

The crystal structure of the starch-binding domain of glucoamylase shows that the conformation of the region surrounding the disulfide bond is highly flexible.
Keywords: starch-binding domain, glucoamylase, β-sheet structure, disulfide bond, Aspergillus niger
Abstract
Glucoamylases are widely used commercially to produce glucose syrup from starch. The starch-binding domain (SBD) of glucoamylase from Aspergillus niger is a small globular protein containing a disulfide bond. The structure of A. niger SBD has been determined by NMR, but the conformation surrounding the disulfide bond was unclear. Therefore, X-ray crystal structural analysis was used to attempt to clarify the conformation of this region. The SBD was purified from an Escherichia coli-based expression system and crystallized at 293 K. The initial phase was determined by the molecular-replacement method, and the asymmetric unit of the crystal contained four protomers, two of which were related by a noncrystallographic twofold axis. Finally, the structure was solved at 2.0 Å resolution. The SBD consisted of seven β-strands and eight loops, and the conformation surrounding the disulfide bond was determined from a clear electron-density map. Comparison of X-ray- and NMR-determined structures of the free SBD showed no significant difference in the conformation of each β-strand, but the conformations of the loops containing the disulfide bond and the L5 loop were different. In particular, the difference in the position of the Cα atom of Cys509 between the X-ray- and NMR-determined structures was 13.3 Å. In addition, the B factors of the amino-acid residues surrounding the disulfide bond are higher than those of other residues. Therefore, the conformation surrounding the disulfide bond is suggested to be highly flexible.
1. Introduction
Glucoamylase (α-1,4-d-glucan glucohydrolase; EC 3.2.1.3) hydrolyzes the α-1,4- and α-1,6-glucosidic linkages of α-1,4-d-glucans such as starch, thereby liberating β-d-glucose (Koshland, 1953 ▸; Pazur & Ando, 1960 ▸; Hiromi, Hamauzu et al., 1966 ▸; Hiromi, Takahashi et al., 1966 ▸; Fierobe et al., 1998 ▸). Glucoamylases are widely used commercially, for example in the production of glucose syrup from starch. The glucoamylases from Aspergillus niger, A. awamori and Rhizopus oryzae are especially important for industrial applications (Reilly, 1999 ▸; Pandey et al., 2000 ▸; Pedersen et al., 2000 ▸; Norouzian et al., 2006 ▸). These glucoamylases contain a starch-binding domain (SBD) in the full-length protein that binds to starch and enhances the amylolytic rate (Hayashida et al., 1989 ▸; Southall et al., 1999 ▸; Rodríguez-Sanoja et al., 2005 ▸). Three-dimensional structures of two glucoamylase SBDs have been analyzed. The first is from R. oryzae, belongs to carbohydrate-binding module (CBM) family 21 and its three-dimensional structure has been determined by NMR and X-ray crystallography, whereas the other is from A. niger, belongs to CBM family 20 and its three-dimensional structure has been determined by NMR (Liu et al., 2007 ▸; Tung et al., 2008 ▸). In both SBDs, two starch-binding sites were observed by structural analysis using bound β-cyclodextrin (β-CD), a cyclic starch analogue.
The full-length A. niger glucoamylase contains a catalytic domain and a C-terminal SBD. Thermal unfolding of both domains was observed by adiabatic differential scanning calorimetry (DSC; Tanaka et al., 1995 ▸). Unfolding of the catalytic domain is irreversible, whereas that of the SBD is reversible. A disulfide bond is formed in the SBD between Cys509 and Cys604 (Sorimachi et al., 1996 ▸). To elucidate the role of this disulfide bond in the SBD, we produced the wild type and several mutants lacking the disulfide bond, and thermodynamic studies were carried out (Tanaka et al., 1998 ▸; Sugimoto et al., 2007 ▸, 2009 ▸). The mutants are stabilized in terms of enthalpy, but a larger change in entropy overwhelms the enthalpic effect, resulting in destabilization. The three-dimensional structure of the glucoamylase SBD from A. niger was obtained by NMR, although the conformations of residues 509–512 and 601–606, which surround the disulfide bond, were unclear owing to a lack of nuclear Overhauser enhancement (NOE) constraints (Sorimachi et al., 1996 ▸). Therefore, the aim of this study was to reveal the detailed conformation surrounding the disulfide bond of the A. niger SBD by X-ray crystal structural analysis.
