

Abstract
Hypoxia is associated with aggressive phenotype and poor prognosis in prostate cancer (PCa) patients suggesting that PCa growth and progression could be controlled via targeting hypoxia-induced signaling and biological effects. Here, we analyzed silibinin (a natural flavonoid) efficacy to target cell growth, angiogenesis and metabolic changes in human PCa, LNCaP and 22Rv1 cells under hypoxic condition. Silibinin treatment inhibited the proliferation, clonogenicity and endothelial cells tube formation by hypoxic (1% O2) PCa cells. Interestingly, hypoxia promoted a lipogenic phenotype in PCa cells via activating acetyl-Co A carboxylase (ACC) and fatty acid synthase (FASN) that was inhibited by silibinin treatment. Importantly, silibinin treatment strongly decreased hypoxia-induced HIF-1α expression in PCa cells together with a strong reduction in hypoxia-induced NADPH oxidase (NOX) activity. HIF-1α overexpression in LNCaP cells significantly increased the lipid accumulation and NOX activity; however, silibinin treatment reduced HIF-1α expression, lipid levels, clonogenicity and NOX activity even in HIF-1α overexpressing LNCaP cells. In vivo, silibinin feeding (200 mg/kg body weight) to male nude mice with 22Rv1 tumors, specifically inhibited tumor vascularity (measured by dynamic contrast-enhanced MRI) resulting in tumor growth inhibition without directly inducing necrosis (as revealed by diffusion-weighted MRI). Silibinin feeding did not significantly affect tumor glucose uptake measured by FDG-PET; however, reduced the lipid synthesis measured by quantitative 1H-NMR metabolomics. IHC analyses of tumor tissues confirmed that silibinin feeding decreased proliferation and angiogenesis as well as reduced HIF-1α, FASN and ACC levels. Together, these findings further support silibinin usefulness against PCa through inhibiting hypoxia-induced signaling.
Keywords: Prostate cancer, Hypoxia, Hypoxia-inducible factor, Silibinin, Lipogenesis
INTRODUCTION
Prostate cancer (PCa) is the most common non-cutaneous malignancy in men, and according to the American Cancer Society reports, 180,890 new cases and 26,120 deaths from PCa are expected in the United States in 2016 [1]. To reduce PCa mortality, better understanding of the biological events that play fundamental role in prostate carcinogenesis, is needed. In this regard, hypoxia (low oxygen conditions) in PCa is associated with an aggressive phenotype, and plays an important role in PCa growth, promotion and metastatic progression as well as disease-relapse [2–4]. In fact, tumor hypoxia status is associated with poor prognosis and is the major reason for treatment failure [3,5–7]. Hypoxia induces genetic, metabolic and proteome reprogramming leading to increased glycolysis, angiogenesis and selection of resistant clones [2,8]. The key regulator of hypoxic response is the transcription factor hypoxia inducible factor 1 (HIF-1) [9–11], consisting of two subunits HIF-1β and HIF-1α. HIF-1β is constitutively expressed and its levels are unaffected by changes in cellular O2; while HIF-1α level is tightly regulated by O2 tension. Under normoxic conditions, HIF-1α is degraded via O2-dependent prolyl hydroxylation by prolyl-hydroxylases (PHDs), targeting the protein for ubiquitinylation by von Hippel-Lindau (VHL), an E3 ligase [11]. Under hypoxic conditions, prolyl hydroxylation of HIF-1α is impaired due to PHDs inactivation, leading to decreased ubiquitination by VHL and increased HIF-1α stability. HIF-1α is overexpressed in cancers as a result of: a) intra-tumoral hypoxia resulting in inhibition of PHDs; b) several metabolic and genetic alterations, such as hyper-activation of Erb-B2, RAS/MAPK, Src, etc.; and c) loss-of-function mutations in tumor-suppressor genes, e.g. VHL, PTEN and succinate dehydrogenase [4,11]. Furthermore, irrespective of oxygenation conditions, HIF-1α expression could also be increased through its enhanced translation via activation of PI3K/Akt/mTOR pathway [4,12]. Few studies have also suggested the role of NADPH oxidase (NOX) in HIF-1α activation [13–15]. Together, these studies suggest that targeting hypoxia-induced signaling and/or molecular regulators of HIF-1α could be an effective strategy towards preventing PCa growth and progression as well as to improve treatment success.
Silibinin, isolated from the Milk Thistle (Silybum marianum) seeds, is a widely-consumed dietary supplement due to its strong hepatoprotective activity [16,17]. Silibinin has shown remarkable efficacy, both in vitro and in vivo, against PCa cells as well as tumor microenvironment components (endothelial cells, cancer-associated fibroblasts, osteoclasts, etc.) [18–23]. Silibinin inhibits PCa growth and progression mainly through targeting cell cycle, apoptosis, epithelial-to-mesenchymal transition (EMT) and metabolism [18,20,24,25]. Earlier, Jung et al. reported that silibinin inhibits HIF-1α protein expression in PCa cells; however, hypoxic conditions in that study were mimicked by using cobalt chloride [26]. Consequently, the effect of silibinin on low oxygen-induced HIF-1α expression and associated biological effects in PCa cells remained mostly undefined. Accordingly, in the present study, we examined the effect of silibinin on hypoxia-induced signaling, proliferation, clonogenicity, angiogenesis and metabolites changes in PCa cells both in cell culture and xenograft model.
An additional important component in PCa cells is that they exhibit significantly higher endogenous lipid biosynthesis, which is linked with increased demand for membranes, energy storage, redox balance, protection from cell death, and activation of several intracellular signaling pathways during uncontrolled cellular proliferation [27–30]. Lipogenesis is a multistep process regulated by several enzymes and factors; however, acetyl-Co A carboxylase (ACC) catalyzes the first key step during fatty acid synthesis via irreversible carboxylation of acetyl-Co A to produce malonyl-Co A, which is the building block for fatty acid synthesis. Another key enzyme in lipogenesis is fatty acid synthase (FASN) that catalyzes the synthesis of fatty acids from malonyl-Co A and acetyl-Co A in the presence of NADPH. Both ACC and FASN are aberrantly expressed in cancer cells and are also considered important druggable targets [31,32]. Recently, we reported that under hypoxic conditions, PCa cells accumulate lipids to support proliferation and invasiveness following reoxygenation [33]. In another study, we reported that silibinin treatment strongly inhibits lipogenic phenotype in PCa cells via decreasing the expression of transcriptional factor sterol regulatory element binding protein 1 (SREBP1) [34]. However, the effect of silibinin on hypoxia-induced lipogenesis and related signaling remains unknown. Therefore, in the present study, we also assessed the effect of silibinin on hypoxia-induced lipid synthesis and lipogenesis-related signaling molecules. Furthermore, we examined the in vivo effects of silibinin on angiogenesis, tumor cellularity and metabolic reprograming in PCa tumor xenografts using dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI), diffusion-weighted MRI (DW-MRI), 18F-fluoro-deoxyglucose positron emission tomography (FDG-PET) and proton nuclear magnetic resonance (1H-NMR) metabolomics. Our results show that silibinin has profound effect on hypoxic signaling and strongly inhibits PCa growth, clonogenicity, angiogenesis, and lipogenesis.
MATERIALS AND METHODS
Cells and reagents
Human prostate carcinoma LNCaP and 22Rv1 cells were obtained from the American Type Culture Collection (Manassas, VA). LNCaP and 22Rv1 cells were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin G and 100 μg/ml streptomycin sulfate at 37°C in a humidified 5% CO2 incubator. Media and other cell culture materials were from Invitrogen Corporation (Gaithersburg, MD). Cells were treated with different concentrations of silibinin (5–200 μM in medium), dissolved originally in dimethyl sulfoxide (DMSO), for descried time periods. An equal amount of DMSO (vehicle) was present in each treatment, including control; DMSO concentration did not exceed 0.1% (v/v) in any treatment. Antibodies for HIF-1α, HIF-1β, PHD1, PHD2, FIH (factor inhibiting HIF), phosphorylated and total ACC, and FASN were from Cell Signaling (Beverly, MA). Ki-67 and CD31 antibodies were from Abcam (Cambridge, MA), cyclin D1 antibody was from Thermo Scientific (Fremont, CA), TUNEL kit was from Promega (Madison, WI), PSA (prostate specific antigen) antibody was from DAKO (Carpinteria, CA), and HIF-1α antibody used for immunohistochemistry (IHC) was from Santa Cruz Biotechnology (Dallas, TX). Enhanced chemiluminescence (ECL) detection system was from GE healthcare (Buckinghamshire, UK). All other reagents were obtained in their commercially available highest purity grade.
Clonogenic assay
PCa cells were seeded in 6-well plates (500–1000 cells/well). After 24 hrs of plating, one set of plate was transferred to hypoxic conditions (1% O2), while other set was maintained under normoxia (21% O2). After 48 hrs, the plate under hypoxia was returned to normoxic condition. Subsequently, plates were treated with silibinin (25 and 50 μM) and cultured for another 5–6 days, after which, the cells were fixed with ice-cold methanol: glacial acetic acid (3:1) for 10 min and stained with 1% crystal violet for 15 min. Number of colonies with greater than 50 cells were counted in each of the treatment groups.
