in signal transduction are alternatively spliced in response to elevated SF2/ASF concentration. A novel SF2/ASF-induced isoform of S6K1 is sufficient to cause a transformation phenotype. Overexpression or knockdown of ASF/SF2 resulted in specific splicing changes in a number of genes involved in these pathways. SF2/ASF overexpression led to cellular transformation resulting in anchorage-independent cell growth in soft-agar and tumour formation in nude mice.[132]. These data argue that SF2/ASF is a proto-oncogene [132]. It is likely that other splicing factors have similar features.
3.2 Mutations of tumour suppressor genes affecting splice site selection can cause cancer
Mutations in splicing regulatory elements that change splice site selection provide a clear link between pre-mRNA processing and cancer development. The mutations ultimately lead to the generation of non-functional tumor suppressor genes, which predisposes to cancer. The identification and understanding of inherited mutations is important for genetic counselling and for follow-up aimed at cancer prevention in affected family members. A list of well-characterized mutations is shown in Table 2.
Table 2. Splicing mutations of tumour suppressor genes.
| Gene | Mutation | Effect | Reference |
|---|---|---|---|
| APC | Donor splice site mutation in intron 4 | Exon 4 skipping → attenuated form of FAP | [133] |
| APC | G→A substitution at the splice acceptor site of intron 7 | Cryptic splice site creation causing a single nucleotide deletion a the beginning of exon 8 | [174] |
| BRCA1 | Non sense mutation in exon 18 | ASF/SF2 ESE disruption → exon 18 skipping | [143] |
| BRCA1 | Intronic point mutation AA →AG (IVS5-12 G→A) | Cryptic 3′ splice site creation →production of a truncated protein | [144] |
| Estrogen receptor | AT →GT point mutation in intron 5 | Cryptic 5′ splice site creation in intron 5 →insertion of a 69 nucleotide cryptic exon into the reading frame | [175] |
| NF1 | Double point mutation in exon 7 | Disruption of ASF/SF2 and SC35 ESE binding sites →In frame deletion | [147] |
| NF2 | Intronic CTAGC →CTAAC | Consensus branch point adenosine creation →cryptic splice site creation →additional exon 5a insertion | [148] |
| MLH1 | Different codon 659 mutations | Exon 17 skipping →internal deletion of 31 aa that abrogates binding to the other mismatch repair protein PMS2 | [140] |
The most frequent early molecular alteration in the initiation of colon cancer involves mutations in the adenomatous polyposis coli (APC) gene, which occur both in rare familial cancer syndromes and in −85% of sporadic cancers. The majority of these mutations result in the truncation of the APC gene product. Germline mutations in APC gene result in familial adenomatous polyposis (FAP). Two mutations that disrupt splicing regulatory elements have been found. The first one affects a splicing donor site in intron 4 resulting in skipping of exon 4 and leading to an attenuated form of FAP [133]. This splicing deregulation was found in liver metastases, where the wild-type allele was deleted [134]. The second mutation is a G to A substitution at the splice acceptor site of intron 7 creating a cryptic splice site and causing a single nucleotide deletion at the beginning of exon 8 [134]. The behaviour of these mutations can be complicated by additional APC transcripts which are generated due to alternative splicing of exons 3-4, 9, 10A/X, and 14 [135] [136, 137]
The MMR MLH1 (MIM 120436) and MSH2 (MIM 609309) genes responsible for hereditary nonpolyposis colorectal cancer (HNPCC or Lynch syndrome; MIM 120435), are prone to mutations. Double exon skipping in MLH1 gives rise to hereditary nonpolyposis colorectal cancer [138, 139]. Also in various hereditary nonpolyposis colorectal cancer, different mutations in codon 659 result in skipping of exon 17 of MLH1 gene. This causes an internal deletion of 31 amino acids that abrogates binding to the other mismatch repair protein PMS2 [140]. A major difficulty in the diagnosis of the Lynch syndrome, like other Mendelian diseases, is the interpretation of numerous variants of unknown clinical and biological significance. In most cases the pathogenic role of the unclassified variants cannot be established from clinical data and segregation analysis, because of the small number of family members available, the incomplete penetrance of MMR mutations and the possibility of additional undetected mutations in the MMR genes. Recently, using an ex vivo functional splicing assay based on a novel splicing reporter minigene to test 87 intronic or exonic variants (20 and 67, respectively) corresponding to 85 alleles found in MLH1 or MSH2 by the French HNPCC consortium, it was possible to show that a high level of mutations alter splicing regulatory elements [141]. However, the biological significance of the six variants is unkown.
Breast cancer gene 1 (BRCA1) is responsible for the majority of hereditary breast and ovarian cancers [142]. Among sporadic cases of breast cancer, expression of BRCA1 is reduced or undetectable in high-grade ductal carcinomas, suggesting the involvement of this gene in the etiology of breast cancer. Germline sequence variations in both splice sites and regulatory elements of BRCA1 gene have been implicated in susceptibility to cancer. For example, an inherited nonsense mutation in exon 18 of the BRCA1 gene disrupts an Exonic Splicing Enhancer. This enhancer binds to the SR protein SF2/ASF, and its mutation causes inappropriate skipping of the constitutive exon 18 [143]. It has also been shown that an AA to AG mutation creates a cryptic 3′ splice site that adds 11 nucleotides to BRCA1 mRNA, which encodes a truncated protein in a breast cancer family [144]. In addition, multiple BRCA1 splice variants are present in different tissues with different expression profiles [145].
The neurofibromatosis 1 (NF1) gene that is linked to development of neurofibromas has one of the highest mutation rates described for any human disorder, and a large proportion of NF1 mutations bear consequences for the correct splicing of the RNA. A systematic study of somatic NF1 mutations [146] demonstrated that most point mutations resulted in splicing defects including exon skipping and the usage of alternative 5′ and 3′ splice sites. Also, a double point mutation near the middle of NF1 exon 7 has been found to cause a high level of in-frame deletion of that exon. These mutations disrupt consensus binding sites for the SR proteins SC35 and ASF/SF2 [147]. Some mutations like a CTAGC to CTAAC in the NF1 gene in the central nervous system, creates a consensus branch point adenosine that activates a cryptic site in the intron leading to an additional exon (exon 5a). Because some normal splicing remains, this mutation is associated with a relatively mild phenotype [148].
These examples illustrate that numerous changes in splicing patterns are characteristic for cancer development and progression. It is currently not clear whether these changes are the cause or the consequence of a malignant phenotype. Gain of function changes in alternative splicing have been observed in several systems and well-characterized examples are summarized in Table 3. These mutations, and the mutations in the tumor supressor genes that cause missplicing and predispose cancer, are strongly suggesting that abarrant processing of some pre-mRNAs is the cause of cancer and could be a therapeutic target.
Table 3. Gain of function of proteins promoting tumour development by alternative splicing.
| Gene | Protein Property/function | Splice variant | Isoform expression in cancer | Cancer type | Reference |
|---|---|---|---|---|---|
| Survivin | Inhibitor of apoptosis | Survivin 2B with pro-apoptotic properties | Down regulated | Breast carcinoma and late stage or metastatic gastric cancer | [176, 177] |
| VEGF | Role in angiogenesis | Isoforms lacking exon 6 | Upregulated | Non-small cell lung cnacer | [178] |
| Cathepsin B | Role in the development and progression of cancers | Certains isoforms | Overexpressed | Colon cancer | [179] |
| FHIT | Tumour suppressor | Aberrant transcripts | Aberrant expression | Gastric, cervical, thyroid and testicular germ-cell tumours | [180-185] |
| Actinin 4 | Actin-binding protein | Variant Va, a mutually exclusive splice variant where exon 8 is replaced by a new exon of the same size that exists in intron 8 | Aberrant expression of the splice isoform | Small cell lung cancer | [186] |
| AIB1 | Hormone coactivator | Isoform lacking exon 3 | Aberrant expression | Breast cancer | [187] |
| RON | Tyrosine kinase receptor | RonΔ165 RonΔ160 RonΔ155 | Overexpressed | Colorectal carcinoma | [188, 189] |
4. Treatment options for missplicing events
The examples discussed here clearly show that a misregulation of alternative splicing plays a large role in numerous human diseases. The identification of molecules capable of correcting and or inhibiting pathological splicing events is therefore an important issue for future therapeutic approaches.