2. Materials and methods
2.1. Crystallization
The purification of an SBD-containing fragment of the wild-type glucoamylase from A. niger was performed as described previously (Tanaka et al., 1998 ▸). The purified protein was concentrated to 10 mg ml−1 and set up for crystallization at 293 K. The final optimized crystals were obtained from hanging-drop conditions using 2.0–2.3 M ammonium sulfate, 1.7%(w/v) PEG 400, 15%(v/v) glycerol, 85 mM HEPES pH 7.5 as the reservoir solution. The crystals grew to maximum dimensions of 0.3 × 0.4 × 0.3 mm in two weeks. For data collection, the crystals were flash-cooled in liquid nitrogen. Crystallization information is given in Table 1 ▸.
Table 1. Crystallization information.
| Method | Hanging-drop vapour diffusion |
| Plate type | 24-well plates |
| Temperature (K) | 293 |
| Protein concentration (mg ml−1) | 10 |
| Buffer composition of protein solution | 20 mM phosphate buffer pH 7.0 |
| Composition of reservoir solution | 2.0–2.3 M ammonium sulfate, 1.7%(w/v) PEG 400, 15%(v/v) glycerol, 85 mM HEPES pH 7.5 |
| Volume and ratio of drop | 4 µl (1:1 protein:reservoir solution) |
| Volume of reservoir (ml) | 0.45 |
2.2. Data collection and processing
Multiple crystals were screened to identify a crystal with reasonable diffraction. The best data set was collected to 2.0 Å resolution under gaseous nitrogen (100 K) on beamline AR-NW12A at the Photon Factory, Tsukuba, Japan using X-rays at a wavelength of 0.978 Å. Data sets were processed and scaled using HKL-2000 (Otwinowski & Minor, 1997 ▸). Data-collection and processing information is shown in Table 2 ▸.
Table 2. Data-collection and processing information.
Values in parentheses are for the outer shell.
| Diffraction source | Beamline AR-NW12A, Photon Factory |
| Wavelength (Å) | 0.978 |
| Temperature (K) | 100 |
| Detector | ADSC Quantum 210 CCD |
| Crystal-to-detector distance (mm) | 150 |
| Rotation range per image (°) | 1 |
| Total rotation range (°) | 200 |
| Exposure time per image (s) | 1 |
| Space group | P4 |
| a, b, c (Å) | 75.36, 75.36, 91.01 |
| α, β, γ (°) | 90, 90, 90 |
| Mosaicity (°) | 0.18–0.29 |
| Resolution range (Å) | 50.00–2.00 (2.03–2.00) |
| Total No. of reflections | 291560 |
| No. of unique reflections | 34452 (1710) |
| Completeness (%) | 99.9 (100) |
| Multiplicity | 8.5 (8.2) |
| 〈I/σ(I)〉 | 79.8 (41.7) |
| R merge † | 0.107 (0.164) |
| Overall B factor from Wilson plot (Å2) | 20.6 |
R
merge = 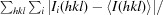
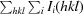 , where Ii(hkl) is the ith measurement and I(hkl) is the weighted mean of all measurements of I (hkl).
, where Ii(hkl) is the ith measurement and I(hkl) is the weighted mean of all measurements of I (hkl).
2.3. Structure solution and refinement
A molecular-replacement search using the program BALBES (Long et al., 2008 ▸) and the FASTA sequence of SBD was performed to obtain the initial phases. Model building was performed using Coot (Emsley & Cowtan, 2004 ▸) in iterative cycles with refinement using REFMAC5 (Murshudov et al., 2011 ▸). Details of the structure solution and model refinement are shown in Table 3 ▸. Figures were prepared using PyMOL (v.1.7; Schrödinger).