Conditioned media collection and Endothelial cell capillary-like tube formation assay
LNCaP cells were grown to 50–60% confluency and were cultured under hypoxic (1% O2) conditions in 0.5% serum RPMI media for 24 hrs. Thereafter, conditioned media was collected, centrifuged and labeled as control conditioned media (CM) and stored at −80°C until further use in tube formation assay. In this assay, growth factor-reduced matrigel was pipetted into pre-chilled 24-well plates (150 μL matrigel per well) and polymerized for 45 min at 37°C. HUVECs (4 × 104 per well) were seeded with LNCaP hypoxic CM with DMSO or different concentration of silibinin (5–200 μM). After 12 hrs of incubation, closed ring tubular structures were counted and photographed. For this assay, we optimized a ratio of 75:25 (vol/vol) for CM and HUVEC EBM media. A mixture of LNCaP cells RPMI media with 0.5% serum (75%) and HUVEC EBM media (25%) served as negative control in these experiments.
Western blotting
At the end of each time-point, total cell lysates or nuclear and cytoplasmic fractions were prepared following previously described methods [35]. Thereafter, 50–70 μg of protein lysate per sample was denatured in 2× sample buffer and subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis on 6–16% Tris–glycine gel (as required based upon the protein molecular weight). The separated proteins were transferred on to nitrocellulose membrane followed by blocking with 5% non-fat milk powder (w/v) in Tris-buffered saline (10 mM Tris–HCl, pH 7.5, 100 mM NaCl, 0.1% Tween 20) for 1 hr at room temperature. Membranes were probed for the protein levels of desired molecules using specific primary antibodies followed by the appropriate peroxidase-conjugated secondary antibody and visualized by ECL detection system. To ensure equal protein loading, each membrane was stripped and re-probed with appropriate loading control. The autoradiograms/bands were scanned and densitometry was performed using Adobe Photoshop 6.0 (Adobe Systems, San Jose, CA).
Oil Red O staining
Approximately 50,000 PCa cells were seeded in 6 well plates, and after 36 hrs, one set of the plates was transferred to hypoxic condition (1% O2), while other set was maintained under normoxia with or without silibinin treatment. After 48 hrs, media were removed, cells washed with PBS and fixed for 20 min with 4% formalin in PBS. Cells were then washed with sterile double distilled water and subsequently with 60% isopropanol for 5 min and stained with 2 ml of filtered Oil Red O solution for 20 min at room temperature. Thereafter, cells were washed with sterile double distilled water till a clear background is obtained; 2 ml of tap water was added to each well and viewed under a phase contrast microscope. Lipid droplets (appeared red in color) were analyzed under a microscope and photomicrographs were captured. Lipid content quantitation was carried out by dye elution using 100% isopropanol and absorbance was measured by spectrophotometer at 500 nm.
NOX activity assay
Cultured cells were collected by trypsinization and washed twice with ice-cold PBS and homogenized in lysis buffer (20 mM KH2PO4 pH 7.0, 1 mM EGTA, 1 mM phenylmethyl sulfonyl fluoride, 10 mg/ml aprotinin, and 0.5 mg/ml leupeptin) by using a Dounce homogenizer (100 strokes on ice). Homogenates were centrifuged at 800 ×g at 4°C for 10 min to remove the unbroken cells and debris. For determining NOX activity, 100 μl aliquots of homogenates were added to 900 μl of 50 mM phosphate buffer containing 1 mM EGTA, 150 mM sucrose, 5 mM lucigenin, and 100 mM NADPH, pH 7.0. Photon emission was measured in a luminometer (Promega, Madison, WI) every 1 min for 15 min. There was no measurable activity in the absence of NADPH. Protein concentration was determined using the DC Protein assay system (Bio Rad, Hercules, CA). Superoxide anion production was expressed as relative light (luminescence) units (RLU)/mg protein.
Generation of LNCaP-vector control and LNCaP-HIF-1α overexpressing stable cell lines
LNCaP cells were transfected with lentiviral particles containing pLX304-HIF-1α or vector control pLX304 (Addgene, Cambridge, MA) purchased from Functional genomics core, University of Colorado (Boulder, CO). Stable transduced cells were selected using blasticidin (8 μg/ml) in complete media for 3–4 weeks. The individual resistant clones were picked and grown separately and maintained in the same selection medium. The over-expressing clone was confirmed by analyzing the mRNA levels of HIF-1α in LNCaP-VC and LNCaP-HIF-1α by semi-quantitative RT-PCR. Total RNA was extracted from LNCaP-VC and LNCaP-HIF-1α cells with Tri reagent (Sigma-Aldrich, St. Louis, MO) following the manufacturer’s instructions. Total RNA was treated with RNase-free DNase (Thermo Scientific, Rockford, IL) and the integrity of RNA was determined by running on agarose gel prior to further downstream application. The RT-PCR was carried out using one-step RT-PCR kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions and total RNA (200 ng) was taken as template for each reaction. HIF-1α oligonucleotide sequences for PCR primers were 5′-CAGAGCAGGAAAAGGAGTCA-3′ and 5′-AGTAGCTGCATGATCGTCTG-3′.
In vivo tumor xenograft study
Athymic (nu/nu) male nude mice were purchased from the NCI (Frederick, MD). The treatment protocol was approved by the Institutional Animal Care and Use Committee of the University of Colorado Denver. About 1 million 22Rv1 cells were suspended in 50 μL of serum free medium, mixed with 50 μL of matrigel, and injected s.c. in flanks of each mouse. Once the xenografts started growing, mice were randomly divided into 2 groups: Group I mice (vehicle control group) orally administered with 200 μL of 0.5% CMC (w/v) in sterile water; Groups II mice orally administered with 200 mg/kg body weight dose of silibinin in 200 μL of 0.5% CMC. Mice were imaged by FDG-PET to assess glucose uptake before the beginning of the silibinin treatment (day 0) as well as on day 4 and day 15. DCE-MRI and DW-MRI were performed for tumor vascularity and cellularity, respectively, on day 1, day 6 and day 16 of the experiment. At the end, tumor tissues were collected and analyzed for metabolites by quantitative 1H-NMR metabolomics. A part of the tumor tissues was also fixed in formalin and analyzed by IHC for various biomarkers. Throughout the experiment, tumor sizes were measured twice weekly using digital caliper and tumor volume was calculated by the formula: 0.5236 L1(L2)2, where L1 is long diameter, and L2 is short diameter.
In Vivo imaging and metabolomics
For FDG-PET studies, the animals were fasted for 4 hrs, blood glucose levels were monitored prior to injection of 250 microCi of FDG (obtained from PetNet) as previously described [36]. After 60 min of awake uptake, the animals were anesthetized with 2% isoflurane and a 10-min PET scan was acquired using Siemens Inveon μPET scanner and Inveon Acquisition Workplace software (IAW v1.5). All PET scans were acquired in a double-sampling mode to improve spatial resolution (1.2 mm). Regions of interest were manually drawn within the tumor lesions and total radioactivity of the ROI determined (in kBq/mL). The normalized uptake values were calculated as tissue activity [kBq/mL])/(corrected injected dose [kBq], where the corrected dose is calculated as C = C0*e(−0.006317*t) (18F decay constant of 0.006317).
All MRI experiments were performed using a Bruker 4.7 Tesla PharmaScan system. The DCE-MRI and DW-MRI protocols have been previously validated at the Animal Imaging Shared Resources (AISR) and published [37,38]. Briefly, animals were anesthetized with an IP dose of ketamine/xylazine (80/15 mg/kg) and catheterized using a 27Ga butterfly catheter. The animals were placed into a home-made holder and inserted into a Bruker 36-mm bird-cage transmitter-received coil tuned. After performing a localizer scan, anatomical proton density MRI, DW-MRI (with six b-values of 0, 150, 300, 600, 800, 1000 s/mm2) and finally gradient-echo DCE-MRI (with 128 time frames) were acquired. During DCE-MRI scans, animals were given a gadolinium contrast via tail vein catheter (0.4 mmol/kg of MultiHance, gadobenate dimeglubine). Apparent diffusion coefficients (ADC) maps were generated in ParaVision based on T2 signal intensity at six b-values from DW-MRI: ADC = −(1/b1)*ln(S1/S0). For DCE-MRI, the T1-signal voxel intensities were calculated for each lesion and a blood vessel on the same anatomical slice as a function of gadolinium injection time. A compartmental pharmacokinetic model, using SAAM program (version 2, University of Washington, Seattle, WA), in which the tissue clearance of gadolinium is related to its transcapillary exchange between tumor and blood vessels (Ktrans), was constructed based on T1-signal intensities.
The snap-frozen tissues were extracted using an acid extraction developed in our lab [39] and lyophilized hydrophilic and lipophilic extracts analyzed by water-suppressed 1H-NMR. The external standard (d2-trimethyl-silyl-propionic acid, 1.2 mmol/L) was used for concentration quantification as well as a chemical shift reverence. Total of 65 endogenous metabolites were quantified from each tumor. All quantitative data sets for each tumor (including metabolite concentrations as well as quantitative imaging end-points) were loaded into MetaboAnalyst software (University of Alabama) and partial least square regression analysis (PLS-DA) was performed for matrix reduction and biomarker identification.