4.1 Antisense strategies
The major challenge in treating splicing disorders is to specifically target one splicing event in a certain pre-mRNA. Since there are several thousand other pre-mRNAs in the cell, the selectivity of splice site intervention is a major problem. One way to address this problem is the use of oligonucleotides that will specifically bind to one sequence.
Special chemistries were devised to prevent RNAseH-mediated cleavage of the RNA and to lower toxicity (reviewed in [149, 150]). A major drawback of the therapeutic use of oligonucleotides is their delivery and uptake in the cells. This problem has been addressed by coupling of oligonucleotides to arginine-rich cell penetrating peptides. [151, 152].
Antisense oligonucleotides have been tested for beta thalassemias [153], Duchenne muscular dystrophy [154, 155], cystic fibrosis [156] cyclophilin transcripts [157], Hutchinson Gilford Progeria Syndrome (HGPS) [158] as well as block HIV replication [159] and alter tau pre-mRNAs [160].
Antisense oligonucleotides can also be used to block splicing enhancers or silencers, which can be found in introns or exons quite far away from the splice sites [161]. Inhibition of splicing silencers (ESSs or ISSs) is of particular interest since it provides a way to activate otherwise repressed exons. This was achieved for the α exon of fibroblast growth factor receptor-1 (FGFR1) transcripts [162] where an antisense morpholino oligonucleotides that blocks silencer elements in glioblastoma cells in culture promotes the inclusion of the α exon. The antisense approach was further developed in ESSENSE (exon-specific splicing enhancement by small chimeric effectors). ESSENSE uses bifunctional reagents that contain a peptide effector domain and an antisense-targeting domain. The effector domains of these protein-nucleic acids were arginine-serine (RS) repeats that mimic the effect of SR proteins [163]. A second incarnation of bifunctional oligomers uses a 2′-Ome-modified binding domain and an effector domain, which is composed of RNA that contains binding sites for known splicing trans-acting factors [164] [165]. The recruited factors mediate activation or silencing of the targeted exon. An example of this type of bifunctional oligonucleotide acts as an ESE and promotes inclusion of the SMN2 exon 7 fibroblasts from SMA patients leading to partial restoration of the SMN function [164].
5.2 Substances that change alternative splicing
The use of RNA-binding molecules as antibiotics, such as gentamicin, chloramphenicol, and tetracycline illustrates that drugs can be targeted against RNA and/or RNA binding proteins. High-throughput screens and testing of substances in model systems have now identified more than 30 substances that change splice site selection. The substances fall into several categories, including HDAC inhibitors, kinase and phosphatase inhibitors, as well as cAMP antagonist and agonists. The currently known substances are reviewed in [166] and updated on the web.
The usefulness of substances that change splice site selection is evident from potential HIV therapies that could be combined with other antiviral strategies. Using an in vitro splicing assay, a chemical screen was performed that identified an indole derivative (IDC16) that interferes with exonic splicing enhancer activity of the SR protein splicing factor SF2/ASF [167]. IDC16 suppresses the production of key viral proteins and inhibits replication of macrophage- and T cell–tropic laboratory strains, clinical isolates, and strains with high-level resistance to inhibitors of viral protease and reverse transcriptase. Thus, human splicing factors represent novel and promising drug targets for the development of antiretroviral therapies, particularly for the inhibition of multidrug-resistant viruses.
Acknowledgments
This work was supported by the EURASNET (European Alternative Splicing Network of Excellence), NIH (National Institutes of Health; P20 RR020171), BMBF (Federal Ministry of Education and Research, Germany) and DFG (Deutsche Forschungsgemeinschaft; SFB 473).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature cited
- 1.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Dov C, Hartmann B, Lundgren J, Valcarcel J. Genome-wide analysis of alternative pre-mRNA splicing. J Biol Chem. 2008;283:1229–33. doi: 10.1074/jbc.R700033200. [DOI] [PubMed] [Google Scholar]
- 3.Kampa D, Cheng J, Kapranov P, Yamanaka M, Brubaker S, Cawley S, Drenkow J, Piccolboni A, Bekiranov S, Helt G, Tammana H, Gingeras TR. Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res. 2004;14:331–42. doi: 10.1101/gr.2094104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006;126:37–47. doi: 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 5.Romero PR, Zaidi S, Fang YY, Uversky VN, Radivojac P, Oldfield CJ, Cortese MS, Sickmeier M, LeGall T, Obradovic Z, Dunker AK. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc Natl Acad Sci U S A. 2006;103:8390–5. doi: 10.1073/pnas.0507916103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stamm S, Ben-Ari S, Rafalska I, Tang Y, Zhang Z, Toiber D, Thanaraj TA, Soreq H. Function of alternative splicing. Gene. 2005;344C:1–20. doi: 10.1016/j.gene.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 7.Hillman RT, Green RE, Brenner SE. An unappreciated role for RNA surveillance. Genome Biol. 2004;5:R8. doi: 10.1186/gb-2004-5-2-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan Q, Saltzman AL, Kim YK, Misquitta C, Shai O, Maquat LE, Frey BJ, Blencowe BJ. Quantitative microarray profiling provides evidence against widespread coupling of alternative splicing with nonsense-mediated mRNA decay to control gene expression. Genes Dev. 2006;20:153–8. doi: 10.1101/gad.1382806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilkie SE, Vaclavik V, Wu H, Bujakowska K, Chakarova CF, Bhattacharya SS, Warren MJ, Hunt DM. Disease mechanism for retinitis pigmentosa (RP11) caused by missense mutations in the splicing factor gene PRPF31. Mol Vis. 2008;14:683–90. [PMC free article] [PubMed] [Google Scholar]
- 10.Vithana EN, Abu-Safieh L, Allen MJ, Carey A, Papaioannou M, Chakarova C, Al-Maghtheh M, Ebenezer ND, Willis C, Moore AT, Bird AC, Hunt DM, Bhattacharya SS. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11) Mol Cell. 2001;8:375–81. doi: 10.1016/s1097-2765(01)00305-7. [DOI] [PubMed] [Google Scholar]
- 11.Boon KL, Grainger RJ, Ehsani P, Barrass JD, Auchynnikava T, Inglehearn CF, Beggs JD. prp8 mutations that cause human retinitis pigmentosa lead to a U5 snRNP maturation defect in yeast. Nat Struct Mol Biol. 2007;14:1077–83. doi: 10.1038/nsmb1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mardon HJ, Sebastio G, Baralle FE. A role for exon sequences in alternative splicing of the human fibronectin gene. Nucl Acids Res. 1987;15:7725–7733. doi: 10.1093/nar/15.19.7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooper TA, Mattox W. The regulation of splice-site selection, and its role in human disease. Am J Hum Genet. 1997;61:259–66. doi: 10.1086/514856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orengo JP, Cooper TA. Alternative splicing in disease. Adv Exp Med Biol. 2007;623:212–23. doi: 10.1007/978-0-387-77374-2_13. [DOI] [PubMed] [Google Scholar]