Table 3. Structure-solution and refinement information.
Values in parentheses are for the outer shell.
| Resolution range (Å) | 45.98–2.00 (2.052–2.000) |
| Completeness (%) | 99.9 |
| No. of reflections, working set | 32650 (2401) |
| No. of reflections, test set | 1734 (106) |
| Final R cryst | 0.178 (0.200) |
| Final R free | 0.229 (0.274) |
| No. of non-H atoms | |
| Protein | 3351 |
| Ion | 20 |
| Ligand | 24 |
| Solvent | 204 |
| Total | 3599 |
| R.m.s. deviations | |
| Bonds (Å) | 0.021 |
| Angles (°) | 2.157 |
| Average B factors (Å2) | |
| Protein | 26.35 |
| Ion | 46.30 |
| Ligand | 54.59 |
| Water | 33.81 |
| Ramachandran statistics† (%) | |
| Favoured | 96.46 |
| Allowed | 2.12 |
| Outliers | 1.42 |
As determined by MolProbity (Chen et al., 2010 ▸).
3. Results and discussion
3.1. Overall structure
The SBD-containing polypeptide (amino acids 509–616) of A. niger glucoamylase was purified from an Escherichia coli-based expression system (Tanaka et al., 1998 ▸) and the purified protein was crystallized at 293 K. The NMR structures of the SBD of A. niger glucoamylase (PDB entries 1kum, 1kul, 1aco and 1acz; Sorimachi et al., 1996 ▸, 1997 ▸) were used as search models for molecular replacement in CCP4 (Winn et al., 2011 ▸) but no phases were derived. A molecular-replacement search using BALBES (Long et al., 2008 ▸) and the FASTA sequence of SBD was then completed, yielding a clear solution with one tetramer in the asymmetric unit. Finally, the structure was solved at 2.0 Å resolution. Four SBD molecules were observed in the asymmetric unit (Fig. 1 ▸ a). The SBD exists as a monomer in solution, but forms a dimer with adjacent molecules in the crystal, two of which are related by a noncrystallographic twofold axis. Thr513 in molecules A and C forms hydrophobic interactions with the corresponding residues in molecules B and D, respectively (Fig. 1 ▸ b). In a similar manner, Pro570 in molecules A and C forms hydrophobic interactions with the corresponding residues in molecules B and D, respectively (Fig. 1 ▸ b). These interactions are considered to be caused by crystal packing. No interactions between the molecules were observed elsewhere. The SBD may have assumed a dimeric structure for crystal packing. The root-mean-square deviation (r.m.s.d.) of each molecule was 0.114–0.119 Å. The four SBD molecules showed almost the same structure in the crystal. The SBD was composed of seven β-strands and eight loops, with one glycerol molecule and one sulfate ion present per SBD molecule (Fig. 1 ▸ c). These ligands are present in the reservoir solution and were therefore considered to be included in the crystal structure.
Figure 1.

Crystal structure of the SBD (PDB entry 5ghl). (a) The asymmetric unit of the SBD is composed of four protein molecules, labelled A–D, which form two dimers (green and blue). (b) Interactions between the protein molecules. Thr513 and Pro570 in molecules A and C form hydrophobic interactions with the corresponding residues in molecules B and D, respectively. (c) Ribbon representation of the SBD, which consists of seven β-strands (β1–β7) and eight loops (L1–L8). A disulfide bond, a glycerol molecule and a sulfate ion are shown in stick representation. (d) Electron-density map surrounding the disulfide bond. The weighted 2F o − F c map (blue) is contoured at 1σ.