IHC analysis
Tumor samples were fixed in 10% buffered-formalin for 12 hrs and processed conventionally. The paraffin-embedded tumor sections (5 μm-thick) were deparaffinized using xylene, and rehydrated in a graded series of ethanol with a final wash in distilled water. Antigen retrieval was performed using 10 mM citrate buffer (pH 6.0) in a decloaking chamber for 15 min. Endogenous peroxidase activity was blocked by immersing the sections in 3.0% H2O2 in methanol (v/v), followed by three changes in 10 mM PBS (pH 7.4). Thereafter, sections were incubated with specific primary antibody, followed by a specific biotinylated secondary antibody, and then conjugated HRP streptavidin and DAB working solution, and counterstained with hematoxylin.
Statistical analysis
Statistical analysis was performed using SigmaStat 2.03 software (Jandel Scientific, San Rafael, CA). Data was analyzed by t-test or one way ANOVA (Tukey test) and a statistically significant difference was considered to be at p<0.05.
RESULTS
Silibinin inhibits growth of human PCa cells under both normoxic and hypoxic conditions
First, we compared the effect of silibinin treatment on growth of PCa cells under normoxic (21% O2) and hypoxic conditions (1% O2). As shown in Figure 1A and 1B, silibinin treatment (50–200 μM) reduced the total cell number, in case of both LNCaP and 22Rv1 LNCaP cells, in a concentration-dependent manner. Similarly, clonogenic assay results showed that silibinin treatment at 25 and 50 μM concentrations strongly reduced the colony formation by LNCaP and 22Rv1 cells in normoxic as well as hypoxia-exposed conditions, with relatively stronger effect observed in LNCaP cells. Both LNCaP and 22Rv1 cells grew relatively slower under hypoxia compared to normoxic conditions.
Figure 1.
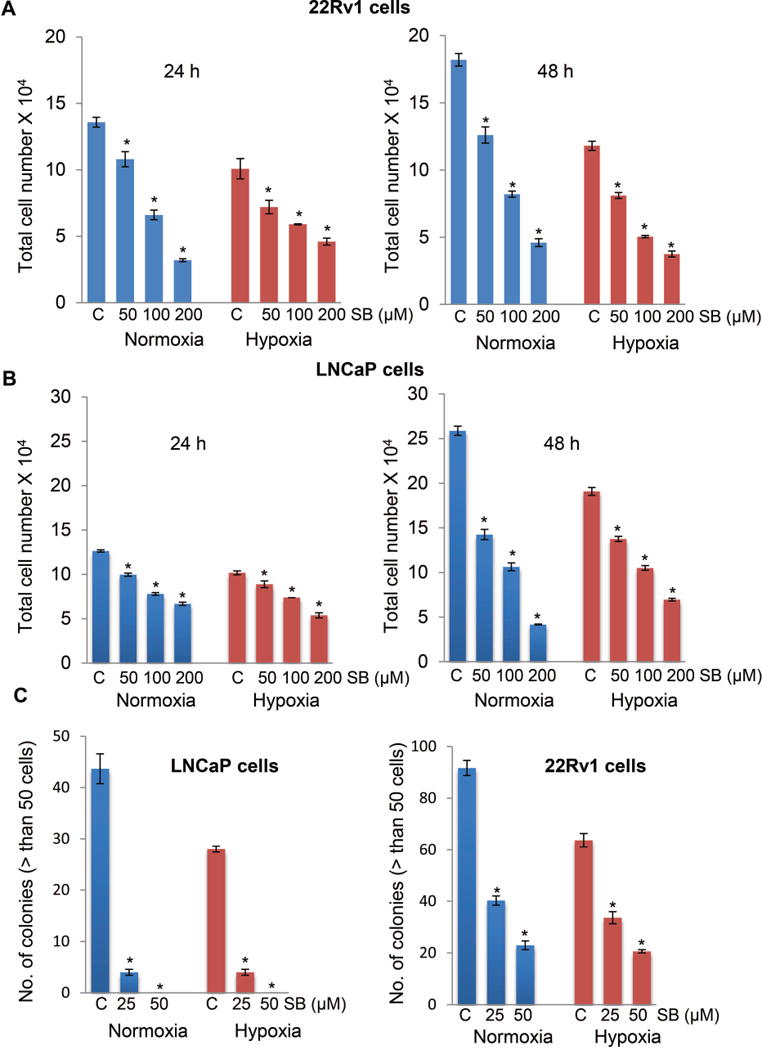
Silibinin inhibits cell viability and clonogenicity of human PCa cells under both normoxic and hypoxic conditions. (A-B) Human PCa LNCaP and 22Rv1 cells were treated with DMSO or silibinin (50–200 μM) under normoxic (21% O2) or hypoxic (1% O2) conditions, and total cell number was determined after 24 and 48 hrs. (C) LNCaP and 22Rv1 cells (500 cells/well) were maintained under normoxic (21% O2) or hypoxic conditions (1% O2) for 48 hrs. Thereafter, plates under hypoxia were returned to normoxic conditions and cultured with or without silibinin (25 and 50 μM). Number of colonies with greater than 50 cells were counted in each of the treatment groups after 6 days. The data shown are mean±SEM of three samples. *, p ≤ 0.001
Silibinin inhibits hypoxic PCa cells-induced capillary tube formation by HUVECs
Hypoxic conditions in tumors are known to induce angiogenesis [8]. In earlier studies, we have reported the direct effect of silibinin on angiogenesis in various cell culture angiogenesis models as well as in mice [40–42]; however, we have not studied the effect of silibinin on hypoxic PCa cells-induced angiogenesis. To answer this question, we first collected the conditioned media (CM) from LNCaP cells cultured under hypoxic conditions (1% O2). Thereafter, HUVECs were cultured in matrigel coated plates in the presence of CM with or without silibinin treatment and tube formation was analyzed. For this assay, we optimized a ratio of 75:25 for CM and HUVEC EBM media. HUVECs cultured in complete media served as positive control while HUVECs cultured in base media (a mixture of LNCaP cells RPMI media with 0.5% serum [75%] and HUVEC EBM media [25%]) served as negative control in these experiments. As shown in Figure 2, CM promoted the closed ring formation by HUVECs, which was inhibited by silibinin in a concentration-dependent manner. These results for the first time showed that silibinin could target angiogenesis induced by secreted factors from hypoxic PCa cells.
Figure 2.
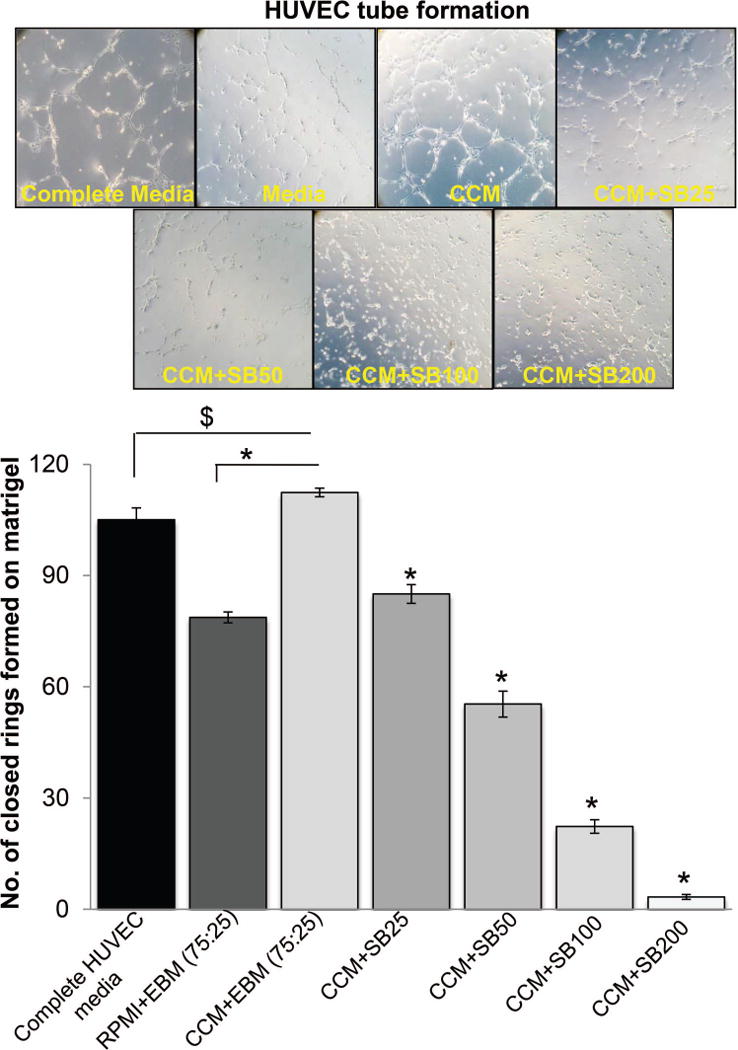
Silibinin inhibits hypoxic PCa cells-induced capillary tube formation by HUVECs. HUVECs (4 × 104 per well) were placed in 24-well plates coated with matrigel with complete HUVEC media, or media mixture (RMPI+EBM) with 0.5% serum, or control conditioned media (CCM) from hypoxic LNCaP cells with DMSO or various concentrations of silibinin (25–200 μM). After 12 hrs, tubular structures were photographed at 100× magnification and number of closed rings formed in each group was counted. Number of closed rings is presented as mean±SEM of three samples for each treatment. *, p ≤ 0.001; #, p ≤ 0.01; $, p ≤ 0.05
Silibinin inhibits hypoxia-induced lipid accumulation in human PCa cells
In general, hypoxic conditions promote glycolysis; however, we recently reported that PCa cells accumulate lipids under hypoxic condition, which was important for their survival and invasiveness [33]. So, next we examined silibinin effect on hypoxia-induced lipid accumulation. As shown in Figures 3A and 3B, LNCaP and 22Rv1 cells accumulated higher amount of lipids (stained by Oil-Red-O) under hypoxia compared to normoxic cells, and silibinin treatment significantly reduced the lipid accumulation in LNCaP and 22Rv1 cells both under normoxic and hypoxic conditions. We next characterized the effect of silibinin on the expression of key regulators of lipogenesis under both normoxic and hypoxic conditions. Silibinin treatment strongly increased the level of inhibitory ACC phosphorylation (Serine 79) in both LNCaP and 22Rv1 cells. Silibinin treatment marginally reduced the total ACC level in LNCaP cells under both normoxic and hypoxic conditions; however, in 22Rv1 cells, silibinin treatment slightly decreased the ACC level under normoxic condition but moderately increased the ACC level under hypoxic condition (Figure 3C). Similarly, silibinin effect on FASN was quite variable and a decrease was observed only in LNCaP cells under hypoxic condition (Figure 3C).