- 15.Nissim-Rafinia M, Kerem B. Splicing regulation as a potential genetic modifier. Trends Genet. 2002;18:123–7. doi: 10.1016/s0168-9525(01)02619-1. [DOI] [PubMed] [Google Scholar]
- 16.Buratti E, Baralle M, Baralle FE. Defective splicing, disease and therapy: searching for master checkpoints in exon definition. Nucleic Acids Res. 2006;34:3494–510. doi: 10.1093/nar/gkl498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17:419–37. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 18.Jeanteur P. Alternative Splicing and Disease. Springer; 2006. [Google Scholar]
- 19.Kim E, Goren A, Ast G. Alternative splicing and disease. RNA Biol. 2008;5:17–9. doi: 10.4161/rna.5.1.5944. [DOI] [PubMed] [Google Scholar]
- 20.House AE, Lynch KW. Regulation of alternative splicing: more than just the ABCs. J Biol Chem. 2008;283:1217–21. doi: 10.1074/jbc.R700031200. [DOI] [PubMed] [Google Scholar]
- 21.Smith CW, Valcarcel J. Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem Sci. 2000;25:381–388. doi: 10.1016/s0968-0004(00)01604-2. [DOI] [PubMed] [Google Scholar]
- 22.Black DL. Mechanisms of Alternative Pre-Messenger RNA Splicing. Annu Rev Biochem. 2003:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 23.Stark H, Luhrmann R. Cryo-electron microscopy of spliceosomal components. Annu Rev Biophys Biomol Struct. 2006;35:435–57. doi: 10.1146/annurev.biophys.35.040405.101953. [DOI] [PubMed] [Google Scholar]
- 24.Valadkhan S, Manley JL. Splicing-related catalysis by protein-free snRNAs. Nature. 2001;413:701–7. doi: 10.1038/35099500. [DOI] [PubMed] [Google Scholar]
- 25.Valadkhan S, Mohammadi A, Wachtel C, Manley JL. Protein-free spliceosomal snRNAs catalyze a reaction that resembles the first step of splicing. Rna. 2007;13:2300–11. doi: 10.1261/rna.626207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008 doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lynch KW, Maniatis T. Assembly of specific SR protein complexes on distinct regulatory elements of the Drosophila doublesex splicing enhancer. Genes Dev. 1996;10:2089–101. doi: 10.1101/gad.10.16.2089. [DOI] [PubMed] [Google Scholar]
- 28.Hertel KJ. Combinatorial control of exon recognition. J Biol Chem. 2008;283:1211–5. doi: 10.1074/jbc.R700035200. [DOI] [PubMed] [Google Scholar]
- 29.Moore MJ. From birth to death: the complex lives of eukaryotic mRNAs. Science. 2005;309:1514–8. doi: 10.1126/science.1111443. [DOI] [PubMed] [Google Scholar]
- 30.Buratti E, Baralle FE. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol Cell Biol. 2004;24:10505–14. doi: 10.1128/MCB.24.24.10505-10514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muro AF, Caputi M, Pariyarath R, Pagani F, Buratti E, Baralle FE. Regulation of fibronectin EDA exon alternative splicing: possible role of RNA secondary structure for enhancer display. Mol Cell Biol. 1999;19:2657–2671. doi: 10.1128/mcb.19.4.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hiller M, Zhang Z, Backofen R, Stamm S. pre-mRNA secondary structure and splice site selection. PLOS Genetics. 2007;3:2147–2155. doi: 10.1371/journal.pgen.0030204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, Kodzius R, Shimokawa K, Bajic VB, Brenner SE, Batalov S, Forrest AR, Zavolan M, Davis MJ, Wilming LG, Aidinis V, Allen JE, Ambesi-Impiombato A, Apweiler R, Aturaliya RN, Bailey TL, Bansal M, Baxter L, Beisel KW, Bersano T, Bono H, Chalk AM, Chiu KP, Choudhary V, Christoffels A, Clutterbuck DR, Crowe ML, Dalla E, Dalrymple BP, de Bono B, Della Gatta G, di Bernardo D, Down T, Engstrom P, Fagiolini M, Faulkner G, Fletcher CF, Fukushima T, Furuno M, Futaki S, Gariboldi M, Georgii-Hemming P, Gingeras TR, Gojobori T, Green RE, Gustincich S, Harbers M, Hayashi Y, Hensch TK, Hirokawa N, Hill D, Huminiecki L, Iacono M, Ikeo K, Iwama A, Ishikawa T, Jakt M, Kanapin A, Katoh M, Kawasawa Y, Kelso J, Kitamura H, Kitano H, Kollias G, Krishnan SP, Kruger A, Kummerfeld SK, Kurochkin IV, Lareau LF, Lazarevic D, Lipovich L, Liu J, Liuni S, McWilliam S, Madan Babu M, Madera M, Marchionni L, Matsuda H, Matsuzawa S, Miki H, Mignone F, Miyake S, Morris K, Mottagui-Tabar S, Mulder N, Nakano N, Nakauchi H, Ng P, Nilsson R, Nishiguchi S, Nishikawa S, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–63. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 34.Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, Helt G, Sementchenko V, Piccolboni A, Bekiranov S, Bailey DK, Ganesh M, Ghosh S, Bell I, Gerhard DS, Gingeras TR. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005;308:1149–54. doi: 10.1126/science.1108625. [DOI] [PubMed] [Google Scholar]
- 35.Kishore S, Stamm S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science. 2006;311:230–2. doi: 10.1126/science.1118265. [DOI] [PubMed] [Google Scholar]
- 36.Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, Garnica A, Cheung SW, Beaudet AL. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40:719–21. doi: 10.1038/ng.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skryabin BV, Gubar LV, Seeger B, Pfeiffer J, Handel S, Robeck T, Karpova E, Rozhdestvensky TS, Brosius J. Deletion of the MBII-85 snoRNA gene cluster in mice results in postnatal growth retardation. PLoS Genet. 2007;3:e235. doi: 10.1371/journal.pgen.0030235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding F, Li HH, Zhang S, Solomon NM, Camper SA, Cohen P, Francke U. SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PLoS ONE. 2008;3:e1709. doi: 10.1371/journal.pone.0001709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding F, Prints Y, Dhar MS, Johnson DK, Garnacho-Montero C, Nicholls RD, Francke U. Lack of Pwcr1/MBII-85 snoRNA is critical for neonatal lethality in Prader-Willi syndrome mouse models. Mamm Genome. 2005;16:424–31. doi: 10.1007/s00335-005-2460-2. [DOI] [PubMed] [Google Scholar]
- 40.Watkins NJ, Lemm I, Luhrmann R. Involvement of Nuclear Import and Export Factors in U8 Box C/D snoRNP Biogenesis. Mol Cell Biol. 2007;27:7018–27. doi: 10.1128/MCB.00516-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu S, Li P, Dybkov O, Nottrott S, Hartmuth K, Luhrmann R, Carlomagno T, Wahl MC. Binding of the human Prp31 Nop domain to a composite RNA-protein platform in U4 snRNP. Science. 2007;316:115–20. doi: 10.1126/science.1137924. [DOI] [PubMed] [Google Scholar]
- 42.Stamm S. Signals and their transduction pathways regulating alternative splicing: a new dimension of the human genome. Hum Mol Genet. 2002;11:2409–2416. doi: 10.1093/hmg/11.20.2409. [DOI] [PubMed] [Google Scholar]
- 43.Shin C, Manley JL. Cell signalling and the control of pre-mRNA splicing. Nat Rev Mol Cell Biol. 2004;5:727–38. doi: 10.1038/nrm1467. [DOI] [PubMed] [Google Scholar]
