

Abstract
The genetic heterogeneity of Mendelian disorders results in a significant proportion of patients that are unable to be assigned a confident molecular diagnosis after conventional exon sequencing and variant interpretation. Here we evaluated how many patients with an inherited retinal disease (IRD) have variants of uncertain significance (VUS's) that are disrupting splicing in a known IRD gene by means other than affecting the canonical dinucleotide splice site. Three in silico splice-affecting variant predictors were leveraged to annotate and prioritize variants for splicing functional validation. An in vitro minigene system was used to assay each variant's effect on splicing. Starting with 745 IRD patients lacking a confident molecular diagnosis we validated 23 VUS's as splicing variants that likely explain disease in 26 patients. Using our results we optimized in silico score cutoffs to guide future variant interpretation. Variants that alter base pairs other than the canonical GT-AG dinucleotide are often not considered for their potential effect on RNA splicing but in silico tools and a minigene system can be utilized for the prioritization and validation of such splice-disrupting variants. These variants can be overlooked causes of human disease but can be identified using conventional exon sequencing with proper interpretation guidelines.
Keywords: Molecular diagnosis, Mendelian disease, variants of uncertain significance (VUS's), non-canonical splicing variants, inherited retinal degenerations, minigene
Introduction
Many Mendelian disorders are genetically heterogeneous having a multitude of different disease genes in which a variety of disease-causing variants have been discovered. This genetic heterogeneity presents an on-going challenge in the field of human genetics in that disease-causing variants are not always obvious and therefore assigning a molecular diagnosis to a patient with a heterogeneous disease can be difficult. A molecular diagnosis is the critical step that unlocks gene-specific therapies (Zhao, et al., 2014), mutation-specific therapies (Gerard, et al., 2016), genetic counseling, family planning, and prognosis management (Ellingford, et al., 2015).
Inherited retinal degenerations (IRDs) are a group of both clinically and genetically heterogeneous Mendelian disorders that are categorized by the predominant cell type affected and the disease's age of onset. Almost all IRDs have both genotypic and phenotypic overlap making both an accurate clinical diagnosis, as well as a confident molecular diagnosis, challenging. IRDs as a group however have made remarkable progress in the past decade when it comes to disease treatment making it all the more crucial for a patient to receive a molecular diagnosis. Multiple IRD genes have active gene therapy clinical trials which a patient could become eligible for after the appropriate molecular diagnosis (Han, et al., 2014; Jacobson, et al., 2012; MacLaren, et al., 2014).
The conventional most cost-effective method for the molecular diagnosis of IRDs involves next-generation sequencing of the protein-coding exons of every gene with an associated retinal phenotype followed by interpreting variants that obviously alter the protein-coding sequence. This results in a molecular diagnosis discovery rate of 66% for Stargardt disease (Zaneveld, et al., 2015), 60% for retinitis pigmentosa (RP)(Zhao, et al., 2014), 75% for Leber congenital amaurosis (LCA)(Wang, et al., 2015a), and 70% for Usher syndrome (Jiang, et al., 2015). The disease with the most molecular diagnosis success, LCA, still has a quarter of its patients lacking a molecular diagnosis after conventional methods. There are three main possibilities for what could be responsible for disease in the remaining proportions: 1) Variants affecting genes not yet linked with an IRD, 2) Variants affecting known IRD genes that we are not sequencing or are failing to detect and interpret as pathogenic, or 3) Novel genetic mechanisms beyond what is classically expected for a Mendelian disease. This study sought to evaluate a potential contribution to the second option - variants that disrupt splicing other than at canonical splice sites.
Mammalian RNA splicing is a delicate process whose precise coordination is not fully understood but its regulation is critical for the proper expression of most genes and their isoforms. In fact 15% of disease-associated SNVs disrupt splicing and 25% of exonic disease-associated variants disrupt splicing regulatory elements (Soukarieh, et al., 2016). The importance of understanding this process and being able to predict which variants alter splicing is therefore essential to understanding human disease. As early as 1992 the field of genetics has known that human disease can be caused by non-canonical splice site variants and these variants account for over 33% of pathogenic donor splice site variants and more than 10% of acceptor splice site variants (Krawczak, et al., 1992).
Many variant annotation pipelines only annotate the two base pairs (bp) directly flanking an exon as “splice site” variants, the canonical GT-AG dinucleotides considered required for splice site recognition. The effect on splicing of variants at other positions around the exon-intron junction can vary depending on the exact substitution because the spliceosome displays a large preference for certain consensus sequences over others (Rosenberg, et al., 2015). Therefore there is no guarantee without further testing that non-canonical splicing variants will affect splicing. In this study we leverage in silico splice-affecting variant predictors to prioritize non-canonical splicing variants for splicing functional validation using an in vitro minigene system in order to evaluate the contribution of non-canonical splicing variants to human disease in our IRD cohort.
Materials and Methods
Clinical diagnosis and patient recruitment
All probands discussed herein were clinically diagnosed with retinitis pigmentosa, Usher syndrome, Leber congenital amaurosis, or Stargardt disease following a thorough ophthalmologic examination by a qualified collaborating ophthalmologist. This study was approved by the institutional ethics boards at each affiliated institution and adhered to the tenets of the declaration of Helsinki. Blood was collected from each proband and family members when available after obtaining informed consent. DNA was extracted using the Qiagen blood genomic DNA extraction kit (Qiagen, Hilden, Germany).
NGS and variant annotation
Library preparation and NGS was performed as previously described (Soens, et al., 2016). Fragmented patient DNA was captured using either the NimbleGen SeqCap EZ Human Exome Library v2.0 (Roche, Basel, Switzerland) or a custom designed SureSelect capture panel (Agilent, Santa Clara, CA) targeting the exons of known IRD genes (Wang, et al., 2015b), the version used covers the 281 genes screened in this study (Supp. Table S1).
NGS data processing, variant calling, and protein-coding variant annotation was performed as previously described with added improvements to the variant filtration step (Soens, et al., 2016). A population frequency threshold of 0.5% was used to filter out common variants which occur too frequently to be the cause of rare IRDs. Four variant frequency databases were merged into our “filtdb” to determine allele frequencies for filtering including ExAC (Lek, et al., 2016), CHARGE (Psaty, et al., 2009), UK10K (Consortium, et al., 2015), and HGVD (Higasa, et al., 2016). Annotation scores for splice-affecting variants were compiled from several sources. Scores for novel splice sites from NNsplice (Reese, et al., 1997) were obtained through an in-house script written to query the NNsplice v0.9 webserver (http://www.fruitfly.org/seq_tools/splice.html), scores from dbscSNV (Jian, et al., 2014) were obtained from dbNSFP v2.9 downloads (https://sites.google.com/site/jpopgen/dbNSFP), additional scores for the splice site loss prediction comparison were obtained from SPIDEX (Xiong, et al., 2015), downloaded from the SPIDEX v1 database (https://www.deepgenomics.com/spidex/), and for Splice Site Finder, Max Ent Scan, Gene Splicer, Human Splicing Finder, and NNsplice, compiled using Alamut Batch v1.4 (http://www.interactive-biosoftware.com/, 2016).
Variant prioritization strategy for splicing functional validation
Our scheme for prioritizing variants for functional validation was as follows (Supp. Figure S1a). Starting with every exon-captured SNV called by Atlas2 from all 745 IRD patients: 1) Removed variants not in one of 281 known IRD genes (Supp. Table S1). 2) Filtered out variants found at an allele frequency greater than 0.5%, variants deemed too common to cause the rare IRDs being studied. 3) Only retained variants annotated by either dbscSNV score to lie within a splicing consensus sequence, or by NNsplice to form a new splice site with a strength >10% of wildtype. 4) For variants in a known recessive disease gene, the candidate splicing variant had to be homozygous or compound heterozygous with a likely pathogenic allele following the ACMG guidelines (Richards, et al., 2015). 5) All variants were confirmed using Sanger sequencing. 6) Compound heterozygous variants must be in trans and segregate with disease when family members are available. 7) Lastly candidates were prioritized for minigene validation based on the dbscSNV scores or their NNsplice score change.