Figure 3.

Silibinin inhibits hypoxia-induced lipid accumulation in human PCa cells. (A-B) LNCaP and 22Rv1 cells were cultured under normoxic or hypoxic conditions with or without silibinin (90 μM) for 48 hrs. Thereafter, lipid content in the cells was measured by Oil Red O staining. Relative absorbance (normalized with relative cell count) as well as corresponding photomicrographs (lipid droplet appears red in color) is presented. *, p ≤ 0.001; #, p ≤ 0.01. (C) LNCaP and 22Rv1 cells were cultured under normoxic (21% O2) or hypoxic (1% O2) conditions in the presence of DMSO or silibinin (50–200 μM) for 6 hrs. At the end, total cell lysates were prepared and analyzed for p-ACC, total ACC and FASN by Western blotting. Membranes were stripped and reprobed for β-actin to assess equal protein loading. Densitometry data presented below the bands are ‘fold change’ as compared with respective control after normalization with respective loading control (β-actin).
Silibinin inhibits hypoxia-induced signaling in PCa cells
Next, we examined the effect of silibinin treatment on hypoxia-induced HIF signaling. As shown in Figure 4A, silibinin treatment reduced the hypoxia-induced HIF-1α expression in both LNCaP and 22Rv1 cells. Silibinin treatment did not affect the level of HIF-1β and PHD1 in both the cell lines; however, PHD2 level was increased by silibinin in both the cell lines under hypoxic condition (Figure 4A). There was no consistent effect of silibinin on FIH expression as we observed a decrease in LNCaP cells but an increase in 22Rv1 cells following silibinin treatment under hypoxic condition (Figure 4A). Interestingly, silibinin treatment did not affect the mRNA level of HIF-1α in both the cell lines (data not shown), suggesting that silibinin-mediated decrease in HIF-1α is probably at post-transcription level.
Figure 4.
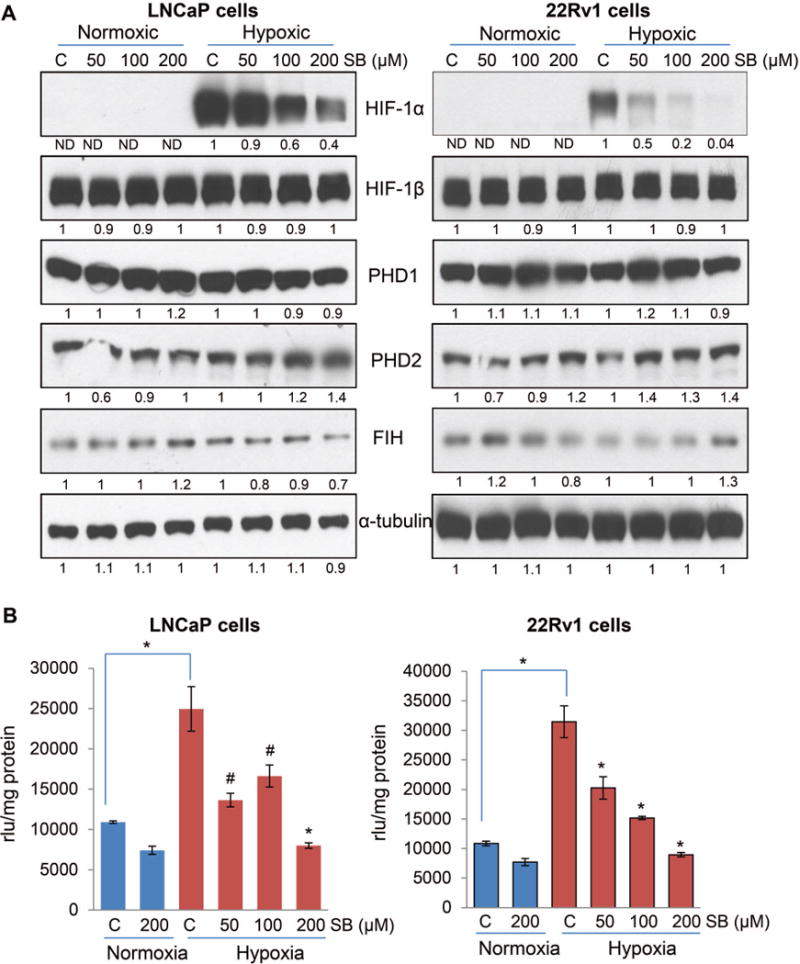
Silibinin inhibits HIF-1α expression and NOX activity in PCa cells under hypoxic conditions. (A) LNCaP and 22Rv1 cells were cultured under normoxic (21% O2) or hypoxic (1% O2) conditions in the presence of DMSO or silibinin (50–200 μM) for 6 hrs. At the end, total cell lysates were prepared and analyzed for HIF-1α, HIF-1β, FIH, PHD1 and 2 by Western blotting. Membranes were stripped and reprobed for α-tubulin to assess equal protein loading. Densitometry data presented below the bands are ‘fold change’ as compared with respective control after normalization with respective loading control (α-tubulin). ND: Not detectable. (B) LNCaP and 22Rv1 cells (4 × 105 cells/60 mm culture dish) were grown under standard culture condition and after 36 hrs of seeding cells were cultured under normoxic (21% O2) or hypoxic (1% O2) conditions in the presence of DMSO or silibinin for 24 hrs. At the end, cells were harvested and NOX activity was measured as mentioned in ‘Materials and Methods’ and represented as rlu/mg protein. Each value represents mean ± SEM of three samples for each treatment. *, p ≤ 0.001; #, p ≤ 0.01
HIF-1α and NOX are known to regulate the expression of each other in a feed-forward manner [15,43], therefore, next we examined the effect of silibinin on hypoxia-induced NOX activation in PCa cells. Interestingly, we observed a strong increase in NOX activity in both LNCaP and 22Rv1 cells under hypoxia compared to normoxia (Figure 4B). More importantly, hypoxia-induced NOX activity was significantly decreased by silibinin treatment in both LNCaP and 22Rv1 cells (Figure 4B).
Silibinin inhibits lipid accumulation, clonogenicity and NOX activity in PCa cells overexpressing HIF-1α under hypoxic conditions
Next, to determine whether the efficacy of silibinin against PCa cells is dependent upon HIF-1α, we overexpressed HIF-1 α in LNCaP cells using lentiviral particles. HIF-1α overexpression was confirmed by RT-PCR (Figure 5A). Interestingly, similar to LNCaP-VC (vector control), silibinin treatment strongly reduced the HIF-1α expression in LNCaP-HIF-1α (HIF-1α overexpressing) cells (Figure 5B and 5C). Moreover, silibinin treatment reduced the lipid content in both LNCaP-VC and LNCaP-HIF-1α cells under normoxic and hypoxic conditions (Figure 5D). Interestingly, in this study, compared to LNCaP-VC cells, LNCaP-HIF-1α cells showed higher lipid accumulation under both normoxic and hypoxic conditions. Silibinin treatment also strongly reduced the clonogenicity in both LNCaP-VC and LNCaP-HIF-1α cells under normoxic and hypoxic conditions (Figure 5E). Furthermore, LNCaP-HIF-1α cells showed higher NOX activity compared to LNCaP-VC cells under hypoxic condition, which was significantly decreased by silibinin treatment (Figure 5F). These results suggest that silibinin treatment has strong efficacy in reducing hypoxia-induced HIF-1α expression in PCa cells. It also seems that lipid accumulation and NOX activity in PCa cells are associated with cellular HIF-1α expression in PCa cells, and silibinin inhibits hypoxia-induced lipogenesis and NOX activity via targeting HIF-1α expression.
Figure 5.