- 44.Blaustein M, Pelisch F, Srebrow A. Signals, pathways and splicing regulation. Int J Biochem Cell Biol. 2007;39:2031–2048. doi: 10.1016/j.biocel.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 45.Stamm S. Regulation of alternative splicing by reversible phosphorylation. J Biol Chem. 2008;283:1223–7. doi: 10.1074/jbc.R700034200. [DOI] [PubMed] [Google Scholar]
- 46.Hagiwara M. Alternative splicing: a new drug target of the post-genome era. Biochim Biophys Acta. 2005;1754:324–31. doi: 10.1016/j.bbapap.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 47.Hernandez F, Perez M, Lucas JJ, Mata AM, Bhat R, Avila J. Glycogen synthase kinase-3 plays a crucial role in tau exon 10 splicing and intranuclear distribution of SC35. Implications for Alzheimer's disease. J Biol Chem. 2004;279:3801–6. doi: 10.1074/jbc.M311512200. [DOI] [PubMed] [Google Scholar]
- 48.Shin C, Feng Y, Manley JL. Dephosphorylated SRp38 acts as a splicing repressor in response to heat shock. Nature. 2004;427:553–8. doi: 10.1038/nature02288. [DOI] [PubMed] [Google Scholar]
- 49.Xiao SH, Manley JL. Phosphorylation of the ASF/SF2 RS domain affects both protein-protein and protein-RNA interactions and is necessary for splicing. Genes Dev. 1997;11:334–44. doi: 10.1101/gad.11.3.334. [DOI] [PubMed] [Google Scholar]
- 50.Huang Y, Yario TA, Steitz JA. A molecular link between SR protein dephosphorylation and mRNA export. Proc Natl Acad Sci U S A. 2004;101:9666–70. doi: 10.1073/pnas.0403533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shen H, Green MR. RS domains contact splicing signals and promote splicing by a common mechanism in yeast through humans. Genes Dev. 2006;20:1755–65. doi: 10.1101/gad.1422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ceulemans H, Bollen M. Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiol Rev. 2004;84:1–39. doi: 10.1152/physrev.00013.2003. [DOI] [PubMed] [Google Scholar]
- 53.Hanks SK. Genomic analysis of the eukaryotic protein kinase superfamily: a perspective. Genome Biol. 2003;4:111. doi: 10.1186/gb-2003-4-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi Y, Reddy B, Manley JL. PP1/PP2A Phosphatases Are Required for the Second Step of Pre-mRNA Splicing and Target Specific snRNP Proteins. Mol Cell. 2006;23:819–29. doi: 10.1016/j.molcel.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 55.Allemand E, Hastings ML, Murray MV, Myers MP, Krainer AR. Alternative splicing regulation by interaction of phosphatase PP2Cgamma with nucleic acid-binding protein YB-1. Nat Struct Mol Biol. 2007;14:630–638. doi: 10.1038/nsmb1257. [DOI] [PubMed] [Google Scholar]
- 56.Cao W, Jamison SF, Garcia-Blanco MA. Both phosphorylation and dephosphorylation of ASF/SF2 are required for pre-mRNA splicing in vitro. Rna. 1997;3:1456–67. [PMC free article] [PubMed] [Google Scholar]
- 57.Cardinali B, Cohen PT, Lamond AI. Protein phosphatase 1 can modulate alternative 5′ splice site selection in a HeLa splicing extract. Febs Lett. 1994;352:276–80. doi: 10.1016/0014-5793(94)00973-2. [DOI] [PubMed] [Google Scholar]
- 58.Ghosh N, Patel N, Jiang K, Watson JE, Cheng J, Chalfant CE, Cooper DR. Ceramide-activated protein phosphatase involvement in insulin resistance via Akt, serine/arginine-rich protein 40, and ribonucleic acid splicing in L6 skeletal muscle cells. Endocrinology. 2007;148:1359–66. doi: 10.1210/en.2006-0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chalfant CE, Rathman K, Pinkerman RL, Wood RE, Obeid LM, Ogretmen B, Hannun YA. De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells. Dependence on protein phosphatase-1. J Biol Chem. 2002;277:12587–95. doi: 10.1074/jbc.M112010200. [DOI] [PubMed] [Google Scholar]
- 60.Kim E, Goren A, Ast G. Alternative splicing: current perspectives. Bioessays. 2008;30:38–47. doi: 10.1002/bies.20692. [DOI] [PubMed] [Google Scholar]
- 61.Lev-Maor G, Sorek R, Shomron N, Ast G. The birth of an alternatively spliced exon: 3′ splice-site selection in Alu exons. Science. 2003;300:1288–91. doi: 10.1126/science.1082588. [DOI] [PubMed] [Google Scholar]
- 62.Lev-Maor G, Goren A, Sela N, Kim E, Keren H, Doron-Faigenboim A, Leibman-Barak S, Pupko T, Ast G. The “Alternative” Choice of Constitutive Exons throughout Evolution. PLoS Genet. 2007;3:e203. doi: 10.1371/journal.pgen.0030203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hasler J, Samuelsson T, Strub K. Useful ‘junk’: Alu RNAs in the human transcriptome. Cell Mol Life Sci. 2007;64:1793–800. doi: 10.1007/s00018-007-7084-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y, Bertolino A, Fazio L, Blasi G, Rampino A, Romano R, Lee ML, Xiao T, Papp A, Wang D, Sadee W. Polymorphisms in human dopamine D2 receptor gene affect gene expression, splicing, and neuronal activity during working memory. Proc Natl Acad Sci U S A. 2007;104:20552–7. doi: 10.1073/pnas.0707106104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pearn J. Classification of spinal muscular atrophies. Lancet. 1980;8174:919–922. doi: 10.1016/s0140-6736(80)90847-8. [DOI] [PubMed] [Google Scholar]
- 66.Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, Dreyfuss G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oprea GE, Krober S, McWhorter ML, Rossoll W, Muller S, Krawczak M, Bassell GJ, Beattie CE, Wirth B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320:524–7. doi: 10.1126/science.1155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wirth B, Brichta L, Hahnen E. Spinal muscular atrophy and therapeutic prospects. Prog Mol Subcell Biol. 2006;44:109–32. doi: 10.1007/978-3-540-34449-0_6. [DOI] [PubMed] [Google Scholar]
- 69.Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossol W, Prior TW, Morris GE, Burghes AH. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9:333–9. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 70.Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70:358–68. doi: 10.1086/338627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Singh NN, Androphy EJ, Singh RN. The regulation and regulatory activities of alternative splicing of the SMN gene. Crit Rev Eukaryot Gene Expr. 2004;14:271–85. doi: 10.1615/critreveukaryotgeneexpr.v14.i4.30. [DOI] [PubMed] [Google Scholar]
- 72.Hofmann Y, Lorson CL, Stamm S, Androphy EJ, Wirth B. Htra2-beta 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2) Proc Natl Acad Sci U S A. 2000;97:9618–23. doi: 10.1073/pnas.160181697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Singh NN, Androphy EJ, Singh RN. In vivo selection reveals combinatorial controls that define a critical exon in the spinal muscular atrophy genes. Rna. 2004;10:1291–305. doi: 10.1261/rna.7580704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Singh NN, Androphy EJ, Singh RN. An extended inhibitory context causes skipping of exon 7 of SMN2 in spinal muscular atrophy. Biochem Biophys Res Commun. 2004;315:381–8. doi: 10.1016/j.bbrc.2004.01.067. [DOI] [PubMed] [Google Scholar]
