
Figure 1.
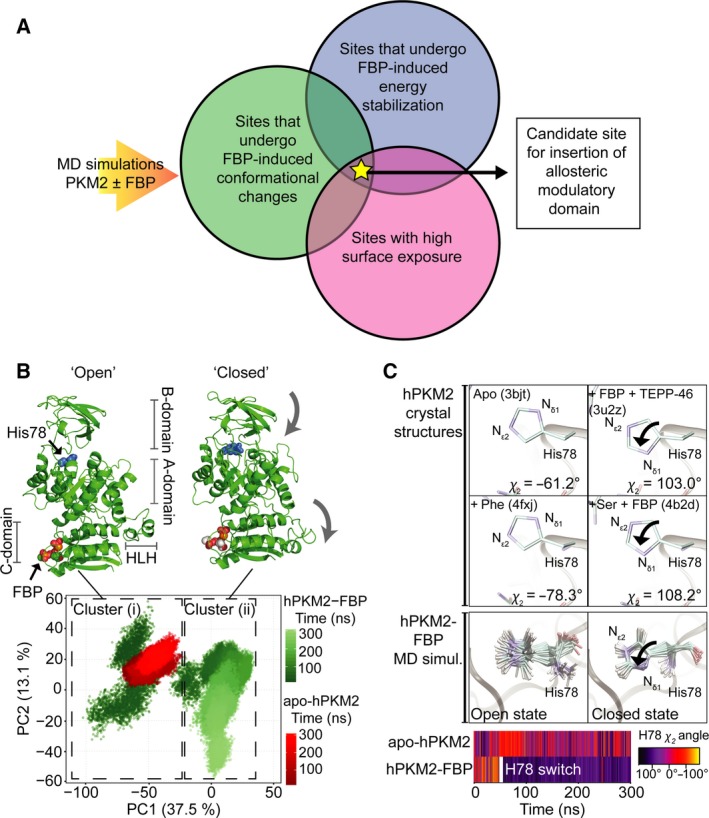
MD reveals conformational changes on PKM2 upon FBP binding. (A) Summary of criteria that led to the identification of a candidate site used for inserting a heterologous allosteric domain to generate a photoswitchable pyruvate kinase. MD simulations of PKM2 with and without the allosteric activator FBP were used to identify residues that undergo FBP‐induced conformational changes (green circle), show energetic stabilisation indicative of allosteric communication with the FBP‐binding pocket (blue circle) and were surface‐exposed (pink circle). A site that fulfilled all three criteria was used to insert a light‐sensory domain. (B) MD simulations of apo‐hPKM2 (red) and hPKM2‐FBP (green) were projected onto the first two principal components (PCs) determined from the PCA of the hPKM2‐FBP trajectory. The time evolutions of both the apo‐ and FBP‐bound hPKM2 MD trajectories, from 0 to 300 ns, are represented as colour gradients. A k‐means clustering of the PCA plot identified two distinct Clusters (i) and (ii) (dashed boxes) that explained 100% of the point variability of the data set. Single most dominant conformations of hPKM2‐FBP were extracted from each of these two clusters and are shown in cartoon representations; FBP and the catalytic histidine (His78, blue) are shown as spacefill representations. Comparison of the structures extracted from Cluster (i) and Cluster (ii) revealed two dominant conformational motions (indicated with the grey arrows) as the protein transitioned from Cluster (i) to Cluster (ii): closure of the B‐domain and flipping downward of the HLH, which led us to assign Clusters (i) and (ii) as ‘open’ and ‘closed’ conformation respectively. (C) A superimposition of hPKM2 active site residues revealed that His78 adopts an altered side‐chain conformation in the crystal structure when the protein is bound to activators fructose 1,6‐bisphosphate (FBP) and TEPP‐46 (PDB ID: 3u2z) and when bound to L‐serine and FBP (PDB ID: 4b2d), compared to crystal structures of apo‐hPKM2 (PDB ID: 3bjt) and hPKM2 in complex with the allosteric inhibitor L‐phenylalanine (PDB ID: 4fxj). In the structures bound to the activators, the Nδ1 nitrogen group is positioned pointing towards the substrate‐binding pocket, and is flipped away from the substrate‐binding pocket in the apo‐ and phenylalanine‐bound structures. Consistent with this change following allosteric ligand binding, the conformation of His78 can provide a read‐out for whether PKM2 is in the catalytically active or inactive state. Below, 18 evenly spaced snap‐shots of the conformation of His78 are shown for a MD simulation of hPKM2‐FBP in the ‘open’ and following the transition to the ‘closed’ states. In the ‘open’ state [Cluster (i) in Fig. 1B], the side chain of His78 is flexible and switches into a conformation seen in that of the active crystal structures following the transition of the simulation into cluster ii [Cluster (ii) in Fig. 1B]. The time evolution of the χ2 dihedral angle of His78 along MD simulations of apo‐hPKM2 and hPKM2‐FBP, colour‐coded according to the scale on the right. The His78 switch occurs after ~ 50 ns (hPKM2‐FBP), while apo‐hPKM2 remains flexible for the duration of the simulation.