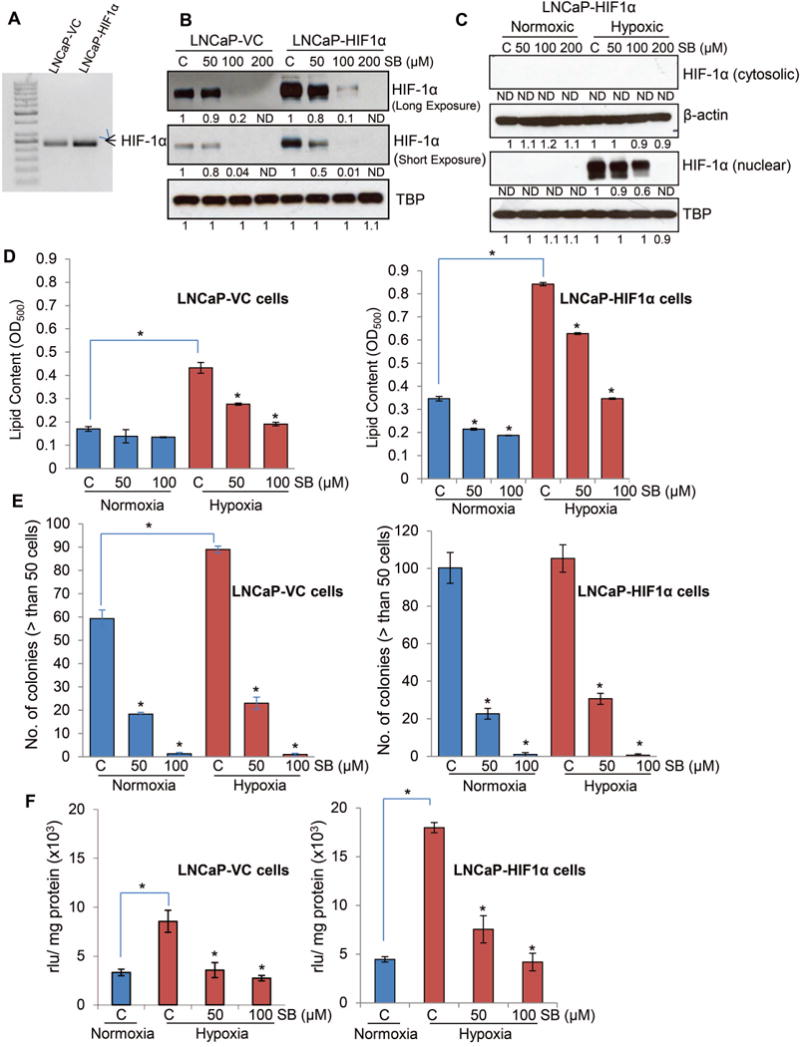
Effect of silibinin on lipid accumulation, clonogenicity and NOX activity in PCa cells overexpressing HIF-1α under hypoxic conditions. (A) HIF-1α was overexpressed in LNCaP cells using lentiviral particles as detailed in the methods. HIF-1α overexpression was confirmed by RT-PCR. (B) LNCaP-VC and LNCaP-HIF1α cell were cultured under hypoxic (1% O2) conditions in the presence of DMSO or silibinin (50–200 μM) for 6 hrs. At the end, nuclear fractions were prepared and analyzed for HIF-1α expression by Western blotting. Membranes were stripped and reprobed for TBP (TATA binding protein) to assess equal protein loading. Densitometry data presented below the bands are ‘fold change’ as compared with respective control after normalization with respective loading control (TBP). ND: Not detectable. (C) LNCaP-HIF1α cells were cultured under normoxic (21% O2) or hypoxic (1% O2) conditions in the presence of DMSO or silibinin (50–200 μM) for 6 hrs. At the end, nuclear and cytoplasmic fraction were prepared and analyzed for HIF-1α by Western blotting. Membranes were stripped and reprobed for β-actin (cytoplasmic fraction) or TBP (nuclear fraction) to assess equal protein loading. Densitometry data presented below the HIF-1α (nuclear) bands are ‘fold change’ as compared with respective control after normalization with respective loading control (TBP). ND: Not detectable. (D) LNCaP-VC and LNCaP-HIF1α cells (5 × 104 cells/well of a six well plate) were grown under standard culture condition and after 36 hrs of seeding cells were cultured under normoxic or hypoxic conditions with or without silibinin for 48 hrs. Thereafter, lipid content in the cells was measured by Oil Red O staining. (E) LNCaP-VC and LNCaP-HIF1 α cell (500 cells/well) were maintained under normoxic or hypoxic conditions for 48 hrs. Thereafter, plates under hypoxia were returned to normoxic conditions and cultured with or without silibinin. Number of colonies with greater than 50 cells was counted in each of the treatment group after 6 days. Media were changed after 3 days supplemented with fresh silibinin at the indicated concentrations. (F) LNCaP-VC and LNCaP-HIF1α cells (4 × 105 cells/60 mm culture dishes) were grown under standard culture condition and after 36 hrs of seeding cells were treated with silibinin and further incubated for 24 hrs under hypoxic conditions. In each case, cells cultured under normoxic condition served as relevant control. At the end of 24 hrs, cells were harvested and NOX activity was measured as mentioned in ‘Materials and Methods’ and represented as rlu/mg protein. Each value represents mean ± SEM of three samples for each treatment.
In vivo efficacy of silibinin to inhibit 22Rv1 xenograft growth and ameliorate hypoxia-induced biological effects
Next, we performed a xenograft study using 22Rv1 cells, where silibinin was administered when tumors attained approximately 400 mm3 size. Mice were imaged regularly by FDG-PET, DW-MRI and DCE-MRI to monitor for metabolic activity, cellularity and blood perfusion as well as tumor volumes. As shown in Figure 6A, silibinin treatment consistently slowed down the xenograft growth, and by the end of the study (16 day of silibinin feeding) we observed a 48% decrease in the tumor volume; however this difference did not achieve statistical significance.
Figure 6.

Silibinin feeding decreases 22Rv1 xenograft growth in male athymic nude mice via targeting the lipid metabolism. (A) 22Rv1 xenografts were initiated in male athymic nude mice. Mice were administered either vehicle (CMC) or 200 mg/kg body weight dose of silibinin for 16 days. Tumor volume was measured on experimental day 0, 5, 9 and 16. Each value in the bar diagram is mean ± SEM of 12 xenografts. (B) Quantitative imaging end-points derived from FDG-PET, DWI, and DCE-MRI on untreated and silibinin-treated animals; *, p ≤ 0.001; $, p ≤ 0.05; (C) ADC maps showing low ADC values for silibinin-treated animals indicating no potential loss of cellularity upon the treatment; (D) decreased gadolinium kinetics on DCE-MRI revealing lower tumor vascularity in silibinin-treated animals; (E) PLS-DA regression analysis on comprehensive imaging and metabolomics date sets; (F) Variable Importance in Projection (VIP) scores from PLS-DA revealing the major discriminative end-points from in vivo silibinin treatment.
Throughout the study, we employed several quantitative imaging end-points (Figure 6B), such as normalized uptake values (NUV) for FDG-PET, apparent diffusion coefficients (ADC) for DW-MRI and gadolinium transport constant (Ktrans) for DCE-MRI, to assess glucose up-take, cell density and tumor vascularity, respectively. Interestingly, unlike classic chemotherapeutic agents, silibinin did not directly affect glucose uptake as seen by an unchanged NUVs for FDG uptake: 242 vs. 259 arbitrary units (n.s., Figure 6B). Also, unlike chemotherapeutics, silibinin did not induce necrosis or loss in cell density as seen by persistently low ADC values on DW-MRI in silibinin-treated groups (Figure 6B and 6C). Loss of cellularity due to the treatment-induced necrosis usually results in increased ADCs above the baseline values (for most tumors, above 1.3×10−3 mm2/s) [44], which was not the case for xenografts in silibinin treated mice. On the other hand, the most pronounced in vivo effect of silibinin observed in our study was significantly decreased tumor perfusion and permeability as seen from DCE-MRI (Figure 6D). The Ktrans as well as initial areas under the curve (IAUCs) for gadolinium uptake were highly decreased in silibinin-treated xenografts (ca. −40% of gadolinium kinetics) (Figure 6B).
At the end of the study, we also analyzed the tissues for metabolite analysis by quantitative 1H-NMR and processed the remaining tissues for IHC analysis. The most prominent metabolic effect of silibinin was on lipid levels. Silibinin treatment led to a dramatic decrease in the tumor levels of total fatty acids, triacylglycerols, phospholipids and cholesterol (Table 1). On the other hand, and confirming the previously seen lack of the effect on glucose uptake, the water-soluble extracts revealed little changes (Table 2). The only changes seen for the hydrophilic metabolites were decreased levels of acetate, ribose and lactate – all of them can be indirectly related to silibinin-related inhibition of lipogenesis and overall cell proliferation.
Table 1.
Effect of silibinin feeding on endogenous metabolites in 22Rv1 xenograft tissues analyzed by 1H-NMR in lipophilic extracts.
| Metabolite | Control (μmol/g) | SB200 (μmol/g) | |
|---|---|---|---|
| 1. | Sphingomyelin | 5.6±3.1 | 4.4±2.1 |
| 2. | MUFA | 68.9±30.0 | 52.8±28.9 |
| 3. | TAG | 55.8±26.4 | 41.3±11.7$ |
| 4. | Glycerol-PL | 96.7±48.7 | 68±21.1$ |
| 5. | Ptd-glycine | 10.2±6.5 | 7.7±2.6 |
| 6. | Ptd-serine | 9.3±5.7 | 6.3±3.1 |
| 7. | Ptd-choline | 41±19.9 | 28.4±6.7$ |
| 8. | Ptd-inositol | 20.2±10.4 | 16.6±6.1 |
| 9. | Cholines | 59.7±29.2 | 41±10.2$ |
| 10. | Ptd-ethanolamine | 15.8±8.6 | 10.7±2.8$ |
| 11. | PUFA | 134.9±58.6 | 105.7±28.9 |
| 12. | Total Fatty acids | 198.9±89.5 | 146.4±39.9$ |
| 13. | (CH2)n | 3217.2±1439.1 | 2333.3±634.8$ |
| 14. | Total Lipids | 705.1±295.6 | 536.1±159.4$ |
| 15. | Cholesterol | 52.7±21.2 | 38.5±13.5$ |
| 16. | PUFA/MUFA | 2.1±0.5 | 2.8±2.5 |
p ≤ 0.05
Table 2.
