

Abstract
In silico methods for linking genomic space to chemical space have played a crucial role in genomics driven discovery of new natural products as well as biosynthesis of altered natural products by engineering of biosynthetic pathways. Here we give an overview of available computational tools and then briefly describe a novel computational framework, namely retro-biosynthetic enumeration of biosynthetic reactions, which can add to the repertoire of computational tools available for connecting natural products to their biosynthetic gene clusters. Most of the currently available bioinformatics tools for analysis of secondary metabolite biosynthetic gene clusters utilize the “Genes to Metabolites” approach. In contrast to the “Genes to Metabolites” approach, the “Metabolites to Genes” or retro-biosynthetic approach would involve enumerating the various biochemical transformations or enzymatic reactions which would generate the given chemical moiety starting from a set of precursor molecules and identifying enzymatic domains which can potentially catalyze the enumerated biochemical transformations. In this article, we first give a brief overview of the presently available in silico tools and approaches for analysis of secondary metabolite biosynthetic pathways. We also discuss our preliminary work on development of algorithms for retro-biosynthetic enumeration of biochemical transformations to formulate a novel computational method for identifying genes associated with biosynthesis of a given polyketide or nonribosomal peptide.
Keywords: Secondary metabolite, Polyketides, Nonribosomal peptides, Genes to metabolites, Metabolites to genes, Genome mining, Retro-biosynthetic enumeration, Biosynthetic gene cluster
1. Introduction
Polyketides and nonribosomal peptides are two major classes of secondary metabolite natural products with enormous diversity in chemical structures and bioactivities.1 Examples of pharmaceutically important polyketides and nonribosomal peptides are lovastatin (a cholesterol-lowering agent),2 erythromycin (an antibiotic), FK506 (an immunosuppressant) and epothilone (anticancer compound).3 These secondary metabolites are biosynthesized by multifunctional megasynthases like polyketide synthase (PKS) and nonribosomal peptide synthetase (NRPS) using a thiotemplate mechanism. The diverse and complex structures of polyketides and nonribosomal peptides arise from assembly line synthesis by these megasynthases. Details of the biosynthetic mechanism have been discussed in a number of earlier reviews.4, 5, 6, 7, 8 Owing to their pharmaceutical and industrial importance, these natural products as well as their biosynthetic mechanisms have been subject of particular interest and extensive characterization.9 Unraveling the “biosynthetic code” of these natural products has opened up the possibilities for identification of novel natural products in various bacterial and fungal organisms and also biosynthetic engineering of rationally designed secondary metabolites for their use as drug molecules.10, 11, 12, 13 The structural diversity arising from combinatorial complexity of their biosynthesis is the reason why these natural products are a great source of drugs. Understanding the mechanisms of their biosynthesis and devising clever strategies to tweak it can potentially yield fruitful results in the form of economically important products.14 The extent of diversity of these natural products has been vastly underestimated and with new niches of microorganisms being explored, the number of novel bioactive metabolites is likely to increase many folds.15, 16 It has been anticipated that novel drugs can be discovered by cultivating and characterizing microorganisms like actinobacteria.17 Therefore, these bacterial strains could be the new unexplored sources of natural products. In addition, the exponential growth of genome sequencing hasunveiled many bacteria containing putative natural product biosynthetic gene clusters with unknown biosynthetic products.18, 19
Linking biosynthetic genes to secondary metabolites and vice versa can potentially help not only in characterization of new secondary metabolites, but also in redesigning known biosynthetic pathways of secondary metabolites to produce novel compounds.4, 20 The problem can in principle be solved using two approaches: Forward (Genes to Metabolites) and Reverse/Retro-biosynthetic (Metabolites to Genes) Approach21, 22 (Fig. 1). In forward approach genomic sequence information is used to predict the chemical structure of the final metabolite. In contrast to forward approach which starts by considering the genes or gene clusters and attempts to predict its biosynthetic product, retro-biosynthetic approach starts from a known metabolite and attempts to identify which gene cluster might be biosynthesizing it.23, 24 Even though traditionally identification of natural products and their biosynthesis have been an area of interest for microbiologists, organic chemists and biochemists, elucidation of the catalytic machinery for biosynthesis of polyketides and nonribosomal peptides by genome encoded PKS and NRPS clusters has opened up the area of genomics driven discovery of new natural products' biosynthetic pathways.13, 25, 26 Bioinformatics has played an important role in in silico identification of new secondarymetabolites by genome mining and several pioneering studies have been successful in experimental characterization of new metabolites predicted by in silico analysis.20, 27, 28 However, majority of the available computational methods for analysis of secondary metabolite biosynthetic pathways utilize forward approach for linking Genes to Metabolites, while automated computational tools for linking secondary metabolites' chemical structures to their biosynthetic gene clusters are not available yet.