Minigene molecular cloning, transfection, and RT-PCR
To assess if our prioritized variants have an effect on splicing we used an established minigene reporter assay the RHCglo minigene (Singh and Cooper, 2006). A genomic region from each patient, consisting of the exon containing or flanked by the candidate splicing variant (the test exon) and between 50-800 base pairs of surrounding intron, was PCR-amplified with the addition of restriction enzyme sites. Forward primers contained the SalI site, reverse primers contained the XbaI site. Using heterozygous patient DNA as a template, a wildtype (WT) and variant (Var) amplicon was obtained, or a wildtype sequence was amplified from control DNA when the patient was homozygous for a variant. RHCglo and the PCR products obtained were digested with SalI and XbaI at 37° Celsius for 2 hours. The minigene and PCR products are purified via QIAquick PCR purification kit (Qiagen), ligated together via an overnight ligation, transformed into competent E.coli and grown on agar plates containing ampicillin. Colony PCR was performed and products were Sanger sequenced to identify colonies carrying the desired inserts. Colonies were selected and grown overnight in 25 mL YPD, 25 μL ampicillin (100 mg/ml). Bacteria were harvested and wildtype and variant plasmids were purified using the PureLink HiPure Plasmid Midiprep Kit (Invitrogen, Carlsbad, CA).
HEK293T cells were seeded in a 24 well plate 0.125 × 106 cells per well. The following day the transfection reagent is prepared with minigene, Lipofectamine2000 (Invitrogen), reduced serum media Opti-MEM, and added to each well. 48 hours after transfection, cells are harvested, and RNA extracted. cDNA was synthesized from 1 μg of RNA. PCR was performed on the synthesized cDNA using primers that anneal specifically to exons 1 and 3 of the RHCglo minigene. PCR products were run on 2% agarose gels. Bands were excised, purified by gel extraction, and Sanger sequenced to determine their exact identity. HEK293T cells are first seeded in a 24 well plate 0.125 × 106 cells per well. The following day the transfection reagent is prepared with DNA, Lipofectamine2000 (Invitrogen), reduced serum media Opti-MEM, and added into each well. 48 hours after transfection, cells are harvested from each well and RNA extracted via the use of QIAshredder (Qiagen) and the RNeasy Mini Kit (Qiagen). cDNA was synthesized from 1 μg of extracted RNA using SuperScript III Reverse Transcriptase (Invitrogen). PCR was performed on the synthesized cDNA using primers that anneal specifically to exons 1 and 3 located in the RHCglo minigene. PCR products were run on 2% agarose gels. Bands were excised from the gel, purified using the QIAquick Gel Extraction Kit (Qiagen), and Sanger sequenced to determine their exact relation to the exon patient DNA.
Sanger sequencing
Sanger sequencing was used to confirm the authenticity of variants identified by NGS, to confirm the variant properly segregated with disease, and to confirm the sequence of gel-extracted RT-PCR bands. Sequencing was performed as previously described (Soens, et al., 2016).
Database submission
All variants that we experimentally tested with the minigene system have been submitted to the ClinVar database and can be browsed at https://www.ncbi.nlm.nih.gov/clinvar/.
Results
Leveraging splice-affecting variant predictors identifies 25 candidate splicing variants
IRDs are genetically heterogeneous so we restricted our candidate variant search to genes previously established to be IRD genes. Therefore, we only considered variants found in one of 281 genes previously reported to cause a retinal phenotype when mutated. Starting with a cohort of 745 IRD patients lacking a molecular diagnosis, we utilized three in silico splice-affecting variant predictors along with other genetic prerequisites (see Variant prioritization strategy in Materials and Methods) to identify 25 initial candidate variants in 28 probands that could be causing an IRD by disrupting splicing (Table 1). Pedigrees displaying variant segregation for nine of the probands are shown in Figure 1. Two variants subsequently failed to segregate with disease within the proband's family but were retained for minigene testing as negative controls. None of these candidate variants alter canonical dinucleotide splice sites and therefore their actual impact on splicing was uncertain.
Table 1. Patient variant information.
| Minigene-validated splicing-disrupted allele | 2nd allele | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient ID | Clinical diagnosis |
Gene | Splicing variant ID |
Sanger validated zygosity |
Chromosomal variant | cDNA variant (position relative to existing splice site or to novel splice site) |
Protein variant | dbscSNV's known splice site-disrupting predictions |
NNsplice's novel splice site prediction |
Chromosomal variant | cDNA variant | Protein variant | Sanger validated zygosity |
Evidence of 2nd allele pathogenicity (PMID) | Splicing allele - 2nd allele segregation status |
||
| ada score | rf score | Variant- created donor |
Variant- created acceptor |
||||||||||||||
| RKK_209 | Leber congenital amaurosis | RPGRIP1 | #1 | Homozygous |
NC_000014.8: g.21802869G>A |
NM_020366.3: c.3339+5G>A |
Intronic | 1.000 | 0.982 | NA | NA | None - minigene-validated splicing variant is homozygous | Homozygous | ||||
| 3674 | Leber congenital | PDE6A | #2 | Heterozygous |
NC_000005.9: g.149264037C>A |
NM_000440.2: c.2027+5G>T |
Intronic | 1.000 | 0.924 | NA | NA |
NC_000005.9: g.149264103C>A |
NM_000440.2: c.1966G>T |
NP_000431.2: p.Glu656Ter |
Heterozygous | In trans with splicing variant, predicted null variant | In trans on NGS reads, Parents not available |
| SRF_611 | Leber congenital amaurosis | AHI1 | #3 | Heterozygous |
NC_000006.11: g.135763715C>A |
NM_001134830.1: c.1912+5G>T |
Intronic | 1.000 | 0.964 | NA | NA |
NC_000006.11: g.135749790A>T |
NM_001134830.1: c.2600T>A |
NP_001128302.1: p.Val867Glu |
Heterozygous | In trans with splicing variant, multiple in s ilico tools predict damaging | Segregation passed |
| 3719 | Leber congenital amaurosis | SLC38A 8 | #4 | Homozygous |
NC_000016.9: g.84070302C>T |
NM_001080442.1: c.388+5G>A |
Intronic | 1.000 | 0.928 | NA | NA | None - minigene-validated splicing variant is homozygous | Homozygous | ||||
| ZPQ_028 | Leber congenital amaurosis | CEP290 | #5 | Heterozygous |
NC_000012.11: g.88508350A>C |
NM_025114.3: c.1910-11T>G |
Intronic | 0.986 | 0.666 | NA | NA |
NC_000012.11: g.88487606_88487607insT |
NM_025114.3: c.3249_3250insA |
NP_079390.3: p.Arg1084ThrfsTer11 |
Heterozygous | In trans with splicing variant, predicted null variant | Segregation passed |
| ZPQ_080 | Leber congenital amaurosis | CEP290 | #5 | Heterozygous |
NC_000012.11: g.88508350A>C |
NM_025114.3: c.1910-11T>G |
Intronic | 0.986 | 0.666 | NA | NA |
NC_000012.11: g.88490671T>A |
NM_025114.3: c.3097A>T |
NP_079390.3: p.Lys1033Ter |
Heterozygous | In trans with splicing variant, predicted null variant | Segregation passed |
| SRF_1724 | Usher syndrome | USH2A | #6 | Homozygous |
NC_000001.10: g.216595191T>A |
NM_206933.2: c.485+3A>T |
Intronic | 1.000 | 0.844 | NA | NA | None - minigene-validated splicing variant is homozygous | Homozygous | ||||
| SRF_692 | Leber congenital amaurosis | SPATA7 | #7 | Heterozygous |
NC_000014.8: g.88903941G>T |
NM_001040428.3: c.1119G>T (First exonic bp) |
NP_001035518.1: p.Glu373Asp |
1.000 | 0.998 | NA | NA |
NC_000014.8: g.88903909C>T |
NM_001040428.3: c.1087C>T |
NP_001035518.1: p.Arg363Ter |
Heterozygous | In trans with splicing variant, predicted null variant, reported disease-causing in literature (19268277, 21602930) | In trans on NGS reads, Parents not available |