3.2. Conformation surrounding the disulfide bond
Cys509 and Cys604 are at the N- and C-termini, respectively, and they are linked by a disulfide bond (Sorimachi et al., 1996 ▸). The presence of the disulfide bond was detected by NMR, but the conformation surrounding the disulfide bonds was unclear because the 15N resonances were too broad (Sorimachi et al., 1996 ▸, 1997 ▸). In this study, the X-ray crystal structure analysis showed electron density corresponding to a disulfide bond between Cys509 and Cys604, and the surrounding structure was revealed (Fig. 1 ▸ d). The average B factor of all amino-acid residues of the SBD was 26.35 Å2, but the B factor of the loop between residues Cys509 and Thr602 was as high as 44.22 Å2. Together with the NMR-determined SBD structure, these results suggest that the conformation surrounding the disulfide bond is highly flexible.
3.3. Comparison of X-ray and NMR structures of the SBD
The X-ray and NMR structures (PDB entry 1kum) of free A. niger glucoamylase SBD were compared, and the r.m.s.d. was 1.91 Å. No significant difference in the conformation of each β-strand was found, but the conformations of the loops (L1 and L7) containing the disulfide bond and the L5 loop were different (Fig. 2 ▸ a). In particular, the difference in the position of the Cα atom of Cys509 between the X-ray and NMR structures was 13.3 Å. In contrast, in comparison with the NMR-determined structure of the SBD–β-CD complex (PDB entry 1ac0) the r.m.s.d. was 2.9 Å, and the conformational change of the L5 loop was smaller than that of the free SBD structure determined by NMR. However, the L3 loop flipped by about 90°, and the difference in position of the Cα atom of Ser559 in the L3 loop between the free X-ray structure and the NMR-determined structure of the SBD–β-CD complex was 15.5 Å (Fig. 2 ▸ b). In both X-ray- and NMR-determined structures of free SBD, the L3 loop showed the same conformation, and thus binding of β-CD to the SBD seems to flip the loop largely owing to interaction with β-CD.
Figure 2.

Overlay of the A. niger free SBD structure determined by X-ray diffraction (cyan) with (a) the A. niger free SBD structure determined by NMR (orange; PDB entry 1kum), (b) the A. niger SBD–β-CD complex structure determined by NMR (yellow; PDB entry 1ac0) and (c) the R. oryzae SBD–β-CD complex structure determined by X-ray diffraction (green; PDB entry 2v8l). The NMR-determined structures of SBD used were minimized average structures.
3.4. Comparison with the glucoamylase SBD from R. oryzae
The three-dimensional structure of only the SBD from R. oryzae glucoamylase has been determined (Liu et al., 2007 ▸; Tung et al., 2008 ▸). Therefore, we compared the A. niger free SBD structure determined by X-ray diffraction and that of the SBD from R. oryzae complexed with β-CD (PDB entry 2v8l; Tung et al., 2008 ▸; Fig. 2 ▸ c). The r.m.s.d. was 5.89 Å owing to the A. niger SBD being composed of seven β-strands and the R. oryzae SBD of nine β-strands. In addition, β-strands β2 and β3 in A. niger SBD were parallel, whereas all of the β-strands in the R. oryzae SBD were antiparallel. Furthermore, the structures surrounding the N- and C-termini were quite different in the two SBDs. Most glucoamylases are composed of a catalytic domain and an SBD; the SBD of A. niger glucoamylase is located at the C-terminus, whereas that of R. oryzae is located at the N-terminus. This difference in the position of the SBD relative to the catalytic domain probably caused the difference in structure near the N- and C-termini. Thus, the position of the β-CD-binding site was probably also different.
In our previous study, thermodynamic analysis of the A. niger SBD and its disulfide-bond-deficient mutants showed that the disulfide bond plays an important thermodynamic role in the stability of the protein. In this study, the conformation surrounding the disulfide bond of A. niger SBD was clarified using X-ray crystal structural analysis. In the future, structural analysis of disulfide-bond-deficient mutants will allow determination of the stabilizing effects of the disulfide bonds in glucoamylase SBDs.