Fig. 1.

Two approaches for deciphering new biosynthetic pathways. (A) “Forward approach”, where information from genes is used to decipher the biological pathways. “Retro-biosynthetic approach” is where a known product is linked to the genes. Some of the available methods belonging to either approach have been mentioned in boxes. (B) Alternative approaches to connecting genes and metabolites. (Left Panel) Use of module organization in comparison of secondary metabolite gene clusters and prediction of the secondary metabolite synthesized. (Right Panel) Retro-biosynthetic approach for prediction of the gene cluster responsible for biosynthesis of a particular secondary metabolite.
In this article, we first give a brief overview of the presently available in silico tools and approaches for analysis of secondary metabolite biosynthetic pathways and identification of novel secondary metabolites by genome mining. Most of the in silico approaches use evolutionary information on sequence/structural features of individual catalytic domains of PKS or NRPS biosynthetic pathways for genome mining of secondary metabolites and for prediction of chemical structures of their putative products. We also discuss the feasibility of devising a retro-biosynthetic approach to link orphan secondary metabolites to their biosynthetic gene cluster. The retro-biosynthetic approach for linking “Metabolites to Genes” involves enumerating the various biochemical transformations or enzymatic reactions which would generate the given secondary metabolite starting from a set of precursor molecules and identifying enzymatic domains which can potentially catalyze the enumerated biochemical transformations.
2. Connecting PKS/NRPS gene clusters to their biosynthetic product
Based on analysis of experimentally characterized PKS and NRPS biosynthetic clusters, a number of bioinformatics resources have been developed as knowledge bases for domain organization and substrate specificities of PKS and NRPS genes. These computational resources can play an important role in genomic mining for novel secondary metabolites and functional analysis of newly identified gene clusters. Some of the major databases which have cataloged very large number of experimentally characterized PKS and NRPS clusters with known biosynthetic products are ClusterMine360, IMG-ABC and MIBiG. Apart from the sequence information and catalytic domain organization, major utility of these databases is to obtain the chemical structures of secondary metabolite products. Recent version of ClusterMine36029 has information on approximately 290 gene clusters involved in biosynthesis of more than 200 polyketides and nonribosomal peptides. In addition to sequence of genes, catalytic domain organization and chemical structure of secondary metabolite product, IMG-ABC30 has also cataloged information on genomic locus for a large number of secondary metabolite gene clusters. The MIBiG31 resource has been developed by a community driven initiative to store secondary metabolite biosynthetic pathways following a minimum information standard and MIBiG-compliant reannotation has been carried out for approximately 400 secondary metabolite biosynthetic gene clusters. Another example of a useful database for secondary metabolites is NORINE,32 which has chemical structures for 1168 nonribosomal peptides. Based on bioinformatics analysis of experimentally characterized PKS and NRPS gene clusters, a number of computational methods have been developed for connecting “genes to metabolites”. In view of the remarkable conservation of overall biosynthetic paradigm for polyketides and nonribosomal peptides, these computational methods have essentially used a knowledge based approach33, 34 for deriving prediction rules based on experimentally characterized PKS and NRPS gene clusters. The tools like NRPS-PKS,35 SBSPKS,36 ASMPKS/MAPSI,37 ClustScan,38 NP.Searcher,39 NRPSpredictor,40 PKS/NRPS41 and PKMiner42 permit semi-automatic identification and annotation of PKS, NRPS or PKS-NRPS hybrid gene clusters. In addition to annotating the domains of multi-domain PKS and NRPS, most of these tools also predict the substrate specificity of adenylation and acyltransferase (AT) domains. Apart from identification of different catalytic domains of NRPS and PKS, SBSPKS can also model three dimensional structures of complete PKS modules and predict the order of substrate channeling in case of PKS clusters consisting of multiple ORFs. Bioinformatics tools have also been developed for analysis of specific class of secondary metabolite gene clusters. SMURF43 allows identification of biosynthetic gene clusters in fungal genome, while PKMiner42 helps in mining of type II PKS gene clusters. Bioinformatics tools for analysis of secondary metabolite biosynthetic genes have also been developed for analysis of metagenomic data. Metagenomic samples can be quickly scanned for novel natural products by using PCR primers specific for secondary metabolite biosynthetic gene clusters.44 This PCR-based sequence tag approach has been coupled with in silico phylogenomic tools to search for putative secondary metabolites. eSNaPD has been specifically developed to analyze large metagenomic sequence tag datasets and aid in the discovery of diverse secondary metabolite gene clusters.45 Another bioinformatics tool which accepts sequence tags from metagenomic datasets along with protein or genomic sequences is NaPDoS.46 It uses phylogenomic information to search and classify NRPS Adenylation and PKS Ketosynthase domains.