Effect of silibinin feeding on endogenous metabolites in 22Rv1 xenograft tissues analyzed by 1H-NMR in hydrophilic extracts.
| Metabolite | Control (μmol/g) | SB200 (μmol/g) | |
|---|---|---|---|
| 1. | Cyclic amino acids | 26.63±9.7 | 33.89±22.5 |
| 2. | Adenosine & Adenine | 16.9±8.1 | 14.7±8.9 |
| 3. | Carbohydrate (Ribose) | 24.8±49.3 | 13.4±5.3$ |
| 4. | Nucleotide | 20.4±12.2 | 14.9±5.7 |
| 5. | Myoinositol | 5.7±2.2 | 5.4±2.1 |
| 6. | Polyols and Sugars | 161.6±55.2 | 162.1±57.5 |
| 7. | Glycine | 4.9±1.9 | 4.2±1.3 |
| 8. | Taurine | 20.9±5.4 | 18.8±6.0 |
| 9. | Total choline | 6.5±2.1 | 5.9±2.2 |
| 10. | Total Creatine | 6.9±1.9 | 6.4±2.1 |
| 11. | Glutathione | 3.1±1.2 | 2.9±1.5 |
| 12. | Aspartate+Citrate+Methionine | 4.4±1.8 | 4.5±2.2 |
| 13. | Total Glutathione | 3.4±1.1 | 3.1±1.4 |
| 14. | Glutamine | 3.2±0.8 | 2.9±1.2 |
| 15. | Succinate | 3.8±1.5 | 3.6±1.8 |
| 16. | Glutamate | 6.6±1.8 | 6.1±1.9 |
| 17. | Total CH2 and CH3 | 49.2 ±19.7 | 49.8±19.6 |
| 18. | Acetate | 2.5±1.3 | 1.3±0.5# |
| 19. | Arginine and Lysine | 8.2±1.8 | 8.2±2.9 |
| 20. | Alanine | 7.8±2.9 | 7.1±2.6 |
| 21. | Lactate | 20.7±4.6 | 14.9±5.4# |
| 22. | Branched chain amino acid | 12.6±5.1 | 12.1±4.6 |
p ≤ 0.01;
p ≤ 0.05
We performed partial least square regression analysis (PLS-DA) on our comprehensive quantitative data sets (including all 69 imaging and metabolomics end-points). We achieved a very good level of group clustering (the untreated versus silibinin-treated data sets, Figure 6E). The highest significance scores (>2) for biomarker identification were assigned for angiogenic inhibition (Ktrans and IAUC from DCE-MRI), followed by decreased acetate and lipid metabolites. All of the distinguished discriminative end-points were statically decreased in silibinin-treated animals (Figure 6F).
IHC analysis of tumor tissues showed that silibinin treatment significantly decreased the expression of biomarkers for proliferation (Ki-67 and cyclin D1), microvessel density (CD31), hypoxia (HIF-1α), and lipogenesis (FASN and ACC) but increased apoptosis (TUNEL) (Figure 7). Since 22Rv1 cells also express functional AR signaling and secrete PSA, we also analyzed the xenograft tissues for PSA expression. As shown in Figure 7, silibinin treatment reduced the PSA level in 22Rv1 tumor tissues.
Figure 7.

Silibinin targets proliferation, apoptosis, hypoxia, lipogenesis, and PSA level in 22Rv1 xenograft tissues. 22Rv1 xenografts were analyzed by IHC for Ki-67, cyclin D1, TUNEL, CD31, HIF-1α, FASN, ACC and PSA. Percentage of Ki-67, cyclin D1, TUNEL, HIF-1α and microvessel density (CD31 positive microvessel) were measured at five arbitrarily selected fields from each tumor at 400× magnification. Immunoreactivity for FASN, ACC and PSA was analyzed in 5 random areas for each tumor tissue and was scored as 0+ (no staining), 1+ (weak staining), 2+ (moderate staining), 3+ (strong staining), 4+ (very strong staining). Each value in the bar diagram is mean ± SEM of 10–12 xenografts. *, p ≤ 0.001
DISCUSSION
Low oxygen conditions (hypoxia) usually persist in most of the growing solid tumors including PCa [6,8,45]. Hypoxic condition promotes cancer cell stemness, EMT, metastasis, angiogenesis, immunosuppression, and radio-resistance [3,8,33,46]. There are several reports supporting that hypoxia in prostate tumor is an early event as well as an independent risk factor for disease progression and treatment failure [3,5–7,47]. Ranasinghe et al showed that HIF-1α expression in PCa tissue is strongly associated with metastasis and CRPC (castrate-resistant prostate cancer) progression, and reported that prostate tumors lacking HIF-1α did not metastasize or develop CRPC [3]. Ragnum et al reported similar finding where hypoxia in the prostate tumors, analyzed by pimonidazole staining, was associated with disease stage and lymph node metastasis [5]. Milosevic et al conducted one of the largest clinical studies of PCa hypoxia with direct measurement of tumor oxygen levels by using ultrasound-guided trans-rectal needle-electrode [6]. This study found that tumor hypoxia is associated with early biochemical relapse after radiotherapy and also predicts local recurrence [6]. Taken together these finds, it is clear that hypoxia and/or the activation of hypoxia-related signaling in prostate tumors could determine treatment success as well as disease progression. These results also suggest that PCa growth and progression could be effectively prevented via targeting hypoxia/hypoxia-related signaling. This rationale is strongly supported by a recent study, where Platz et al reported that compared with nonusers, men who regularly used digoxin, a cardiac glycoside, (a non-specific HIF-1α inhibitor) had a 25% lower risk of PCa [48]. In a retrospective review of prospectively collected medical records, Ranasinghe et al concluded that nonspecific HIF-1α inhibitors (digoxin, metformin and angiotensin-2 receptor blocker) increase the progression-free survival and decrease the risk of developing CRPC and metastasis in PCa patients on continuous androgen deprivation therapy [49]. In this regard, the results from the present study are significant as we observed that natural non-toxic flavonolignan silibinin possesses strong efficacy in decreasing hypoxia-induced HIF-1α expression as well as in inhibiting hypoxia-induced biological effects in PCa cells both in vitro and in vivo. Importantly, silibinin is widely consumed as a dietary supplement for its hepatoprotective benefits, and is already being tested in several clinical trials including one in PCa patients for its safety and pharmacokinetics [22,23,50]. Therefore, silibinin should be further evaluated for its efficacy to target hypoxia/hypoxia-related signaling molecules in PCa patients to fully exploit its clinical usefulness.
Earlier, we have reported that HIF-1α expression correlates with the PCa stage in TRAMP mice, and silibinin feeding (1% in diet) significantly reduced the HIF-1α expression in TRAMP PCa tissues [20]. Furthermore, silibinin-mediated decrease in HIF-1α expression was associated with strong inhibition of angiogenesis and metastasis to distant tissues [20]. So far, two studies have examined the mechanisms through which silibinin could decrease HIF-1α expression in cancer cells. Garcia-Maceira and Mateo showed that silibinin treatment decreased HIF-1α levels under hypoxic conditions (2% O2) in HeLa and Hep3B cells but without affecting its transcript level and PHD activity [12]. This study also found that silibinin treatment inhibited cap-dependent translation of HIF-1α via suppression of mTOR/p70S6K/4E-BP1 signaling [12]. In another study, Jung et al showed that silibinin treatment decreased the cobalt chloride-induced HIF-1α expression in PCa cells via inhibiting the global protein synthesis involving mTOR/p70S6K/4E-BP1/eIF4 [26]. In the present study, for the first time we showed that silibinin possesses strong efficacy against hypoxia-induced HIF-1α protein expression even in PCa cells overexpressing HIF-1α. Interestingly, we also observed that silibinin treatment strongly inhibited the hypoxia-induced NOX activity in PCa cells. Earlier, Nanduri et al have reported that HIF-1α activation by intermittent hypoxia requires NOX [13]. In another study, Moon et al also showed that NOX-mediated reactive oxygen (ROS) production activates HIF-1α after hyperthermia treatment [14]. Therefore, it is plausible that NOX activation under chronic hypoxic conditions could promote HIF-1α expression in PCa cells. Recently, we have also reported that hypoxia-induced HIF-1α expression in PCa cells is reversed by NOX inhibitors (diphenyleneiodonium chloride and Graviola pulp extract) [15]. Furthermore, it is possible that under hypoxic conditions, NOX activates HIF-1α indirectly by activating mitogenic signaling pathways including PI3K/mTOR. Importantly, HIF-1α is also known to regulate expression of various NOX subunits [51,52], and we have also observed an increase in NOX activity in LNCaP cells overexpressing HIF-1α under hypoxic conditions, which further suggests a feed-forward loop between these two molecules. However, further studies are needed to clearly establish the role of NOX in hypoxia-induced HIF-1α as well as the mechanism through which silibinin inhibits NOX activity in PCa cells.