- 75.Cartegni L, Hastings ML, Calarco JA, de Stanchina E, Krainer AR. Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am J Hum Genet. 2006;78:63–77. doi: 10.1086/498853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30:377–84. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 77.Kashima T, Rao N, David CJ, Manley JL. hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Hum Mol Genet. 2007;16:3149–59. doi: 10.1093/hmg/ddm276. [DOI] [PubMed] [Google Scholar]
- 78.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003;34:460–3. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 79.Novoyatleva T, Heinrich B, Tang Y, Benderska N, Butchbach ME, Lorson CL, Lorson MA, Ben-Dov C, Fehlbaum P, Bracco L, Burghes AH, Bollen M, Stamm S. Protein phosphatase 1 binds to the RNA recognition motif of several splicing factors and regulates alternative pre-mRNA processing. Hum Mol Genet. 2008:52–70. doi: 10.1093/hmg/ddm284. [DOI] [PubMed] [Google Scholar]
- 80.Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochem Biophys Acta. 2005;1739:91–103. doi: 10.1016/j.bbadis.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 81.Gallo JM, Noble W, Martin TR. RNA and protein-dependent mechanisms in tauopathies: consequences for therapeutic strategies. Cell Mol Life Sci. 2007;64:1701–14. doi: 10.1007/s00018-007-6513-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Andreadis A. Misregulation of tau alternative splicing in neurodegeneration and dementia. Prog Mol Subcell Biol. 2006;44:89–107. doi: 10.1007/978-3-540-34449-0_5. [DOI] [PubMed] [Google Scholar]
- 83.Hasegawa M, Smith MJ, Iijima M, Tabira T, Goedert M. FTDP-17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS lett. 1999;443:93–96. doi: 10.1016/s0014-5793(98)01696-2. [DOI] [PubMed] [Google Scholar]
- 84.Jiang Z, Tang H, Havlioglu N, Zhang X, Stamm S, Yan R, Wu JY. Mutations in tau gene exon 10 associated with FTDP-17 alter the activity of an exonic splicing enhancer to interact with Tra2-beta1. J Biol Chem. 2003;278:18997–19007. doi: 10.1074/jbc.M301800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang J, Gao QS, Wang Y, Lafyatis R, Stamm S, Andreadis A. Tau Exon 10, Whose Missplicing Causes Frontotemporal Dementia, is Regulated by an Intricate Interplay of Cis Elements and Trans Factors. J Neurochem. 2004;88:1078–1090. doi: 10.1046/j.1471-4159.2003.02232.x. [DOI] [PubMed] [Google Scholar]
- 86.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–7. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 87.Dawson HN, Cantillana V, Chen L, Vitek MP. The tau N279K exon 10 splicing mutation recapitulates frontotemporal dementia and parkinsonism linked to chromosome 17 tauopathy in a mouse model. J Neurosci. 2007;27:9155–68. doi: 10.1523/JNEUROSCI.5492-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Spillantini MG, Goedert M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998;21:428–33. doi: 10.1016/s0166-2236(98)01337-x. [DOI] [PubMed] [Google Scholar]
- 89.Connell JW, Rodriguez-Martin T, Gibb GM, Kahn NM, Grierson AJ, Hanger DP, Revesz T, Lantos PL, Anderton BH, Gallo JM. Quantitative analysis of tau isoform transcripts in sporadic tauopathies. Brain Res Mol Brain Res. 2005;137:104–9. doi: 10.1016/j.molbrainres.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 90.Boutajangout A, Boom A, Leroy K, Brion JP. Expression of tau mRNA and soluble tau isoforms in affected and non-affected brain areas in Alzheimer's disease. FEBS Lett. 2004;576:183–9. doi: 10.1016/j.febslet.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 91.Ingelsson M, Ramasamy K, Cantuti-Castelvetri I, Skoglund L, Matsui T, Orne J, Kowa H, Raju S, Vanderburg CR, Augustinack JC, de Silva R, Lees AJ, Lannfelt L, Growdon JH, Frosch MP, Standaert DG, Irizarry MC, Hyman BT. No alteration in tau exon 10 alternative splicing in tangle-bearing neurons of the Alzheimer's disease brain. Acta Neuropathol. 2006;112:439–49. doi: 10.1007/s00401-006-0095-3. [DOI] [PubMed] [Google Scholar]
- 92.Glatz DC, Rujescu D, Tang Y, Berendt FJ, Hartmann AM, Faltraco F, Rosenberg C, Hulette C, Jellinger K, Hampel H, Riederer P, Moller HJ, Andreadis A, Henkel K, Stamm S. The alternative splicing of tau exon 10 and its regulatory proteins CLK2 and TRA2-BETA1 changes in sporadic Alzheimer's disease. J Neurochem. 2006;96:635–644. doi: 10.1111/j.1471-4159.2005.03552.x. [DOI] [PubMed] [Google Scholar]
- 93.De Sandre-Giovannoli A, Levy N. Altered splicing in prelamin A-associated premature aging phenotypes. Prog Mol Subcell Biol. 2006;44:199–232. doi: 10.1007/978-3-540-34449-0_9. [DOI] [PubMed] [Google Scholar]
- 94.Zhu H, Tucker HM, Grear KE, Simpson JF, Manning AK, Cupples LA, Estus S. A common polymorphism decreases low-density lipoprotein receptor exon 12 splicing efficiency and associates with increased cholesterol. Hum Mol Genet. 2007;16:1765–72. doi: 10.1093/hmg/ddm124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zou F, Gopalraj RK, Lok J, Zhu H, Ling IF, Simpson JF, Tucker HM, Kelly JF, Younkin SG, Dickson DW, Petersen RC, Graff-Radford NR, Bennett DA, Crook JE, Estus S. Sex-dependent Association of a Common Low Density Lipoprotein Receptor Polymorphism with RNA Splicing Efficiency in the Brain and Alzheimers Disease. Hum Mol Genet. 2007 doi: 10.1093/hmg/ddm365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cooper DN, Stenson PD, Chuzhanova NA. The Human Gene Mutation Database (HGMD) and its exploitation in the study of mutational mechanisms. Curr Protoc Bioinformatics. 2006;Chapter 1 doi: 10.1002/0471250953.bi0113s12. Unit 1 13. [DOI] [PubMed] [Google Scholar]
- 97.Birgens H, Ljung R. The thalassaemia syndromes, Scand. J Clin Lab Invest. 2007;67:11–25. doi: 10.1080/00365510601046417. [DOI] [PubMed] [Google Scholar]
- 98.Maayan C, Kaplan E, Shachar S, Peleg O, Godfrey S. Incidence of familial dysautonomia in Israel 1977-1981. Clin Genet. 1987;32:106–8. doi: 10.1111/j.1399-0004.1987.tb03334.x. [DOI] [PubMed] [Google Scholar]
- 99.Carmel I, Tal S, Vig I, Ast G. Comparative analysis detects dependencies among the 5′ splice-site positions. Rna. 2004;10:828–40. doi: 10.1261/rna.5196404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ibrahim el C, Hims MM, Shomron N, Burge CB, Slaugenhaupt SA, Reed R. Weak definition of IKBKAP exon 20 leads to aberrant splicing in familial dysautonomia. Hum Mutat. 2007;28:41–53. doi: 10.1002/humu.20401. [DOI] [PubMed] [Google Scholar]
- 101.Cheishvili D, Maayan C, Smith Y, Ast G, Razin A. IKAP/hELP1 deficiency in the cerebrum of familial dysautonomia patients results in down regulation of genes involved in oligodendrocyte differentiation and in myelination. Hum Mol Genet. 2007;16:2097–104. doi: 10.1093/hmg/ddm157. [DOI] [PubMed] [Google Scholar]