Majority of the tools mentioned above identify the PKS and NRPS catalytic domains, whereas NP.searcher can also indentify auxiliary and tailoring domains in PKS and NRPS gene clusters. Based on the predicted substrate specificities of adenylation and acyltransferase domains in NRPS and PKS clusters, NP.searcher appends monomers to the growing chain of polyketide or nonribosomal peptide and then the predicted chemical structure is further modified based on all possible combinations of predicted tailoring and cyclization steps. NP.searcher hence outputs chemical structures for a list of putative secondary metabolites and focuses specially on nonribosomal peptides.
Recently developed antiSMASH47 pipeline can identify the biosynthetic loci covering the whole range of known secondary metabolite compound classes (polyketides, nonribosomal peptides, terpenes, aminoglycosides, aminocoumarins, indolocarbazoles, lantibiotics, bacteriocins, nucleosides, beta-lactams, butyrolactones, siderophores, melanins and others). antiSMASH48 is also integrated with tools like ClusterFinder49 which allows identification of putative secondary metabolite gene clusters encoding novel class of secondary metabolites. It uses the PFAM domain50 definition to search for enzymes involved in synthesis of secondary metabolites. It also allows comparison of identified clusters with experimentally characterized clusters using clusterBLAST. Latest update of antiSMASH can identify active site residues of core PKS domains like AT, KS, DH, KR, ACP, TE and tailoring domains like cytochrome P450 oxygenase using ‘Active Site Finder’ module. antiSMASH also uses domain information of modular PKS and NRPS to predict the linear polyketides produced by the query cluster. Although the chemical structure prediction feature includes effect of reductive domains KR, DH and ER on the polyketide structure, predictions of post-PKS/NRPS modifications and cyclizations are not yet available in antiSMASH.
Another web-based tool that connects secondary metabolite gene cluster to the chemical structures of secondary metabolites is PRISM (PRediction Informatics for Secondary Metabolomes).51 It uses a library of 479 HMM models for the identification of these gene clusters. These HMM models include HMMs for thiotemplate domains, substrate specific adenylation and acyltransferase domains, domains catalyzing a number of tailoring reactions, and acyl-adenylating domains, among others. The PRISM algorithm identifies putative PKS/NRPS modules along with the specific substrate monomers. Based on permutation of open reading frames (ORF), the position of loading and termination modules and principle of co-linearity the order of substrate channeling is predicted. After deciphering the chemical structure of the linear polyketide or nonribosomal peptide based on co-linearity rule, PRISM carries out pseudo-random enumeration of a number of different tailoring reactions and all combination of cyclization patterns to generate a combinatorial library of chemical structures of putative secondary metabolites.
The aforementioned computational methods have been designed to relate sequences of secondary metabolite gene clusters to the chemical structures of the unknown metabolites by using the forward approach. They essentially use various sequence and structure based bioinformatics approaches to predict the catalytic reaction a given enzyme would catalyze in the biosynthetic pathway, its substrates and products. In biochemical pathways consisting of multiple catalytic reactions, it is also necessary to predict the precise order in which these reactions will be catalyzed; otherwise it will lead to a combinatorial explosion of possible chemical structures of the final metabolic product. Most of the above mentioned computational tools predict the order of biochemical transformations by the so called co-linearity rule52 or based on inter subunit interactions in the limited context of modular PKS clusters. However, there are significant deviations from co-linearity rule in many PKS/NRPS clusters and also occurrence of complex tailoring enzymes and cyclization patterns make prediction of the correct order of catalytic reactions an enormously difficult task. Hence, despite reports of successes in general identification of new secondary metabolites by forward approach are extremely difficult, none of the above mentioned computational tools permit a completely automated prediction of chemical structures of secondary metabolites based on genome analysis.