Based upon the published literature, it appears that signaling pathways related to hypoxia, lipogenesis and CRPC are quite interwoven and together promote PCa progression [33,34,53–56]. For example, androgen ablation by castration or anti-androgen treatment promotes hypoxic response in prostate/PCa cells [53–55], while hypoxia promotes AR transcriptional activity under low androgen conditions, mimicking the androgen independence stage [56]. We have previously reported that transcriptional regulators of lipogenesis (SREBP1/2) play an important role in achieving androgen-independent growth in LNCaP cells [34]. Furthermore, we have recently reported that PCa cells accumulate lipids under hypoxic conditions that play important role in their proliferation and invasiveness following reoxygenation [33]. Results from present study showed that silibinin treatment strongly reduced the hypoxia-induced lipogenesis. Also importantly, we observed higher lipogenesis in LNCaP cells overexpressing HIF-1α suggesting that HIF-1α could be an important player in controlling lipid metabolism. It is quite plausible that lipogenesis or lipid accumulation under hypoxic conditions could be diverted towards de novo androgen biogenesis and activation of AR signaling. Therefore, it is clear that we need to target the signaling nexus involving hypoxia, lipogenesis and CRPC to effectively control PCa. In the present study, we observed that silibinin treatment decreased the HIF-1α level, decreased the level of several lipid metabolites as well as inhibited the AR signaling associated with inhibition of 22Rv1 xenograft growth, clearly suggesting that silibinin treatment could disrupt this nexus and could prevent the PCa progression.
Our quantitative imaging and metabolomics endpoints in tumor xenograft study clearly showed that silibinin treatment leads to a significant inhibition of angiogenesis (DCE-MRI) and lipogenesis. This vascular and metabolic reprograming occurs in the absence of direct cytotoxic effects since FDG uptakes (PET) and cell density (ADCs in DWI) were not affected by in vivo silibinin treatment. These findings provide a direct evidence for the specific targeted action of silibinin without exhibiting the conventional cytotoxic profile attributed to classical chemotherapeutics.
Overall, results from the present study suggest that silibinin strongly inhibits HIF-1α expression in PCa cells and tumors as well as strongly inhibits hypoxia-induced biological effects. These results are clinically significant as human consumption of silibinin is safe and hepatoprotective against various toxic conditions. Therefore, silibinin could be useful either alone or in combination with other treatments such as radiotherapy where hypoxia is the major reason for treatment failure.
Acknowledgments
Department of Biotechnology, Government of India is acknowledged for providing Overseas Associateship to Dr. Anand Ramteke.
Grant Support: This work was supported by NCI R01 grant CA102514 (to RA) and R21 grant CA199628 (to GD). The Animal Imaging Shared Resources (to NJS) received direct funding support from NCI through Cancer Center Support Grant (P30CA046934) and NIH through the CCTSI Grant (UL1TR001082).
List of Abbreviations
- ACC
Acetyl-Co A carboxylase
- ADC
Apparent diffusion coefficients
- ANOVA
Analysis of variance
- CM
Conditioned media
- DCE-MRI
Dynamic contrast-enhanced magnetic resonance imaging
- DMSO
dimethyl sulfoxide
- 4E-BP1
4E-binding protein1
- DW-MRI
Diffusion-weighted magnetic resonance imaging
- ECL
Enhanced chemiluminescence
- eIF4
Eukaryotic initiation factor 4
- EMT
Epithelial-to-mesenchymal transition
- FASN
Fatty acid synthase
- FBS
Fetal bovine serum
- FDG-PET
18F-fluoro-deoxyglucose positron emission tomography
- FIH
Factor inhibiting HIF
- HIF-1
Hypoxia inducible factor 1
- 1H-NMR
Proton nuclear magnetic resonance
- HRP
Horseradish peroxidase, IHC: Immunohistochemistry
- mTOR
Mammalian target of rapamycin
- NOX
NADPH oxidase
- ORO
Oil Red O
- PCa
Prostate cancer
- PHDs
Prolyl-hydroxylases
- PSA
Prostate specific antigen
- ROS
Reactive oxygen species
- RT-PCR
Reverse transcription polymerase chain reaction
- SEM
Standard error of the mean
- SREBP1
Sterol regulatory element binding protein 1
- TRAMP
Transgenic adenocarcinoma of mouse prostate
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- VHL
Von Hippel-Lindau
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Fraga A, Ribeiro R, Principe P, Lopes C, Medeiros R. Hypoxia and Prostate Cancer Aggressiveness: A Tale With Many Endings. Clin Genitourin Cancer. 2015;13:295–301. doi: 10.1016/j.clgc.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Ranasinghe WK, Xiao L, Kovac S, et al. The role of hypoxia-inducible factor 1alpha in determining the properties of castrate-resistant prostate cancers. PLoS One. 2013;8:e54251. doi: 10.1371/journal.pone.0054251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu X, Kang Y. Hypoxia and hypoxia-inducible factors: master regulators of metastasis. Clin Cancer Res. 2010;16:5928–5935. doi: 10.1158/1078-0432.CCR-10-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ragnum HB, Vlatkovic L, Lie AK, et al. The tumour hypoxia marker pimonidazole reflects a transcriptional programme associated with aggressive prostate cancer. Br J Cancer. 2015;112:382–390. doi: 10.1038/bjc.2014.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milosevic M, Warde P, Menard C, et al. Tumor hypoxia predicts biochemical failure following radiotherapy for clinically localized prostate cancer. Clin Cancer Res. 2012;18:2108–2114. doi: 10.1158/1078-0432.CCR-11-2711. [DOI] [PubMed] [Google Scholar]
- 7.Vergis R, Corbishley CM, Norman AR, et al. Intrinsic markers of tumour hypoxia and angiogenesis in localised prostate cancer and outcome of radical treatment: a retrospective analysis of two randomised radiotherapy trials and one surgical cohort study. Lancet Oncol. 2008;9:342–351. doi: 10.1016/S1470-2045(08)70076-7. [DOI] [PubMed] [Google Scholar]
- 8.Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26:605–622. doi: 10.1016/j.ccell.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harris AL. Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 10.Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26:281–290. doi: 10.1007/s10555-007-9066-y. [DOI] [PubMed] [Google Scholar]
- 11.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Maceira P, Mateo J. Silibinin inhibits hypoxia-inducible factor-1alpha and mTOR/p70S6K/4E-BP1 signalling pathway in human cervical and hepatoma cancer cells: implications for anticancer therapy. Oncogene. 2009;28:313–324. doi: 10.1038/onc.2008.398. [DOI] [PubMed] [Google Scholar]
- 13.Nanduri J, Vaddi DR, Khan SA, et al. HIF-1alpha activation by intermittent hypoxia requires NADPH oxidase stimulation by xanthine oxidase. PLoS One. 2015;10:e0119762. doi: 10.1371/journal.pone.0119762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moon EJ, Sonveaux P, Porporato PE, et al. NADPH oxidase-mediated reactive oxygen species production activates hypoxia-inducible factor-1 (HIF-1) via the ERK pathway after hyperthermia treatment. Proc Natl Acad Sci U S A. 2010;107:20477–20482. doi: 10.1073/pnas.1006646107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deep G, Kumar R, Jain AK, et al. Graviola inhibits hypoxia-induced NADPH oxidase activity in prostate cancer cells reducing their proliferation and clonogenicity. Sci Rep. 2016;6:23135. doi: 10.1038/srep23135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agarwal R, Agarwal C, Ichikawa H, Singh RP, Aggarwal BB. Anticancer potential of silymarin: from bench to bed side. Anticancer Res. 2006;26:4457–4498. [PubMed] [Google Scholar]
- 17.Pradhan SC, Girish C. Hepatoprotective herbal drug, silymarin from experimental pharmacology to clinical medicine. Indian J Med Res. 2006;124:491–504. [PubMed] [Google Scholar]
- 18.Deep G, Singh RP, Agarwal C, Kroll DJ, Agarwal R. Silymarin and silibinin cause G1 and G2-M cell cycle arrest via distinct circuitries in human prostate cancer PC3 cells: a comparison of flavanone silibinin with flavanolignan mixture silymarin. Oncogene. 2006;25:1053–1069. doi: 10.1038/sj.onc.1209146. [DOI] [PubMed] [Google Scholar]
- 19.Singh RP, Raina K, Deep G, Chan D, Agarwal R. Silibinin suppresses growth of human prostate carcinoma PC-3 orthotopic xenograft via activation of extracellular signal-regulated kinase 1/2 and inhibition of signal transducers and activators of transcription signaling. Clin Cancer Res. 2009;15:613–621. doi: 10.1158/1078-0432.CCR-08-1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raina K, Rajamanickam S, Singh RP, Deep G, Chittezhath M, Agarwal R. Stage-specific inhibitory effects and associated mechanisms of silibinin on tumor progression and metastasis in transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2008;68:6822–6830. doi: 10.1158/0008-5472.CAN-08-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh RP, Raina K, Sharma G, Agarwal R. Silibinin inhibits established prostate tumor growth, progression, invasion, and metastasis and suppresses tumor angiogenesis and epithelial-mesenchymal transition in transgenic adenocarcinoma of the mouse prostate model mice. Clin Cancer Res. 2008;14:7773–7780. doi: 10.1158/1078-0432.CCR-08-1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flaig TW, Gustafson DL, Su LJ, et al. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Invest New Drugs. 2007;25:139–146. doi: 10.1007/s10637-006-9019-2. [DOI] [PubMed] [Google Scholar]