| FBP_54 | Leber congenital amaurosis | CRB1 | #8 | Homozygous |
NC_000001.10: g.197398744T>C |
NM_001193640.1: c.2506T>C (First exonic bp) |
NP_001180569.1: p.Cys836Arg |
0.090 | 0.334 | NA | NA | None - minigene-validated splicing variant is homozygous | Homozygous | ||||
| FBP_353 | Leber congenital amaurosis | RPGRIP1 | #9 | Heterozygous |
NC_000014.8: g.21789561G>A |
NM_020366.3: c.1611G>A (First exonic bp) |
NP_065099.3: p.Gln537Gln |
1.000 | 1.000 | NA | NA |
NC_000014.8: g.21795830_21795831insT |
NM_020366.3: c.2759_2760insT |
NP_065099.3: p.Gln920HisfsTer14 |
Heterozygous | Compound heterozygous with splicing variant, predicted null variant, reported disease-causing in literature (11283794) | Parents not available |
| SRF_268 | Stargardt disease | ABCA4 | #10 | Heterozygous |
NC_000001.10: g.94496547C>T |
NM_000350.2: c.4253+5G>A |
Intronic | 0.998 | 0.858 | NA | NA |
NC_000001.10: g.94487505T>C |
NM_000350.2: c.4670A>G |
NP_000341.2: p.Tyr1557Cys |
Heterozygous | Compound heterozygous with splicing variant, multiple in silico tools predict damaging | Parents not available |
| FBP_CBA | Stargardt disease | ABCA4 | #11 | Heterozygous |
NC_000001.10: g.94476351C>T |
NM_000350.2: c.5714+5G>A |
Intronic | 1.000 | 0.988 | NA | NA |
NC_000001.10: g.94528266G>A |
NM_000350.2: c.1804C>T |
NP_000341.2: p.Arg602Trp |
Heterozygous | In trans with splicing variant, reported disease-causing in literature (9973280) | Segregation passed |
| SRF_175 | Leber congenital amaurosis | RPE65 | #12 | Heterozygous |
NC_000001.10: g.68896965C>A |
NM_000329.2: c.1338G>T (First exonic bp) |
NP_000320.1: p.Arg446Ser |
1.000 | 0.940 | NA | NA |
NC_000001.10: g.68912438A>C |
NM_000329.2: c.200T>G |
NP_000320.1: p.Leu67Arg |
Heterozygous | In trans with splicing variant, reported disease-causing in literature (22509104, 23661369) | Segregation passed |
| SRF_1582 | Leber congenital amaurosis | RPE65 | #12 | Homozygous |
NC_000001.10: g.68896965C>A |
NM_000329.2: c.1338G>T (First exonic bp) |
NP_000320.1: p.Arg446Ser |
1.000 | 0.940 | NA | NA | None - minigene-validated splicing variant is homozygous | Homozygous | ||||
| 1302 | Leber congenital amaurosis | KCNJ13 | #13 | Homozygous |
NC_000002.11: g.233635615G>A |
NM_001172417.1: c.218C>T (Third exonic bp) |
NP_001165888.1: p.Thr73Ile |
0.999 | 0.922 | NA | NA | None - minigene-validated splicing variant is homozygous | Homozygous | ||||
| 285 | Leber congenital amaurosis | CEP290 | #14 | Heterozygous |
NC_000012.11: g.88512415C>T |
NM_025114.3: c.1623+5G>A |
Intronic | 1.000 | NA | NA | NA |
NC_000012.11: g.88494960T>C |
NM_025114.3: c.2991+1655A>G |
Intronic | Heterozygous | Compound heterozygous with splicing variant, reported disease-causing by cryptic exon formation in literature (16909394, 19823873, 23591405) | Parents not available |
| SRF_1065 | Retinitis pigmentosa | RPGR | #15 | Hemizygous |
NC_000023.10: g.38150209T>A |
NM_000328.2: c.1572+3A>T |
Intronic | 1.000 | 0.926 | NA | NA | None - minigene-validated splicing variant is hemizygous | Segregation passed | ||||
| RKK_212 | Retinitis pigmentosa | ABCA4 | #16 | Heterozygous |
NC_000001.10: g.94473856G>T |
NM_000350.2: c.5836-3C>A |
Intronic | 1.000 | 0.852 | NA | NA |
NC_000001.10: g.94564350C>A |
NM_000350.2: c.768G>T |
NP_000341.2: p.Val256Val |
Heterozygous | Compound heterozygous with splicing variant, reported disease-causing in literature (10090887, 19074458, 22264887) | Parents not available |
| SRF_1694 | Usher syndrome | USH2A | #17 | Heterozygous |
NC_000001.10: g.215931934T>A |
NM_206933.2: c.11389+3A>T |
Intronic | 0.982 | 0.638 | NA | NA |
NC_000001.10: g.215963400C>T |
NM_206933.2: c.10182+1G>A |
Splicing | Heterozygous | In trans with splicing variant, predicted null variant | Segregation passed |
| SRF_1687 | Usher syndrome | USH2A | #17 | Heterozygous |
NC_000001.10: g.215931934T>A |
NM_206933.2: c.11389+3A>T |
Intronic | 0.982 | 0.638 | NA | NA |
NC_000001.10: g.216051224T>C |
NM_206933.2: c.8559-2A>G |
Splicing | Heterozygous | Compound heterozygous with splicing variant, predicted null variant | Parents not available |
| JMS_010 | Leber congenital amaurosis | RPE65 | #18 | Heterozygous |
NC_000001.10: g.68903897T>C |
NM_000329.2: c.1101A>G (First intronic bp) |
NP_000320.1: p.Arg367Arg |
NA | NA | 1.00 1bp away | None |
NC_000001.10: g.68910540C>T |
NM_000329.2: c.272G>A |
NP_000320.1: p.Arg91Gln |
Heterozygous | Compound heterozygous with splicing variant, reported disease-causing in literature (11095629, 19431183) | Parents not available |
| DGB_032 | Leber congenital amaurosis | RPGRIP1 | #19 | Heterozygous |
NC_000014.8: g.21770720A>G |
NM_020366.3: c.564A>G (Fifth intronic bp) |
NP_065099.3: p.Glu188Glu |
NA | NA | 0.94 5bp away | None |
NC_000014.8: g.21795785_21795786insT |
NM_020366.3: c.2714_2715insT |
NP_065099.3: p.Asn907Ter |
Heterozygous | In trans with splicing variant, predicted null variant | Segregation passed |
| SRF_1099 | Unspecified retinal dystrophy | ADGRA3 | #20 | Heterozygous |
NC_000004.11: g.22422659C>T |
NM_145290.3: c.1659G>A (Second intronic bp) |
NP_660333.2: p.Thr553Thr |
NA | NA | None | 0.66 2bp away |
NC_000004.11: g.22390451C>T |
NM_145290.3: c.2843G>A |
NP_660333.2: p.Arg948His |
Heterozygous | Compound heterozygous with splicing variant, multiple in silico tools predict damaging | Parents not available |
| 3772 | Leber congenital amaurosis | RP2 | #21 | Hemizygous |
NC_000023.10: g.46696640A>C |
NM_006915.2: c.102+3A>C |
Intronic | 0.999 | 0.890 | NA | NA | None - minigene-validated splicing variant is hemizygous | Parents not available | ||||
| JMS_011 | Leber congenital amaurosis | RP2 | #22 | Heterozygous |
NC_000023.10: g.46696637G>A |
NM_006915.2: c.102G>A (First exonic bp) |
NP_008846.2: p.Lys34Lys |
1.000 | 0.992 | NA | NA | None - minigene-validated splicing variant is X-linked | Parents not available | ||||
| ZPQ_055 | Leber congenital amaurosis | SPATA7 | #23 | Heterozygous |
NC_000014.8: g.88852181G>A |
NM_001040428.3: c.19G>A (First exonic bp) |
NP_001035518.1: p.Val7Ile |
1.000 | 1.000 | NA | NA |
NC_000014.8: g.88903909C>T |
NM_001040428.3: c.1087C>T |
NP_001035518.1: p.Arg363Ter |
Heterozygous | In trans with splicing variant, predicted null variant, reported disease-causing in literature (19268277, 21602930) | Segregation passed |
| Two patients with a candidate splicing variant that failed segregation - minigene negative controls | |||||||||||||||||
| ZBL-33 | Retinitis pigmentosa | KIAA1549 | Negative Control 1 | Heterozygous |
NC_000007.13: g.138546029G>A |
NM_001164665.1: c.5103C>T (First exonic bp) |
NP_001158137.1: p.Ser1701Ser |
NA | NA | None | 0.58 0bp away |
NC_000007.13: g.138603068G>A |
NM_001164665.1: c.1304C>T |
NP_001158137.1: p.Thr435Met |
Heterozygous | Likely not pathogenic | Segregation failed |
| SRF_1037 | Retinitis pigmentosa | CACNA1F | Negative Control 2 | Hemizygous |
NC_000023.10: g.49084860C>T |
NM_001256789.1: c.867G>A (Second exonic bp) |
NP_001243718.1: p.Gly289Gly |
NA | NA | 0.66 1bp away | None | None - variant is X-linked | Segregation failed | ||||
| One patient with a homozygous frameshift in a second known recessive disease gene potentially contributing to their phenotype | |||||||||||||||||
| 3719 | Leber congenital amaurosis | PLA | 2G6 | Homozygous |
NC_000022.10: g.38522454delG |
NM_001004426.1: c.1189delC |
NP_001004426.1: p.Leu397TyrfsTer2 |
Reported in literature (16783378, 24628580) to cause infantile neuroaxonal dystrophy. Mutations in PLA2G6 cause optic atrophy with nystagmus on the same phenotypic spectrum as infantile neuroaxonal dystrophy. | |||||||||
Figure 1. Pedigrees for nine of the probands passing segregation for their candidate splicing variant.