Supplementary Material
PDB reference: starch-binding domain of glucoamylase, 5ghl
Acknowledgments
The authors are very grateful to Professor Akiyoshi Tanaka of Mie University for continued support of this study. This work was performed under the approval of the Photon Factory Program Advisory Committee (Proposal No. 2005G289).
References
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fierobe, H. P., Clarke, A. J., Tull, D. & Svensson, B. (1998). Biochemistry, 37, 3753–3759. [DOI] [PubMed]
- Hayashida, S., Nakahara, K., Kanlayakrit, W., Hara, T. & Teramoto, Y. (1989). Agric. Biol. Chem. 53, 143–149.
- Hiromi, K., Hamauzu, Z. I., Takahashi, K. & Ono, S. (1966). J. Biochem. 59, 411–418. [DOI] [PubMed]
- Hiromi, K., Takahashi, K., Hamauzu, Z. I. & Ono, S. (1966). J. Biochem. 59, 469–475. [DOI] [PubMed]
- Koshland, D. E. Jr (1953). Biol. Rev. 28, 416–436.
- Liu, Y.-N., Lai, Y.-T., Chou, W.-I., Chang, M. D.-T. & Lyu, P.-C. (2007). Biochem. J. 403, 21–30. [DOI] [PMC free article] [PubMed]
- Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. (2008). Acta Cryst. D64, 125–132. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Norouzian, D., Akbarzadeh, A., Scharer, J. M. & Moo Young, M. (2006). Biotechnol. Adv. 24, 80–85. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Pandey, A., Nigam, P., Soccol, C. R., Soccol, V. T., Singh, D. & Mohan, R. (2000). Biotechnol. Appl. Biochem. 31, 135–152. [DOI] [PubMed]
- Pazur, J. H. & Ando, T. (1960). J. Biol. Chem. 235, 297–302. [PubMed]
- Pedersen, H., Beyer, M. & Nielsen, J. (2000). Appl. Microbiol. Biotechnol. 53, 272–277. [DOI] [PubMed]
- Reilly, P. J. (1999). Starch/Stärke, 51, 269–274.
- Rodríguez-Sanoja, R., Oviedo, N. & Sánchez, S. (2005). Curr. Opin. Microbiol. 8, 260–267. [DOI] [PubMed]
- Sorimachi, K., Jacks, A. J., Le Gal-Coëffet, M. F., Williamson, G., Archer, D. B. & Williamson, M. P. (1996). J. Mol. Biol. 259, 970–987. [DOI] [PubMed]
- Sorimachi, K., Le Gal-Coëffet, M. F., Williamson, G., Archer, D. B. & Williamson, M. P. (1997). Structure, 5, 647–661. [DOI] [PubMed]
- Southall, S. M., Simpson, P. J., Gilbert, H. J., Williamson, G. & Williamson, M. P. (1999). FEBS Lett. 447, 58–60. [DOI] [PubMed]
- Sugimoto, H., Nakaura, M., Kosuge, Y., Imai, K., Miyake, H., Karita, S. & Tanaka, A. (2007). Biosci. Biotechnol. Biochem. 71, 1535–1541. [DOI] [PubMed]
- Sugimoto, H., Nakaura, M., Nishimura, S., Karita, S., Miyake, H. & Tanaka, A. (2009). Protein Sci. 18, 1715–1723. [DOI] [PMC free article] [PubMed]
- Tanaka, A., Fukada, H. & Takahashi, K. (1995). J. Biochem. 117, 1024–1028. [DOI] [PubMed]
- Tanaka, A., Karita, S., Kosuge, Y., Senoo, K., Obata, H. & Kitamoto, N. (1998). Biosci. Biotechnol. Biochem. 62, 2127–2132. [DOI] [PubMed]
- Tung, J.-Y., Chang, M. D.-T., Chou, W.-I., Liu, Y.-Y., Yeh, Y.-H., Chang, F.-Y., Lin, S.-C., Qiu, Z.-L. & Sun, Y.-J. (2008). Biochem. J. 416, 27–36. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: starch-binding domain of glucoamylase, 5ghl