- 23.Flaig TW, Glode M, Gustafson D, et al. A study of high-dose oral silybin-phytosome followed by prostatectomy in patients with localized prostate cancer. Prostate. 2010;70:848–855. doi: 10.1002/pros.21118. [DOI] [PubMed] [Google Scholar]
- 24.Deep G, Agarwal R. Antimetastatic efficacy of silibinin: molecular mechanisms and therapeutic potential against cancer. Cancer Metastasis Rev. 2010;29:447–463. doi: 10.1007/s10555-010-9237-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deep G, Gangar SC, Agarwal C, Agarwal R. Role of E-cadherin in antimigratory and antiinvasive efficacy of silibinin in prostate cancer cells. Cancer Prev Res (Phila) 2011;4:1222–1232. doi: 10.1158/1940-6207.CAPR-10-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung HJ, Park JW, Lee JS, et al. Silibinin inhibits expression of HIF-1alpha through suppression of protein translation in prostate cancer cells. Biochem Biophys Res Commun. 2009;390:71–76. doi: 10.1016/j.bbrc.2009.09.068. [DOI] [PubMed] [Google Scholar]
- 27.Zadra G, Photopoulos C, Loda M. The fat side of prostate cancer. Biochim Biophys Acta. 2013;1831:1518–1532. doi: 10.1016/j.bbalip.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suburu J, Chen YQ. Lipids and prostate cancer. Prostaglandins Other Lipid Mediat. 2012;98:1–10. doi: 10.1016/j.prostaglandins.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flavin R, Zadra G, Loda M. Metabolic alterations and targeted therapies in prostate cancer. J Pathol. 2011;223:283–294. doi: 10.1002/path.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rysman E, Brusselmans K, Scheys K, et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010;70:8117–8126. doi: 10.1158/0008-5472.CAN-09-3871. [DOI] [PubMed] [Google Scholar]
- 31.Wang C, Rajput S, Watabe K, Liao DF, Cao D. Acetyl-CoA carboxylase-a as a novel target for cancer therapy. Front Biosci (Schol Ed) 2010;2:515–526. doi: 10.2741/s82. [DOI] [PubMed] [Google Scholar]
- 32.Pandey PR, Liu W, Xing F, Fukuda K, Watabe K. Anti-cancer drugs targeting fatty acid synthase (FAS) Recent Pat Anticancer Drug Discov. 2012;7:185–197. doi: 10.2174/157489212799972891. [DOI] [PubMed] [Google Scholar]
- 33.Schlaepfer IR, Nambiar DK, Ramteke A, et al. Hypoxia induces triglycerides accumulation in prostate cancer cells and extracellular vesicles supporting growth and invasiveness following reoxygenation. Oncotarget. 2015;6:22836–22856. doi: 10.18632/oncotarget.4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nambiar DK, Deep G, Singh RP, Agarwal C, Agarwal R. Silibinin inhibits aberrant lipid metabolism, proliferation and emergence of androgen-independence in prostate cancer cells via primarily targeting the sterol response element binding protein 1. Oncotarget. 2014;5:10017–10033. doi: 10.18632/oncotarget.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deep G, Inturi S, Agarwal R. Methods to analyze chemopreventive effect of silibinin on prostate cancer biomarkers protein expression. In: Bode A, Dong Z, editors. Cancer Prevention: Dietary Factors and Pharmacology. Humana Press; 2013. pp. 85–105. [Google Scholar]
- 36.Schlaepfer IR, Glode LM, Hitz CA, et al. Inhibition of Lipid Oxidation Increases Glucose Metabolism and Enhances 2-Deoxy-2-[(18)F]Fluoro-D-Glucose Uptake in Prostate Cancer Mouse Xenografts. Mol Imaging Biol. 2015;17:529–538. doi: 10.1007/s11307-014-0814-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raina K, Ravichandran K, Rajamanickam S, Huber KM, Serkova NJ, Agarwal R. Inositol hexaphosphate inhibits tumor growth, vascularity, and metabolism in TRAMP mice: a multiparametric magnetic resonance study. Cancer Prev Res (Phila) 2013;6:40–50. doi: 10.1158/1940-6207.CAPR-12-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frey L, Lepkin A, Schickedanz A, Huber K, Brown MS, Serkova N. ADC mapping and T1-weighted signal changes on post-injury MRI predict seizure susceptibility after experimental traumatic brain injury. Neurol Res. 2014;36:26–37. doi: 10.1179/1743132813Y.0000000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Serkova NJ, Glunde K. Metabolomics of cancer. Methods Mol Biol. 2009;520:273–295. doi: 10.1007/978-1-60327-811-9_20. [DOI] [PubMed] [Google Scholar]
- 40.Deep G, Gangar SC, Rajamanickam S, et al. Angiopreventive efficacy of pure flavonolignans from milk thistle extract against prostate cancer: targeting VEGF-VEGFR signaling. PLoS One. 2012;7:e34630. doi: 10.1371/journal.pone.0034630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh RP, Sharma G, Dhanalakshmi S, Agarwal C, Agarwal R. Suppression of advanced human prostate tumor growth in athymic mice by silibinin feeding is associated with reduced cell proliferation, increased apoptosis, and inhibition of angiogenesis. Cancer Epidemiol Biomarkers Prev. 2003;12:933–939. [PubMed] [Google Scholar]
- 42.Singh RP, Dhanalakshmi S, Agarwal C, Agarwal R. Silibinin strongly inhibits growth and survival of human endothelial cells via cell cycle arrest and downregulation of survivin, Akt and NF-kappaB: implications for angioprevention and antiangiogenic therapy. Oncogene. 2005;24:1188–1202. doi: 10.1038/sj.onc.1208276. [DOI] [PubMed] [Google Scholar]
- 43.Block K, Gorin Y, Hoover P, et al. NAD(P)H oxidases regulate HIF-2alpha protein expression. J Biol Chem. 2007;282:8019–8026. doi: 10.1074/jbc.M611569200. [DOI] [PubMed] [Google Scholar]
- 44.Padhani AR, Koh DM. Diffusion MR imaging for monitoring of treatment response. Magn Reson Imaging Clin N Am. 2011;19:181–209. doi: 10.1016/j.mric.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 45.Stewart GD, Ross JA, McLaren DB, Parker CC, Habib FK, Riddick AC. The relevance of a hypoxic tumour microenvironment in prostate cancer. BJU Int. 2010;105:8–13. doi: 10.1111/j.1464-410X.2009.08921.x. [DOI] [PubMed] [Google Scholar]
- 46.Ramteke A, Ting H, Agarwal C, et al. Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol Carcinog. 2015;54:554–565. doi: 10.1002/mc.22124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhong H, Semenza GL, Simons JW, De Marzo AM. Up-regulation of hypoxia-inducible factor 1alpha is an early event in prostate carcinogenesis. Cancer Detect Prev. 2004;28:88–93. doi: 10.1016/j.cdp.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 48.Platz EA, Yegnasubramanian S, Liu JO, et al. A novel two-stage, transdisciplinary study identifies digoxin as a possible drug for prostate cancer treatment. Cancer Discov. 2011;1:68–77. doi: 10.1158/2159-8274.CD-10-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ranasinghe WK, Sengupta S, Williams S, et al. The effects of nonspecific HIF1alpha inhibitors on development of castrate resistance and metastases in prostate cancer. Cancer Med. 2014;3:245–251. doi: 10.1002/cam4.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoh C, Boocock D, Marczylo T, et al. Pilot study of oral silibinin, a putative chemopreventive agent, in colorectal cancer patients: silibinin levels in plasma, colorectum, and liver and their pharmacodynamic consequences. Clin Cancer Res. 2006;12:2944–2950. doi: 10.1158/1078-0432.CCR-05-2724. [DOI] [PubMed] [Google Scholar]
- 51.Diebold I, Petry A, Hess J, Gorlach A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol Biol Cell. 2010;21:2087–2096. doi: 10.1091/mbc.E09-12-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol. 2011;226:2925–2933. doi: 10.1002/jcp.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Halin S, Hammarsten P, Wikstrom P, Bergh A. Androgen-insensitive prostate cancer cells transiently respond to castration treatment when growing in an androgen-dependent prostate environment. Prostate. 2007;67:370–377. doi: 10.1002/pros.20473. [DOI] [PubMed] [Google Scholar]
- 54.Shabsigh A, Ghafar MA, de la Taille A, et al. Biomarker analysis demonstrates a hypoxic environment in the castrated rat ventral prostate gland. J Cell Biochem. 2001;81:437–444. [PubMed] [Google Scholar]
- 55.Rothermund CA, Gopalakrishnan VK, Eudy JD, Vishwanatha JK. Casodex treatment induces hypoxia-related gene expression in the LNCaP prostate cancer progression model. BMC Urol. 2005;5:5. doi: 10.1186/1471-2490-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mitani T, Yamaji R, Higashimura Y, Harada N, Nakano Y, Inui H. Hypoxia enhances transcriptional activity of androgen receptor through hypoxia-inducible factor-1alpha in a low androgen environment. J Steroid Biochem Mol Biol. 2011;123:58–64. doi: 10.1016/j.jsbmb.2010.10.009. [DOI] [PubMed] [Google Scholar]