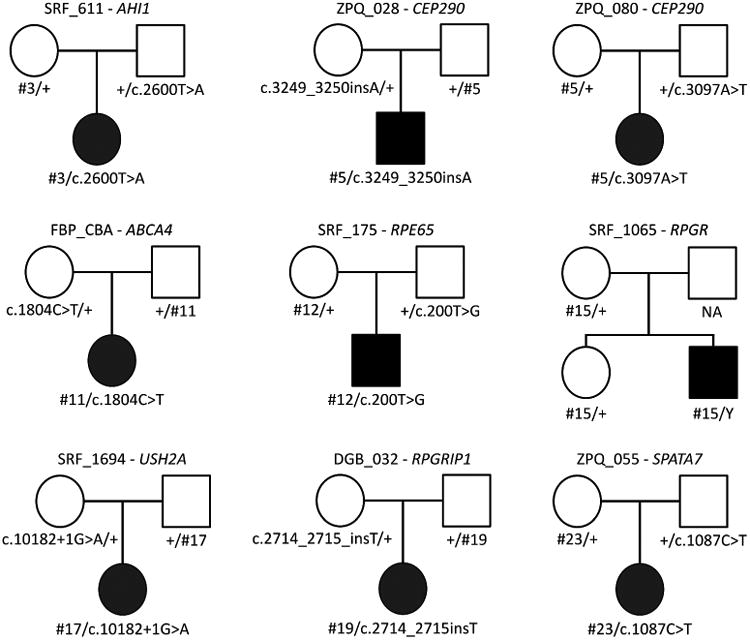
Circles represent females, squares males. Solid shapes indicated the affected proband while empty shapes are unaffected relatives. The NA indicates a father whose DNA was unavailable. The candidate splicing variant is indicated by its number (#) ID which can be compared to Table 1. cDNA change annotations are taken from Table 1 where corresponding gene models are noted.
A minigene functional validation system reveals abnormal splicing caused by 23 variants
To confirm that each candidate splicing variant has a functional impact on RNA splicing we utilized an in vitro minigene system, the RHCglo vector, as a splicing assay. RHCglo contains a single gene with only three exons and functional splicing. The middle exon is flanked by restriction enzyme sites that can be used to substitute in an exon of interest (the “test” exon) with or without the variant to be assayed (Supp. Figure S1b). Every variant evaluated in this study is a single nucleotide substitution and every gel band that could be cleanly excised was sequenced to confirm its composition (Figure 2). The 23 variants can be organized into four groups based on their location within the gene and their consequence on RNA splicing. The two variants assayed as potential negative controls showed no effect on splicing (NC1) and an increase in test exon inclusion (NC2) and therefore are likely not pathogenic as expected.
Figure 2. A minigene splicing assay reveals variant-induced aberrant splicing.

Gel electrophoresis of RT-PCR products for all tested minigenes. The control minigene is unmodified RHCglo containing a ∼50 base pair exon that displays partial exon inclusion. Numbers (#) refer to the splicing variant ID while NC1 and NC2 refer to the two negative control variants tested. The differences between the respective wildtype (WT) and variant (Var) band composition reveal the variant-induced changes in splicing. The adjacent diagrams, which are not to scale, are provided as a schematic of the variant-induced changes in transcript configuration. Dark gray exons correspond to the first and last exon of the minigene, light gray exons correspond to the exons cloned in for variant testing, a line corresponds to the flanking introns included for variant testing. A band labeled ART indicates an artefact of the minigene construction process which does not correspond to a possible endogenous splicing event.
Eleven variants (Variant IDs #1-11) located in the splicing consensus sequence of a middle exon (an exon that is neither the first nor last) resulted in clear changes in minigene splicing. All of these variants cause a shift in splicing towards transcripts that do not contain the test exon, evidence that each variant is resulting in a loss of splice site recognition and increased exon skipping. Three variants (Variant IDs #12-14) were located in the splicing consensus sequence of a middle exon with a neighboring adjacent exon (<100bp away). The adjacent exon, due to cloning limitations, was therefore included in the minigene transcripts in both wildtype and variant contexts adjacent to the test exon. Evidence for each variant's disruption of the consensus sequence it is located in is apparent and a corresponding loss of the test exon can be observed upon introduction of each variant.
Six variants (Variant IDs #15-20) were located either within a splicing consensus sequence or deep within the exon of a middle exon, but distinctly resulted in alternative splice site usage rather than loss of the test exon. All three bands resulting from variant #15 correspond to the usage of different weak intronic cryptic donor splice sites located nearby in the downstream intron of RPGR. The major band caused by variant #16 corresponds to an elongation of the exon by 30bp due to the presence of a cryptic acceptor splice site in intron 41 of ABCA4 that is activated upon disruption of the acceptor splice site where variant #16 is located. The major band for variant #17, which displays an obvious downwards shift compared to wildtype, corresponds to a truncation of 43bp due to the usage of a cryptic donor splice site found within exon 58 of USH2A that is utilized upon variant introduction. The three variants #18-20 are the only three variants found deep within an exon rather than at a known splicing consensus sequence. These SNVs, which are synonymous on the protein-coding level, were predicted by NNsplice to create new splice sites as opposed to disrupting a known splice site. The variant-containing transcripts all show an out-of-frame truncation of the exon corresponding to the use of the variant-created novel splice sites. #18 and #19 coincidentally both result in a 28bp truncation while #20 causes a 55bp truncation.
Three variants (Variant IDs #21-23) were found in the donor splice site consensus sequence of a gene's first exon and therefore there was no characterized acceptor splice site upstream to evaluate the variant in our minigene system. We decided to proceed with minigene testing but modified our cloning design to include a large portion of the upstream 5′ UTR with the hypothesis that there may be a cryptic acceptor splice site that is not used in vivo but that we could use to evaluate the impact of our variant on the recognition of the donor splice site in question (see Supp. Figure S1c). This strategy proved successful and multiple viable acceptor splice sites were discovered as determined by mapping the resulting RT-PCR bands back to their genomic locus. Each of these three variants resulted in the destruction of the donor splice site as evident from the loss of exon-including splicing products once the SNV was introduced. We expect the in vivo consequence would however be the inclusion of a proportion or all of intron 1 in the mRNA transcript.