- 102.Andresen BS, Dobrowolski SF, O'Reilly L, Muenzer J, McCandless SE, Frazier DM, Udvari S, Bross P, Knudsen I, Banas R, Chace DH, Engel P, Naylor EW, Gregersen N. Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am J Hum Genet. 2001;68:1408–18. doi: 10.1086/320602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nielsen KB, Sorensen S, Cartegni L, Corydon TJ, Doktor TK, Schroeder LD, Reinert LS, Elpeleg O, Krainer AR, Gregersen N, Kjems J, Andresen BS. Seemingly neutral polymorphic variants may confer immunity to splicing-inactivating mutations: a synonymous SNP in exon 5 of MCAD protects from deleterious mutations in a flanking exonic splicing enhancer. Am J Hum Genet. 2007;80:416–32. doi: 10.1086/511992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69:3584–96. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Acharya KK, Govind CK, Shore AN, Stoler MH, Reddi PP. cis-requirement for the maintenance of round spermatid-specific transcription. Dev Biol. 2006;295:781–90. doi: 10.1016/j.ydbio.2006.04.443. [DOI] [PubMed] [Google Scholar]
- 106.Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008;13:867–78. doi: 10.2741/2727. [DOI] [PubMed] [Google Scholar]
- 107.Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. Embo J. 2001;20:1774–84. doi: 10.1093/emboj/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ayala YM, Pantano S, D'Ambrogio A, Buratti E, Brindisi A, Marchetti C, Romano M, Baralle FE. Human, Drosophila, and C.elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol. 2005;348:575–88. doi: 10.1016/j.jmb.2005.02.038. [DOI] [PubMed] [Google Scholar]
- 109.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 110.Zhang YJ, Xu YF, Dickey CA, Buratti E, Baralle F, Bailey R, Pickering-Brown S, Dickson D, Petrucelli L. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci. 2007;27:10530–4. doi: 10.1523/JNEUROSCI.3421-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ayala YM, Misteli T, Baralle FE. TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc Natl Acad Sci U S A. 2008;105:3785–9. doi: 10.1073/pnas.0800546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–72. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Huh GS, Hynes RO. Regulation of alternative pre-mRNA splicing by a novel repeated hexanucleotide element. Genes Dev. 1994;8:1561–74. doi: 10.1101/gad.8.13.1561. [DOI] [PubMed] [Google Scholar]
- 114.Hui J, Bindereif A. Alternative pre-mRNA splicing in the human system: unexpected role of repetitive sequences as regulatory elements. Biol Chem. 2005;386:1265–71. doi: 10.1515/BC.2005.143. [DOI] [PubMed] [Google Scholar]
- 115.Laule M, Meisel C, Prauka I, Cascorbi I, Malzahn U, Felix SB, Baumann G, Roots I, Stangl K, Stangl V. Interaction of CA repeat polymorphism of the endothelial nitric oxide synthase and hyperhomocysteinemia in acute coronary syndromes: evidence of gender-specific differences. J Mol Med. 2003;81:305–9. doi: 10.1007/s00109-003-0433-z. [DOI] [PubMed] [Google Scholar]
- 116.Hung LH, Heiner M, Hui J, Schreiner S, Benes V, Bindereif A. Diverse roles of hnRNP L in mammalian mRNA processing: a combined microarray and RNAi analysis. Rna. 2008;14:284–96. doi: 10.1261/rna.725208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ranum LP, Cooper TA. RNA-Mediated Neuromuscular Disorders. Annu Rev Neurosci. 2006 doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- 118.Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sorek R, Ast G, Graur D. Alu-containing exons are alternatively spliced. Genome Res. 2002;12:1060–7. doi: 10.1101/gr.229302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Knebelmann B, Forestier L, Drouot L, Quinones S, Chuet C, Benessy F, Saus J, Antignac C. Splice-mediated insertion of an Alu sequence in the COL4A3 mRNA causing autosomal recessive Alport syndrome. Hum Mol Genet. 1995;4:675–9. doi: 10.1093/hmg/4.4.675. [DOI] [PubMed] [Google Scholar]
- 121.Varon R, Gooding R, Steglich C, Marns L, Tang H, Angelicheva D, Yong KK, Ambrugger P, Reinhold A, Morar B, Baas F, Kwa M, Tournev I, Guerguelcheva V, Kremensky I, Lochmuller H, Mullner-Eidenbock A, Merlini L, Neumann L, Burger J, Walter M, Swoboda K, Thomas PK, von Moers A, Risch N, Kalaydjieva L. Partial deficiency of the C-terminal-domain phosphatase of RNA polymerase II is associated with congenital cataracts facial dysmorphism neuropathy syndrome. Nat Genet. 2003;35:185–9. doi: 10.1038/ng1243. [DOI] [PubMed] [Google Scholar]
- 122.Vervoort R, Gitzelmann R, Lissens W, Liebaers I. A mutation (IVS8+0.6kbdelTC) creating a new donor splice site activates a cryptic exon in an Alu-element in intron 8 of the human beta-glucuronidase gene. Hum Genet. 1998;103:686–93. doi: 10.1007/pl00008709. [DOI] [PubMed] [Google Scholar]
- 123.Venables JP. Unbalanced alternative splicing and its significance in cancer. Bioessays. 2006;28:378–86. doi: 10.1002/bies.20390. [DOI] [PubMed] [Google Scholar]
- 124.Skotheim RI, Nees M. Alternative splicing in cancer: noise, functional, or systematic? Int J Biochem Cell Biol. 2007;39:1432–49. doi: 10.1016/j.biocel.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 125.Stickeler E, Kittrell F, Medina D, Berget SM. Stage-specific changes in SR splicing factors and alternative splicing in mammary tumorigenesis. Oncogene. 1999;18:3574–82. doi: 10.1038/sj.onc.1202671. [DOI] [PubMed] [Google Scholar]
- 126.Fischer DC, Noack K, Runnebaum IB, Watermann DO, Kieback DG, Stamm S, Stickeler E. Expression of splicing factors in human ovarian cancer. Oncol Rep. 2004;11:1085–1090. [PubMed] [Google Scholar]
- 127.Forch P, Valcarcel J. Molecular mechanisms of gene expression regulation by the apoptosis-promoting protein TIA-1. Apoptosis. 2001;6:463–8. doi: 10.1023/a:1012441824719. [DOI] [PubMed] [Google Scholar]
- 128.Izquierdo JM, Valcarcel J. Fas-activated serine/threonine kinase (FAST K) synergizes with TIA-1/TIAR proteins to regulate Fas alternative splicing. J Biol Chem. 2007;282:1539–43. doi: 10.1074/jbc.C600198200. [DOI] [PubMed] [Google Scholar]
- 129.Zerbe LK, Pino I, Pio R, Cosper PF, Dwyer-Nield LD, Meyer AM, Port JD, Montuenga LM, Malkinson AM. Relative amounts of antagonistic splicing factors, hnRNP A1 and ASF/SF2, change during neoplastic lung growth: implications for pre-mRNA processing. Mol Carcinog. 2004;41:187–96. doi: 10.1002/mc.20053. [DOI] [PubMed] [Google Scholar]
- 130.Ghigna C, Moroni M, Porta C, Riva S, Biamonti G. Altered Expression of Heterogeneous Nuclear Ribonucleoproteins and SR Factors in Human Colon Adenocarcinomas. Cancer Research. 1998;58:5818–5824. [PubMed] [Google Scholar]
- 131.Ghigna C, Giordano S, Shen H, Benvenuto F, Castiglioni F, Comoglio PM, Green MR, Riva S, Biamonti G. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell. 2005;20:881–90. doi: 10.1016/j.molcel.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 132.Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007;14:185–93. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Neklason DW, Solomon CH, Dalton AL, Kuwada SK, Burt RW. Intron 4 mutation in APC gene results in splice defect and attenuated FAP phenotype. Fam Cancer. 2004;3:35–40. doi: 10.1023/B:FAME.0000026824.85766.22. [DOI] [PubMed] [Google Scholar]
- 134.Kurahashi H, Takami K, Oue T, Kusafuka T, Okada A, Tawa A, Okada S, Nishisho I. Biallelic inactivation of the APC gene in hepatoblastoma. Cancer Res. 1995;55:5007–11. [PubMed] [Google Scholar]