Specific clinical features support the minigene-validated patient genotypes
Although the minigene results support a disruption of splicing in the 23 variant alleles reported herein, abnormal splicing alone doesn't guarantee pathogenicity. To further support the pathogenic nature of the tested variants we examined the specific clinical phenotype of each disease-affected retina, in Figure 3 we show five probands that matched genotype-phenotype observations reported previously. Diseased retinas resulting from mutations in CRB1 are unusually thickened and lack proper lamination (Jacobson, et al., 2003), both features which are evident in optical coherence tomography images of FBP_54 (Figure 3a). The fundus of proband JMS_010 exhibits narrowed retinal vessels, a mottled pattern of pigmentation in the RPE, and a confluent macular atrophy, all features which have been previously observed in retinas affected by mutations in RPE65 (Figure 3b). Stargardt disease is a monogenic disorder caused by mutations in ABCA4. SRF_268 was diagnosed with Stargardt disease after reporting impaired color vision and presenting a characteristic atrophic maculopathy with retinal flecks in both eyes (Figure 3c). SRF_1065's splicing variant is in RPGR on the X chromosome. IRD caused by mutations in RPGR manifest a severe retinal phenotype consistent with SRF_1065 including poor vision and night blindness from early childhood, severe astigmatism, and a visual field restricted to tunnel vision with flat electroretinogram waves. In addition, the mother of SRF_1065 is highly myopic which is typical for a female carrier of X-linked RP (Figure 3d). The phenotype of SRF_1694 lends support to the disease causality of their USH2A variants due to the specific combination and progression of hearing and vision deficits that lead to the clinical diagnosis of Usher syndrome type 2. Usher II is only known to be caused by mutations in three genes and USH2A is the most commonly affected. SRF_1694 noticed deafness and night blindness at the age of 3 years old where after her visual acuity progressively deteriorated. Her fundus showed diffuse grayish pigment clumps involving the macula and thinned retinal blood vessels (Figure 3e).
Figure 3. Select ophthalmologic data supportive of the splicing variant's pathogenicity based on known genotype-phenotype associations.
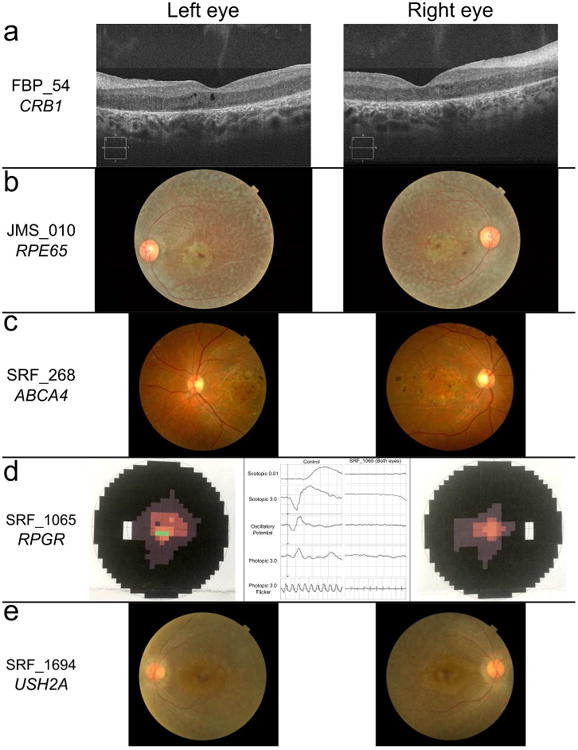
(a) Optical coherence tomography images of FBP_54's retina compatible with CRB1 disease-causing variants. (b) Fundoscopy images of JMS_010 compatible with RPE65 disease-causing variants. (c) Fundoscopy images of SRF_268 compatible with ABCA4 disease-causing variants. (d) Visual field tests from either eye of SRF_1065 as well as a flat electroretinogram recording which was present in both eyes, features compatible with RPGR disease-causing variants. (e) Fundoscopy images of SRF_1694 compatible with USH2A disease-causing variants.
In contrast to the above known genotype-phenotype correlations which support our findings, we also identified and validated splice-affecting variants in genes not previously linked with the clinical diagnoses of probands 3674, SRF_611, 3719, SRF_1099, 3772, and JMS_011. Due to the overlapping phenotypic spectrum that is known to exist among IRDs, and the absence of detectable variation in genes directly linked with their clinical diagnosis, we propose these variants as the most likely cause of the reported phenotype in these probands. Two cases are particularly interesting, proband 3719 has biallelic truncating variants (considering variant #4 as a full exon loss) in two genes that already have a Mendelian phenotype, SLC38A8 and PLA2G6. Neither of which are considered established IRD genes, but both of which report visual defects as a feature. We propose this rare combination of mutations manifested as infantile visual impairment preceding other complications leading to an LCA diagnosis. In proband SRF_1099 we identified compound heterozygous variants in ADGRA3 (GPR125), a candidate gene with only a single previous report of its connection to IRDs (Abu-Safieh, et al., 2013). Our findings provide an independent case of ADGRA3 variants identified in an IRD patient.
Comparison of in silico splice-affecting variant predictors and ideal score cutoff determination
29 tools that can provide a splicing prediction annotation score were considered and eight scores were selected for assessment based on the usability of the tool to obtain hundreds of predictions. The eight scores were also chosen based on recommendations of performance drawn from 11 different publications that utilized, reported on, or compared the performance of these tools (Colombo, et al., 2013; Houdayer, et al., 2012; Hunt, et al., 2014; Jian, et al., 2013; Jian, et al., 2014; Lelieveld, et al., 2016; Mort, et al., 2014; Neveling, et al., 2012; Sharma, et al., 2014; Soukarieh, et al., 2016; Tang, et al., 2016). The final eight scores chosen for comparison were NNsplice, Splice Site Finder, Max Ent Scan, Gene Splicer, Human Splicing Finder, SPIDEX's dPSI, and the rf and ada scores from dbscSNV. Our comparison focused on predictions for variants found within known splicing consensus sequences since 20 out of 23 of our validated variants fell within a known consensus sequence and caused a disruption of that splice site.
To perform the comparison we needed a set of negative control variants found within known splicing consensus sequences that do not disrupt splicing. We compiled 245 variants from our variant frequency filtering database “filtdb” which includes variants from over 80,000 exomes (ExAC, CHARGE, UK10K, HGVD) of individuals without an IRD. All negative control variants are located in a splicing consensus sequence of one of the 281 genes screened in our study and have an MAF > 5%. Raw predictive values from each tool for the 265 (20+245) variants used in this comparison can be found in Supp. Table S2.
Since many molecular diagnostic centers do not have the resources necessary to functionally validate splicing variants, a high degree of specificity is critical for in silico prediction tools to limit the time spent validating variants that are not truly splice-affecting. To this end we desired score cutoffs for each tool necessary to achieve a specificity of 95%, which translates to a prediction tool's score threshold correctly predicting 233 out of 245 negative control variants as having no effect on splicing. On the opposite end of the spectrum are research labs that study splicing and the effect variants have on splicing. These labs may want to identify all splice-affecting variants for use in large studies. To accommodate such searches we also wanted a score cutoff necessary to achieve a sensitivity of 90%, translating to the correct prediction that 18 out of 20 of our validated variants are indeed altering splicing. For both our 95% specific and 90% sensitive targets, we also wanted to know what the corresponding tradeoff was in sensitivity and specificity.
To visualize the score cutoff determination we plotted the predicted score of each variant, or the change in score induced by the variant (WT-Var), depending on the tool, for each variant in both groups of positive and negative controls. The variant score distributions for the three tools we initially utilized for variant identification are shown in Figure 4a while the score distributions for the other five assessed tools can be found in Supp. Figure S2. The more readily that the two control score distributions can be separated, the better the tool is at correctly predicting true positive and true negative splice-affecting variants. Graphing the sensitivity-specificity tradeoff at our desired targets for each score, we can visually compare the predictive performance of each method on our control variant sets (Figure 4b).
Figure 4. Determination and comparison of the score cutoff necessary to obtain a specificity of 95% or sensitivity of 90% for eight splice-affecting variant predictors when predicting variants that disrupt known splicing consensus sequences.
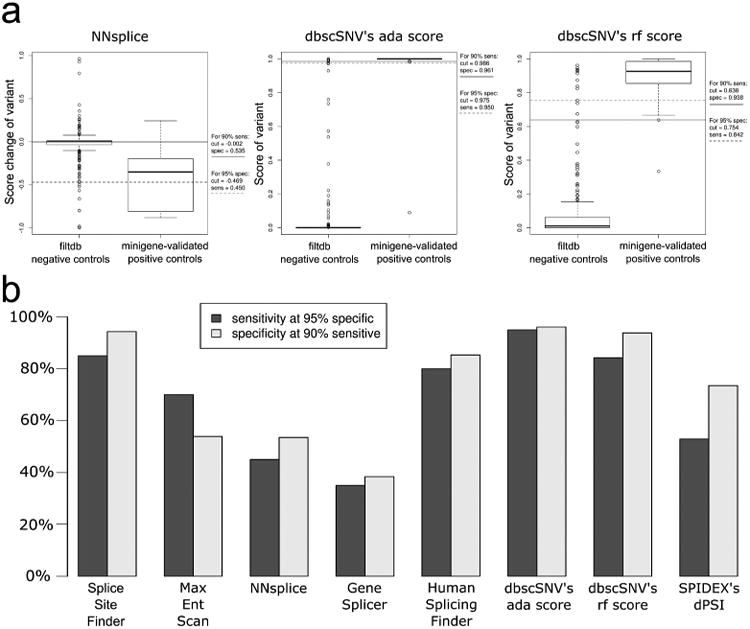
(a) Variant score distribution and the corresponding cutoffs needed for 95% specificity and 90% sensitivity for the ada_score, rf_score, and NNsplice predictors used in this study. (b) Histogram comparing the performance of the eight predictors by comparing the sensitivity and specificity that is achieved at the corresponding desired cutoffs of 95% specific and 90% sensitive.