- 135.Samowitz WS, Thliveris A, Spirio LN, White R. Alternatively spliced adenomatous polyposis coli (APC) gene transcripts that delete exons mutated in attenuated APC. Cancer Res. 1995;55:3732–4. [PubMed] [Google Scholar]
- 136.Xia L, St Denis KA, Bapat B. Evidence for a novel exon in the coding region of the adenomatous polyposis coli (APC) gene. Genomics. 1995;28:589–91. doi: 10.1006/geno.1995.1195. [DOI] [PubMed] [Google Scholar]
- 137.Bala S, Kraus C, Wijnen J, Meera Khan P, Ballhausen WG. Multiple products in the protein truncation test due to alternative splicing in the adenomatous polyposis coli (APC) gene. Hum Genet. 1996;98:528–33. doi: 10.1007/s004390050254. [DOI] [PubMed] [Google Scholar]
- 138.Tanko Q, Franklin B, Lynch H, Knezetic J. A hMLH1 genomic mutation and associated novel mRNA defects in a hereditary non-polyposis colorectal cancer family. Mutat Res. 2002;503:37–42. doi: 10.1016/s0027-5107(02)00031-3. [DOI] [PubMed] [Google Scholar]
- 139.Clarke LA, Veiga I, Isidro G, Jordan P, Ramos JS, Castedo S, Boavida MG. Pathological exon skipping in an HNPCC proband with MLH1 splice acceptor site mutation. Genes Chromosomes Cancer. 2000;29:367–70. [PubMed] [Google Scholar]
- 140.Nystrom-Lahti M, Holmberg M, Fidalgo P, Salovaara R, de la Chapelle A, Jiricny J, Peltomaki P. Missense and nonsense mutations in codon 659 of MLH1 cause aberrant splicing of messenger RNA in HNPCC kindreds. Genes Chromosomes Cancer. 1999;26:372–5. doi: 10.1002/(sici)1098-2264(199912)26:4<372::aid-gcc12>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 141.Tournier I, Vezain M, Martins A, Charbonnier F, Baert-Desurmont S, Olschwang S, Wang Q, Buisine MP, Soret J, Tazi J, Frébourg T, Tosi M. A large fraction of unclassified variants of the mismatch repair genes MLH1 and MSH2 is associated with splicing defects. Hum Mut. 2008 doi: 10.1002/humu.20796. in press. [DOI] [PubMed] [Google Scholar]
- 142.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 143.Liu HX, Cartegni L, Zhang MQ, Krainer AR. A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat Genet. 2001;27:55–8. doi: 10.1038/83762. [DOI] [PubMed] [Google Scholar]
- 144.Hoffman JD, Hallam SE, Venne VL, Lyon E, Ward K. Implications of a novel cryptic splice site in the BRCA1 gene. Am J Med Genet. 1998;80:140–4. [PubMed] [Google Scholar]
- 145.Orban TI, Olah E. Emerging roles of BRCA1 alternative splicing. Mol Pathol. 2003;56:191–7. doi: 10.1136/mp.56.4.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Serra E, Ars E, Ravella A, Sanchez A, Puig S, Rosenbaum T, Estivill X, Lazaro C. Somatic NF1 mutational spectrum in benign neurofibromas: mRNA splice defects are common among point mutations. Hum Genet. 2001;108:416–29. doi: 10.1007/s004390100514. [DOI] [PubMed] [Google Scholar]
- 147.Colapietro P, Gervasini C, Natacci F, Rossi L, Riva P, Larizza L. NF1 exon 7 skipping and sequence alterations in exonic splice enhancers (ESEs) in a neurofibromatosis 1 patient. Hum Genet. 2003;113:551–4. doi: 10.1007/s00439-003-1009-2. [DOI] [PubMed] [Google Scholar]
- 148.De Klein A, Riegman PH, Bijlsma EK, Heldoorn A, Muijtjens M, den Bakker MA, Avezaat CJ, Zwarthoff EC. A G--> A transition creates a branch point sequence and activation of a cryptic exon, resulting in the hereditary disorder neurofibromatosis 2. Hum Mol Genet. 1998;7:393–8. doi: 10.1093/hmg/7.3.393. [DOI] [PubMed] [Google Scholar]
- 149.Sazani P, Kole R. Therapeutic potential of antisense oligonucleotides as modulators of alternative splicing. J Clin Invest. 2003;112:481–6. doi: 10.1172/JCI19547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.van Ommen GJ, van Deutekom J, Aartsma-Rus A. The therapeutic potential of antisense-mediated exon skipping. Curr Opin Mol Ther. 2008;10:140–9. [PubMed] [Google Scholar]
- 151.Lebleu B, Moulton HM, Abes R, Ivanova GD, Abes S, Stein DA, Iversen PL, Arzumanov AA, Gait MJ. Cell penetrating peptide conjugates of steric block oligonucleotides. Adv Drug Deliv Rev. 2008;60:517–29. doi: 10.1016/j.addr.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Abes R, Arzumanov A, Moulton H, Abes S, Ivanova G, Gait MJ, Iversen P, Lebleu B. Arginine-rich cell penetrating peptides: Design, structure-activity, and applications to alter pre-mRNA splicing by steric-block oligonucleotides. J Pept Sci. 2008;14:455–60. doi: 10.1002/psc.979. [DOI] [PubMed] [Google Scholar]
- 153.Lacerra G, Sierakowska H, Carestia C, Fucharoen S, Summerton J, Weller D, Kole R. Restoration of hemoglobin A synthesis in erythroid cells from peripheral blood of thalassemic patients. Proc Natl Acad Sci U S A. 2000;97:9591–6. doi: 10.1073/pnas.97.17.9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mann CJ, Honeyman K, Cheng AJ, Ly T, Lloyd F, Fletcher S, Morgan JE, Partridge TA, Wilton SD. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc Natl Acad Sci U S A. 2001;98:42–7. doi: 10.1073/pnas.011408598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Aartsma-Rus A, van Ommen GJ. Antisense-mediated exon skipping: a versatile tool with therapeutic and research applications. Rna. 2007;13:1609–24. doi: 10.1261/rna.653607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Friedman KJ, Kole J, Cohn JA, Knowles MR, Silverman LM, Kole R. Correction of aberrant splicing of the cystic fibrosis transmembrane conductance regulator (CFTR) gene by antisense oligonucleotides. J Biol Chem. 1999;274:36193–36199. doi: 10.1074/jbc.274.51.36193. [DOI] [PubMed] [Google Scholar]
- 157.Asparuhova M, Kole R, Schumperli D. Antisense derivatives of U7 and other small nuclear RNAs as tools to modify pre-mRNA splicing patterns. Gene Ther Regualtion. 2005 in press. [Google Scholar]
- 158.Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005;11:440–5. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu S, Asparuhova M, Brondani V, Ziekau I, Klimkait T, Schumperli D. Inhibition of HIV-1 multiplication by antisense U7 snRNAs and siRNAs targeting cyclophilin A. Nucleic Acids Res. 2004;32:3752–9. doi: 10.1093/nar/gkh715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kalbfuss B, Mabon SA, Misteli T. Correction of alternative splicing of tau in frontotemporal dementia and parkinsonism linked to chromosome 17. J Biol Chem. 2001;276:42986–93. doi: 10.1074/jbc.M105113200. [DOI] [PubMed] [Google Scholar]
- 161.Takeshima Y, Wada H, Yagi M, Ishikawa Y, Minami R, Nakamura H, Matsuo M. Oligonucleotides against a splicing enhancer sequence led to dystrophin production in muscle cells from a Duchenne muscular dystrophy patient. Brain Dev. 2001;23:788–90. doi: 10.1016/s0387-7604(01)00326-6. [DOI] [PubMed] [Google Scholar]
- 162.Bruno IG, Jin W, Cote GJ. Correction of aberrant FGFR1 alternative RNA splicing through targeting of intronic regulatory elements. Hum Mol Genet. 2004;13:2409–20. doi: 10.1093/hmg/ddh272. [DOI] [PubMed] [Google Scholar]