Discussion
Many inherited disorders are genetically heterogeneous and there remains a significant fraction of patients in which the underlying molecular etiology of disease remains unknown. The impact that variants outside the canonical GU-AG dinucleotides may have on splicing is sometimes not considered, primarily due to the challenge of functional validation needed to be certain of a definitive effect. For this reason it is likely that a portion of patients lacking a molecular diagnosis may be due to non-canonical splice-altering variants that are currently considered VUS's. We here show that focusing only on variants that alter the canonical GU-AG dinucleotides as “splicing variants” may cause a patient with pathogenic variants in a known disease gene to be considered unsolved. The results described in this study have supported the molecular diagnosis of 26 individual probands showing that non-canonical splicing variants are prevalent in our Mendelian disease cohorts as 3.5% (26/745) of our patients carry them. Excitingly, six probands with variants in either RPE65 or ABCA4 are now directly eligible for on-going gene therapy trials to remedy their retinal disease.
The most logical method to probe a variant's effect on splicing is to perform RT-PCR directly on the transcript(s) in question using RNA from the patient's affected tissue. Unfortunately for a substantial number of human diseases, including IRDs, obtaining a sample of the tissue where the transcript is expressed requires an invasive, undesirable procedure, and therefore is not feasible. Also problematic is the knowledge that human cell lines and animal models of the same tissue are known to have differences in splicing (Garanto, et al., 2015). To circumvent this obstacle we leveraged a minigene system with the understanding that changes in minigene splicing are evidence of variant-induced splicing alterations but the exact in vivo impact of the variant on splicing may not be truly represented. All patient variants that passed our prioritization scheme and are reported herein had an obvious influence on the spliceosomal recognition of nearby splice sites resulting in deviations from wildtype RNA splicing that we believe is evidence of variant pathogenicity.
The locations of the splicing variants identified in our study show a remarkable concordance with positions that have been reported to have the largest effect on splice site definition, and those shown to be under the strongest conservation (Rivas, et al., 2015; Rosenberg, et al., 2015). Specifically our variants are enriched at the third and fifth intronic position of donor splice sites (4/23 and 8/23 respectively), as well as at the first exonic basepair of either type of splice site (6/23). Of particular interest, due to its distance from the exon-intron boundary, Rivas et al. notes the 11th intronic position of acceptor splice sites having an unusually strong effect on splicing. Splicing variant #5 is located at this precise position and resulted in a complete loss of exon inclusion using our minigene system. Our remaining variants were located, one each, at the third exonic base pair of a donor splice, the third intronic base pair of an acceptor splice site, and the last two both created novel splice sites by contributing to canonical positions at the new first and second intronic base pairs.
The loss of a splice site often results in exon skipping which will lead to a shortened protein-coding reading frame and frequently a frameshift, both events with probable deleterious consequences on protein function. In unusual occasions, nearby cryptic splice sites may be activated if at an appropriate distance and orientation. The overall aberrant splicing results combined with known genotype-phenotype associations and familial genetic evidence allows us increased confidence that each variant with minigene evidence in our study is indeed contributing to the patient's phenotype.
Although the dbscSNV scores have been cited in more than 10 publications since 2014, no independent study has evaluated the performance of the scores using a novel set of splice-altering variants (Bernardis, et al., 2016; Cheng, et al., 2017; Geoffroy, et al., 2015; Huffman, et al., 2015; Lelieveld, et al., 2016; Li and Wang, 2017; McLaren, et al., 2016; Nishio and Usami, 2017; Petersen, et al., 2017; Piovesan, et al., 2015; Vallee, et al., 2016; Xu, et al., 2017; Xue, et al., 2016; Zou, et al., 2017). In this study we show that both the ada and rf scores outperform most other predictive tools while also being freely available for academic use and precomputed for every splicing consensus sequence in the genome. Since these scores are readily obtained and can be easily used to annotate the thousands of variants produced by modern NGS, they are the scores we recommend other groups leverage when attempting similar studies of non-canonical splicing variants. We also recommend the score cutoffs determined during our comparative assessment (Figure 4a) but encourage other groups to fine tune their own variant prioritization pipelines.
We hope that geneticists realize the importance of considering the impact that all VUS's may have on splicing, particularly those that fall within a known splicing consensus sequence, but also those that fall deep within exons. The necessity to consider these variants is amplified by the fact that additional sequencing is not needed in most cases to identify such variants since most exon-targeted NGS has sufficient coverage of the flanking introns to call variants in the intronic portions of consensus sequences. The preexistence of the data needed for such studies combined with the available splice-affecting variant predictors and the effectiveness of an in vitro minigene system for validation means that the future will doubtlessly hold an abundance of pathogenic non-canonical splicing variants being identified as the cause of human disease.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the contributions of Kaylie Jones to this work.
This work is supported by the following grants: the National Eye Institute (R01EY022356, R01EY018571, EY002520), Retinal Research Foundation, Foundation Fighting Blindness (BR-GE-0613-0618-BCM), NIH shared instrument grant 1S10RR026550 to R.C.; the Foundation Fighting Blindness USA (CD-CL-0214-0631-PUMCH), the National Natural Science Foundation of China (81470669), Beijing Natural Science Foundation (7152116) and CAMS Innovation Fund for Medical Sciences (CIFMS 2016-12M-1-002) to R.S.; National Eye Institute (R01EY09076) to D.B.; R.K.K. acknowledges Foundation Fighting Blindness Canada, Canadian Institutes of Health Research, and Fonds de recherché Santé Quebéc et Réseau Vision. Z. Soens is supported by NEI training grant 5T32EY007001-40.
Grant Sponsors: National Eye Institute (R01EY022356, R01EY018571, EY002520, and R01EY09076); NIH shared instrument (1S10RR026550); Foundation Fighting Blindness (BR-GE-0613-0618-BCM and CD-CL-0214-0631-PUMCH); Retinal Research Foundation; National Natural Science Foundation of China (81470669); Beijing Natural Science Foundation (7152116); CAMS Innovation Fund for Medical Sciences (CIFMS 2016-12M-1-002); Canadian Institutes of Health Research; Fonds de recherché Santé Quebéc et Réseau Vision
Footnotes
The authors declare no conflict of interest.
References
- Abu-Safieh L, Alrashed M, Anazi S, Alkuraya H, Khan AO, Al-Owain M, Al-Zahrani J, Al-Abdi L, Hashem M, Al-Tarimi S, Sebai MA, Shamia A, Ray-Zack MD, Nassan M, Al-Hassnan ZN, Rahbeeni Z, Waheeb S, Alkharashi A, Abboud E, Al-Hazzaa SA, Alkuraya FS. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013;23(2):236–47. doi: 10.1101/gr.144105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardis I, Chiesi L, Tenedini E, Artuso L, Percesepe A, Artusi V, Simone ML, Manfredini R, Camparini M, Rinaldi C, Ciardella A, Graziano C, Balducci N, Tranchina A, Cavallini GM, Pietrangelo A, Marigo V, Tagliafico E. Unravelling the Complexity of Inherited Retinal Dystrophies Molecular Testing: Added Value of Targeted Next-Generation Sequencing. Biomed Res Int. 2016;2016:6341870. doi: 10.1155/2016/6341870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SJ, Shi FY, Liu H, Ding Y, Jiang S, Liang N, Gao G. Accurately annotate compound effects of genetic variants using a context-sensitive framework. Nucleic Acids Res. 2017;45(10):e82. doi: 10.1093/nar/gkx041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M, Vecchi G, Caleca L, Foglia C, Ripamonti CB, Ficarazzi F, Barile M, Varesco L, Peissel B, Manoukian S, Radice P. Comparative in vitro and in silico analyses of variants in splicing regions of BRCA1 and BRCA2 genes and characterization of novel pathogenic mutations. PloS one. 2013;8(2) doi: 10.1371/journal.pone.0057173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium UK, Walter K, Min JL, Huang J, Crooks L, Memari Y, McCarthy S, Perry JR, Xu C, Futema M, Lawson D, Iotchkova V, Schiffels S, Hendricks AE, Danecek P, Li R, Floyd J, Wain LV, Barroso I, Humphries SE, Hurles ME, Zeggini E, Barrett JC, Plagnol V, Richards JB, Greenwood CM, Timpson NJ, Durbin R, Soranzo N. The UK10K project identifies rare variants in health and disease. Nature. 2015;526(7571):82–90. doi: 10.1038/nature14962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellingford JM, Sergouniotis PI, Lennon R, Bhaskar S, Williams SG, Hillman KA, O'Sullivan J, Hall G, Ramsden SC, Lloyd CI, Woolf AS, Black GCM. Pinpointing clinical diagnosis through whole exome sequencing to direct patient care: a case of Senior-Loken syndrome. Lancet (London, England) 2015;385(9980):1916. doi: 10.1016/S0140-6736(15)60496-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garanto A, Duijkers L, Collin RWJ. Species-Dependent Splice Recognition of a Cryptic Exon Resulting from a Recurrent Intronic CEP290 Mutation that Causes Congenital Blindness. International Journal of Molecular Sciences. 2015;16(3):5285–5298. doi: 10.3390/ijms16035285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy V, Pizot C, Redin C, Piton A, Vasli N, Stoetzel C, Blavier A, Laporte J, Muller J. VaRank: a simple and powerful tool for ranking genetic variants. PeerJ. 2015;3:e796. doi: 10.7717/peerj.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard X, Garanto A, Rozet JM, Collin RW. Antisense Oligonucleotide Therapy for Inherited Retinal Dystrophies. Adv Exp Med Biol. 2016;854:517–24. doi: 10.1007/978-3-319-17121-0_69. [DOI] [PubMed] [Google Scholar]
- Han Z, Conley SM, Naash MI. Gene therapy for Stargardt disease associated with ABCA4 gene. Adv Exp Med Biol. 2014;801:719–24. doi: 10.1007/978-1-4614-3209-8_90. [DOI] [PubMed] [Google Scholar]
- Higasa K, Miyake N, Yoshimura J, Okamura K, Niihori T, Saitsu H, Doi K, Shimizu M, Nakabayashi K, Aoki Y, Tsurusaki Y, Morishita S, Kawaguchi T, Migita O, Nakayama K, Nakashima M, Mitsui J, Narahara M, Hayashi K, Funayama R, Yamaguchi D, Ishiura H, Ko WY, Hata K, Nagashima T, Yamada R, Matsubara Y, Umezawa A, Tsuji S, Matsumoto N, Matsuda F. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet. 2016;61(6):547–53. doi: 10.1038/jhg.2016.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houdayer C, Caux-Moncoutier V, Krieger S, Barrois M, Bonnet F, Bourdon V, Bronner M, Buisson M, Coulet F, Gaildrat P, Lefol C, Léone M, Mazoyer S, Muller D, Remenieras A, Révillion F, Rouleau E, Sokolowska J, Vert JPP, Lidereau R, Soubrier F, Sobol H, Sevenet N, Bressac-de Paillerets B, Hardouin A, Tosi M, Sinilnikova OM, Stoppa-Lyonnet D. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Human mutation. 2012;33(8):1228–1238. doi: 10.1002/humu.22101. [DOI] [PubMed] [Google Scholar]
- Alamut ® batch. Interactive Biosoftware 2016 http://www.interactive-biosoftware.com/
- Huffman JE, de Vries PS, Morrison AC, Sabater-Lleal M, Kacprowski T, Auer PL, Brody JA, Chasman DI, Chen MH, Guo X, Lin LA, Marioni RE, Muller-Nurasyid M, Yanek LR, Pankratz N, Grove ML, de Maat MP, Cushman M, Wiggins KL, Qi L, Sennblad B, Harris SE, Polasek O, Riess H, Rivadeneira F, Rose LM, Goel A, Taylor KD, Teumer A, Uitterlinden AG, Vaidya D, Yao J, Tang W, Levy D, Waldenberger M, Becker DM, Folsom AR, Giulianini F, Greinacher A, Hofman A, Huang CC, Kooperberg C, Silveira A, Starr JM, Strauch K, Strawbridge RJ, Wright AF, McKnight B, Franco OH, Zakai N, Mathias RA, Psaty BM, Ridker PM, Tofler GH, Volker U, Watkins H, Fornage M, Hamsten A, Deary IJ, Boerwinkle E, Koenig W, Rotter JI, Hayward C, Dehghan A, Reiner AP, O'Donnell CJ, Smith NL. Rare and low-frequency variants and their association with plasma levels of fibrinogen, FVII, FVIII, and vWF. Blood. 2015;126(11):e19–29. doi: 10.1182/blood-2015-02-624551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RC, Simhadri VL, Iandoli M, Sauna ZE, Kimchi-Sarfaty C. Exposing synonymous mutations. Trends in Genetics. 2014;30(7) doi: 10.1016/j.tig.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Aleman TS, Pianta MJ, Sumaroka A, Schwartz SB, Smilko EE, Milam AH, Sheffield VC, Stone EM. Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum Mol Genet. 2003;12(9):1073–8. doi: 10.1093/hmg/ddg117. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Ratnakaram R, Heon E, Schwartz SB, Roman AJ, Peden MC, Aleman TS, Boye SL, Sumaroka A, Conlon TJ, Calcedo R, Pang JJ, Erger KE, Olivares MB, Mullins CL, Swider M, Kaushal S, Feuer WJ, Iannaccone A, Fishman GA, Stone EM, Byrne BJ, Hauswirth WW. Gene Therapy for Leber Congenital Amaurosis Caused by RPE65 Mutations: Safety and Efficacy in 15 Children and Adults Followed Up to 3 Years. Archives of Ophthalmology. 2012;130(1):9–24. doi: 10.1001/archophthalmol.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian X, Boerwinkle E, Liu X. In silico tools for splicing defect prediction: a survey from the viewpoint of end users. Genetics in Medicine. 2013;16(7):497–503. doi: 10.1038/gim.2013.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian X, Boerwinkle E, Liu X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Research. 2014;42(22):13534–13544. doi: 10.1093/nar/gku1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Liang X, Li Y, Wang J, Zaneveld JE, Wang H, Xu S, Wang K, Wang B, Chen R, Sui R. Comprehensive molecular diagnosis of 67 Chinese Usher syndrome probands: high rate of ethnicity specific mutations in Chinese USH patients. Orphanet J Rare Dis. 2015;10:110. doi: 10.1186/s13023-015-0329-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90(1-2):41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG Exome Aggregation C. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelieveld SH, Veltman JA, Gilissen C. Novel bioinformatic developments for exome sequencing. Human Genetics. 2016;135(6):1–12. doi: 10.1007/s00439-016-1658-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Wang K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am J Hum Genet. 2017;100(2):267–280. doi: 10.1016/j.ajhg.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLaren RE, Groppe M, Barnard AR, Cottriall CL, Tolmachova T, Seymour L, Clark KR, During MJ, Cremers FP, Black GC, Lotery AJ, Downes SM, Webster AR, Seabra MC. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet. 2014;383(9923):1129–37. doi: 10.1016/S0140-6736(13)62117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17(1):122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mort M, Sterne-Weiler T, Li B, Ball EV, Cooper DN, Radivojac P, Sanford JR, Mooney SD. MutPred Splice: machine learning-based prediction of exonic variants that disrupt splicing. Genome Biology. 2014;15(1) doi: 10.1186/gb-2014-15-1-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neveling K, Collin R, Gilissen C, van Huet R, Visser L, Kwint MP, Gijsen SJ, Zonneveld MN, Wieskamp N, de Ligt J, Siemiatkowska AM, Hoefsloot LH, Buckley MF, Kellner U, Branham KE, den Hollander AI, Hoischen A, Hoyng C, Klevering JB, van den Born IL, Veltman JA, Cremers F, Scheffer H. Next-generation genetic testing for retinitis pigmentosa. Human Mutation. 2012;33(6):963–972. doi: 10.1002/humu.22045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio SY, Usami SI. The Clinical Next-Generation Sequencing Database: A Tool for the Unified Management of Clinical Information and Genetic Variants to Accelerate Variant Pathogenicity Classification. Hum Mutat. 2017;38(3):252–259. doi: 10.1002/humu.23160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen BS, Fredrich B, Hoeppner MP, Ellinghaus D, Franke A. Opportunities and challenges of whole-genome and -exome sequencing. BMC Genet. 2017;18(1):14. doi: 10.1186/s12863-017-0479-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piovesan A, Caracausi M, Ricci M, Strippoli P, Vitale L, Pelleri MC. Identification of minimal eukaryotic introns through GeneBase, a user-friendly tool for parsing the NCBI Gene databank. DNA Res. 2015;22(6):495–503. doi: 10.1093/dnares/dsv028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psaty BM, O'Donnell CJ, Gudnason V, Lunetta KL, Folsom AR, Rotter JI, Uitterlinden AG, Harris TB, Witteman JC, Boerwinkle E Consortium C. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2(1):73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. Journal of Computational Biology. 1997;4(3):232–240. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL Committee ALQA. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas MA, Pirinen M, Conrad DF, Lek M, Tsang EK, Karczewski KJ, Maller JB, Kukurba KR, DeLuca DS, Fromer M, Ferreira PG, Smith KS, Zhang R, Zhao F, Banks E, Poplin R, Ruderfer DM, Purcell SM, Tukiainen T, Minikel EV, Stenson PD, Cooper DN, Huang KH, Sullivan TJ, Nedzel J, Consortium GT, Geuvadis C, Bustamante CD, Li JB, Daly MJ, Guigo R, Donnelly P, Ardlie K, Sammeth M, Dermitzakis ET, McCarthy MI, Montgomery SB, Lappalainen T, MacArthur DG. Human genomics. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science. 2015;348(6235):666–9. doi: 10.1126/science.1261877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg AB, Patwardhan RP, Shendure J, Seelig G. Learning the Sequence Determinants of Alternative Splicing from Millions of Random Sequences. Cell. 2015;163(3):698–711. doi: 10.1016/j.cell.2015.09.054. [DOI] [PubMed] [Google Scholar]
- Sharma N, Sosnay PR, Ramalho AS, Douville C, Franca A, Gottschalk LB, Park J, Lee M, Vecchio - Pagan B, Raraigh KS, Amaral MD, Karchin R, Cutting GR. Experimental Assessment of Splicing Variants Using Expression Minigenes and Comparison with In Silico Predictions. Human Mutation. 2014;35(10):1249–1259. doi: 10.1002/humu.22624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Cooper TA. Minigene reporter for identification and analysis of cis elements and trans factors affecting pre-mRNA splicing. Biotechniques. 2006;41(2):177–81. doi: 10.2144/000112208. [DOI] [PubMed] [Google Scholar]
- Soens ZT, Li Y, Zhao L, Eblimit A, Dharmat R, Li Y, Chen Y, Naqeeb M, Fajardo N, Lopez I, Sun Z, Koenekoop RK, Chen R. Hypomorphic mutations identified in the candidate Leber congenital amaurosis gene CLUAP1. Genet Med. 2016;18(10):1044–51. doi: 10.1038/gim.2015.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukarieh O, Gaildrat P, Hamieh M, Drouet A, Baert-Desurmont S, Frébourg T, Tosi M, Martins A. Exonic Splicing Mutations Are More Prevalent than Currently Estimated and Can Be Predicted by Using In Silico Tools. PLOS Genetics. 2016;12(1) doi: 10.1371/journal.pgen.1005756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang R, Prosser DO, Love DR. Evaluation of Bioinformatic Programmes for the Analysis of Variants within Splice Site Consensus Regions. Advances in Bioinformatics. 2016;2016:5614058. doi: 10.1155/2016/5614058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee MP, Di Sera TL, Nix DA, Paquette AM, Parsons MT, Bell R, Hoffman A, Hogervorst FB, Goldgar DE, Spurdle AB, Tavtigian SV. Adding In Silico Assessment of Potential Splice Aberration to the Integrated Evaluation of BRCA Gene Unclassified Variants. Hum Mutat. 2016;37(7):627–39. doi: 10.1002/humu.22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang X, Zou X, Xu S, Li H, Soens Z, Wang K, Li Y, Dong F, Chen R, Sui R. Comprehensive Molecular Diagnosis of a Large Chinese Leber Congenital Amaurosis CohortMolecular Diagnosis in a Chinese LCA Cohort. Investigative Ophthalmology & Visual Science. 2015a;56(6):3642–3655. doi: 10.1167/iovs.14-15972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang X, Zou X, Xu S, Li H, Soens ZT, Wang K, Li Y, Dong F, Chen R, Sui R. Comprehensive Molecular Diagnosis of a Large Chinese Leber Congenital Amaurosis Cohort. Invest Ophthalmol Vis Sci. 2015b;56(6):3642–55. doi: 10.1167/iovs.14-15972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, Morris Q, Barash Y, Krainer AR, Jojic N, Scherer SW, Blencowe BJ, Frey BJ. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347(6218):1254806. doi: 10.1126/science.1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Xie YA, Abouzeid H, Gordon CT, Fiorentino A, Sun Z, Lehman A, Osman IS, Dharmat R, Riveiro-Alvarez R, Bapst-Wicht L, Babino D, Arno G, Busetto V, Zhao L, Li H, Lopez-Martinez MA, Azevedo LF, Hubert L, Pontikos N, Eblimit A, Lorda-Sanchez I, Kheir V, Plagnol V, Oufadem M, Soens ZT, Yang L, Bole-Feysot C, Pfundt R, Allaman-Pillet N, Nitschke P, Cheetham ME, Lyonnet S, Agrawal SA, Li H, Pinton G, Michaelides M, Besmond C, Li Y, Yuan Z, von Lintig J, Webster AR, Le Hir H, Stoilov P, Consortium UKIRD. Amiel J, Hardcastle AJ, Ayuso C, Sui R, Chen R, Allikmets R, Schorderet DF. Mutations in the Spliceosome Component CWC27 Cause Retinal Degeneration with or without Additional Developmental Anomalies. Am J Hum Genet. 2017;100(4):592–604. doi: 10.1016/j.ajhg.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C, Raveendran M, Harris RA, Fawcett GL, Liu X, White S, Dahdouli M, Rio Deiros D, Below JE, Salerno W, Cox L, Fan G, Ferguson B, Horvath J, Johnson Z, Kanthaswamy S, Kubisch HM, Liu D, Platt M, Smith DG, Sun B, Vallender EJ, Wang F, Wiseman RW, Chen R, Muzny DM, Gibbs RA, Yu F, Rogers J. The population genomics of rhesus macaques (Macaca mulatta) based on whole-genome sequences. Genome Res. 2016;26(12):1651–1662. doi: 10.1101/gr.204255.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaneveld J, Siddiqui S, Li H, Wang X, Wang H, Wang K, Li H, Ren H, Lopez I, Dorfman A, Khan A, Wang F, Salvo J, Gelowani V, Li Y, Sui R, Koenekoop R, Chen R. Comprehensive analysis of patients with Stargardt macular dystrophy reveals new genotype-phenotype correlations and unexpected diagnostic revisions. Genet Med. 2015;17(4):262–70. doi: 10.1038/gim.2014.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Wang F, Wang H, Li Y, Alexander S, Wang K, Willoughby CE, Zaneveld JE, Jiang L, Soens ZT, Earle P, Simpson D, Silvestri G, Chen R. Next-generation sequencing-based molecular diagnosis of 82 retinitis pigmentosa probands from Northern Ireland. Human Genetics. 2014;134(2):217230. doi: 10.1007/s00439-014-1512-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou WB, Wu H, Boulling A, Cooper DN, Li ZS, Liao Z, Chen JM, Ferec C. In silico prioritization and further functional characterization of SPINK1 intronic variants. Hum Genomics. 2017;11(1):7. doi: 10.1186/s40246-017-0103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.