- 163.Cartegni L, Krainer AR. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat Struct Biol. 2003;10:120–5. doi: 10.1038/nsb887. [DOI] [PubMed] [Google Scholar]
- 164.Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc Natl Acad Sci U S A. 2003;100:4114–9. doi: 10.1073/pnas.0633863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Villemaire J, Dion I, Elela SA, Chabot B. Reprogramming alternative pre-messenger RNA splicing through the use of protein-binding antisense oligonucleotides. J Biol Chem. 2003;278:50031–9. doi: 10.1074/jbc.M308897200. [DOI] [PubMed] [Google Scholar]
- 166.Sumanasekera C, Watt DS, Stamm S. Substances that can change alternative splice-site selection. Biochem Soc Trans. 2008;36:483–490. doi: 10.1042/BST0360483. [DOI] [PubMed] [Google Scholar]
- 167.Bakkour N, Lin YL, Maire S, Ayadi L, Mahuteau-Betzer F, Nguyen CH, Mettling C, Portales P, Grierson D, Chabot B, Jeanteur P, Branlant C, Corbeau P, Tazi J. Small-molecule inhibition of HIV pre-mRNA splicing as a novel antiretroviral therapy to overcome drug resistance. PLoS Pathog. 2007;3:1530–9. doi: 10.1371/journal.ppat.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Slaugenhaupt SA, Blumenfeld A, Gill SP, Leyne M, Mull J, Cuajungco MP, Liebert CB, Chadwick B, Idelson M, Reznik L, Robbins C, Makalowska I, Brownstein M, Krappmann D, Scheidereit C, Maayan C, Axelrod FB, Gusella JF. Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am J Hum Genet. 2001;68:598–605. doi: 10.1086/318810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, Li D, Payami H, Awert F, Markopoulou K, Andreadis A, D'Souza I, Lee VM, Reed L, Trojanowski JQ, Zhukareva V, Bird T, Schellenberg G, Wilhelmsen KC. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci U S A. 1998;95:13103–7. doi: 10.1073/pnas.95.22.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rizzu P, Van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A, Niermeijer MF, Hillebrand M, Ravid R, Oostra BA, Goedert M, van Duijn CM, Heutink P. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet. 1999;64:414–21. doi: 10.1086/302256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.D'Souza I, Poorkai P, Hong M, Nochlin D, Lee VMY, Bird TD, Schellenberg GD. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA. 1999;96:5598–5603. doi: 10.1073/pnas.96.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Iijima M, Tabira T, Poorkaj P, Schellenberg GD, Trojanowski JQ, Lee VM, Schmidt ML, Takahashi K, Nabika T, Matsumoto T, Yamashita Y, Yoshioka S, Ishino H. A distinct familial presenile dementia with a novel missense mutation in the tau gene. Neuroreport. 1999;10:497–501. doi: 10.1097/00001756-199902250-00010. [DOI] [PubMed] [Google Scholar]
- 173.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene [see comments] Cell. 1995;80:155–65. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 174.Charames GS, Cheng H, Gilpin CA, Hunter AG, Berk T, Bapat B. A novel aberrant splice site mutation in the APC gene. J Med Genet. 2002;39:754–7. doi: 10.1136/jmg.39.10.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wang M, Dotzlaw H, Fuqua SA, Murphy LC. A point mutation in the human estrogen receptor gene is associated with the expression of an abnormal estrogen receptor mRNA containing a 69 novel nucleotide insertion. Breast Cancer Res Treat. 1997;44:145–51. doi: 10.1023/a:1005753117205. [DOI] [PubMed] [Google Scholar]
- 176.Krieg A, Mahotka C, Krieg T, Grabsch H, Muller W, Takeno S, Suschek CV, Heydthausen M, Gabbert HE, Gerharz CD. Expression of different survivin variants in gastric carcinomas: first clues to a role of survivin-2B in tumour progression. Br J Cancer. 2002;86:737–43. doi: 10.1038/sj.bjc.6600153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Vegran F, Boidot R, Oudin C, Riedinger JM, Lizard-Nacol S. Distinct expression of Survivin splice variants in breast carcinomas. Int J Oncol. 2005;27:1151–7. [PubMed] [Google Scholar]
- 178.Cheung N, Wong MP, Yuen ST, Leung SY, Chung LP. Tissue-specific expression pattern of vascular endothelial growth factor isoforms in the malignant transformation of lung and colon. Hum Pathol. 1998;29:910–4. doi: 10.1016/s0046-8177(98)90195-2. [DOI] [PubMed] [Google Scholar]
- 179.Keppler D, Sloane BF. Cathepsin B: multiple enzyme forms from a single gene and their relation to cancer. Enzyme Protein. 1996;49:94–105. doi: 10.1159/000468619. [DOI] [PubMed] [Google Scholar]
- 180.McIver B, Grebe SK, Wang L, Hay ID, Yokomizo A, Liu W, Goellner JR, Grant CS, Smith DI, Eberhardt NL. FHIT and TSG101 in thyroid tumours: aberrant transcripts reflect rare abnormal RNA processing events of uncertain pathogenetic or clinical significance. Clin Endocrinol (Oxf) 2000;52:749–57. [PubMed] [Google Scholar]
- 181.Lee SH, Kim CJ, Park HK, Koh JW, Cho MH, Baek MJ, Lee MS. Characterization of aberrant FHIT transcripts in gastric adenocarcinomas. Exp Mol Med. 2001;33:124–30. doi: 10.1038/emm.2001.22. [DOI] [PubMed] [Google Scholar]
- 182.Lee SH, Kim HY, Kim TJ, Park HK, Kim WH, Woo KM, Cho MH. Aberrant splicing of FHIT transcripts in human gastric cancer cell lines. Res Commun Mol Pathol Pharmacol. 2002;112:39–49. [PubMed] [Google Scholar]
- 183.Huiping C, Kristjansdottir S, Bergthorsson JT, Jonasson JG, Magnusson J, Egilsson V, Ingvarsson S. High frequency of LOH, MSI and abnormal expression of FHIT in gastric cancer. Eur J Cancer. 2002;38:728–35. doi: 10.1016/s0959-8049(01)00432-4. [DOI] [PubMed] [Google Scholar]
- 184.Kraggerud SM, Aman P, Holm R, Stenwig AE, Fossa SD, Nesland JM, Lothe RA. Alterations of the fragile histidine triad gene, FHIT, and its encoded products contribute to testicular germ cell tumorigenesis. Cancer Res. 2002;62:512–7. [PubMed] [Google Scholar]
- 185.Terry G, Ho L, Londesborough P, Cuzick J. Abnormal FHIT expression profiles in cervical intraepithelial neoplastic (CIN) lesions. Br J Cancer. 2002;86:376–81. doi: 10.1038/sj.bjc.6600077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Honda K, Yamada T, Seike M, Hayashida Y, Idogawa M, Kondo T, Ino Y, Hirohashi S. Alternative splice variant of actinin-4 in small cell lung cancer. Oncogene. 2004;23:5257–62. doi: 10.1038/sj.onc.1207652. [DOI] [PubMed] [Google Scholar]
- 187.Reiter R, Wellstein A, Riegel AT. An isoform of the coactivator AIB1 that increases hormone and growth factor sensitivity is overexpressed in breast cancer. J Biol Chem. 2001;276:39736–41. doi: 10.1074/jbc.M104744200. [DOI] [PubMed] [Google Scholar]
- 188.Collesi C, Santoro MM, Gaudino G, Comoglio PM. A splicing variant of the RON transcript induces constitutive tyrosine kinase activity and an invasive phenotype. Mol Cell Biol. 1996;16:5518–26. doi: 10.1128/mcb.16.10.5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhou YQ, He C, Chen YQ, Wang D, Wang MH. Altered expression of the RON receptor tyrosine kinase in primary human colorectal adenocarcinomas: generation of different splicing RON variants and their oncogenic potential. Oncogene. 2003;22:186–97. doi: 10.1038/sj.onc.1206075. [DOI] [PubMed] [Google Scholar]