3. Connecting secondary metabolites to their biosynthetic gene clusters using probabilistic matching
In contrast to the large number of software for linking genes to metabolites, Pep2Path53 is the only software package currently available for linking chemical structures of nonribosomal peptides to gene clusters. It helps in matching of tandem mass spectra of nonribosomal peptides to their gene clusters. It accepts either MS-derived NRP mass shift sequence or a short stretch of amino acid and genome sequences. When the input is mass shifts it is first converted into amino acid tag. The genome sequence, on the other hand, is scanned for putative NRPS gene cluster using antiSMASH. Then Pep2Path uses Bayesian algorithm to predict the chances of an amino acid in the tag to be synthesized by the predicted NRPS modules. Using this probability a final score for complete gene cluster is then calculated. Pep2Path is also designed to identify gene clusters corresponding to ribosomally synthesized post-translationally-modified peptides (RiPPs).
4. Retro-biosynthetic approach
Here, we discuss our preliminary work toward development of a retro-biosynthetic approach for linking chemical structures of secondary metabolites to succession of reactions that potentially produce it. With correct enumeration of biochemical transformation it will be possible to link the enumerated biochemical reactions to genes containing enzymatic domains which can catalyze such reactions. Hence, this computational method can be further developed in future as an alternative to probabilistic matching method for linking secondary metabolites to gene clusters.
There are several organisms for which complete genome sequences are available and many secondary metabolites have also been experimentally characterized in the corresponding organisms.54 However, the genes responsible for the biosynthesis of the corresponding metabolites are not known.4 Therefore, a reverse or retro-biosynthetic approach can in principle be applied in such cases. Retro-biosynthetic approach starts from a known metabolite and attempts to identify which gene cluster might be biosynthesizing it. Using the knowledge of enzymatic reactions and logic of chemical transformation the immediate precursor molecule(s) are predicted. The predicted precursor is used for another round of retro-biosynthetic enumeration to predict precursors of the precursor. This cycle of reaction enumeration is continued until a known starting product is obtained. After E.J. Corey illustrated the concept of retrosynthesis, the approach has helped in delineating biochemical pathways too.23, 24 The benefits of the approach in reconstruction of pathways have been discussed earlier.55, 56 This approach is beneficial in cases where the mass spectrometric or similar analysis has revealed the chemical structure of final metabolite but its biosynthetic gene has not been characterized. Retro-biosynthetic tools are available for predicting metabolic routes between two metabolites57, 58, 59, 60 and predicting biosynthetic routes of plant secondary metabolites.61 Similar automated in silico tools have been also developed mainly for the prediction of biodegradation pathways.61, 62, 63 These approaches are reaction rule based, where generalized reactions are applied to final metabolite to enumerate precursor metabolites. Application of all possible generalized reactions at each stage of precursor enumeration can lead to prediction of huge number of possible pathways – combinatorial explosion.60 To avoid such combinatorial explosion, these tools rank the possibility of enumerated reaction based on available enzymatic and chemical knowledge. Also, focusing on a smaller set of reactions like xenobiotic degradation or chemical transformations relevant for plant secondary metabolites helps in decreasing the false positive hits. The essential task for developing retro-biosynthetic approach is to predict all possible enzymatic reactions which can lead to the final secondary metabolite of known chemical structure starting from certain precursor molecules. In the next step, potential enzymes that can catalyze each of these enzymatic reactions can be identified by sequence or structure based bioinformatics methods. In recent years few computational tools like ReBit,63 FMM59 and PathPred61 have been developed for retro-biosynthetic enumeration of biochemical reactions and have been applied for biosynthesis of novel natural products by synthetic biology approach. Even though PathPred focuses on predicting pathway for plant secondary metabolites, the focus of most retro-biosynthesis related computational tool development has been on primary metabolites and chemical degradation pathways, because information about these pathways is well documented in databases like KEGG.64, 65 In contrast, information about natural product biosynthesis is still dispersed in scientific literature. PathPred and ReBit are the only two servers that predict biosynthetic reactions. PathPred predicts multistep reaction pathway for degradation of xenobiotic compounds and biosynthesis of plant secondary metabolites. It uses a database of Biochemical transformation patterns for substrate-products called RPAIR.66 ReBit predicts a set of enzymes capable of using the given query either for biosynthesis or biodegradation.
Since biosynthesis of polyketides and nonribosomal peptides involves a limited number of reactions compared to metabolic pathways in general, they are amenable to retro-biosynthetic approach for predicting which gene clusters in a given genome might be making a known secondary metabolite. Our group has attempted to develop a computational protocol for reconstructing the biosynthetic pathways of polyketides and nonribosomal peptides using retro-biosynthetic approach. Fig. 2 shows a schematic depiction of various steps involved in retro-biosynthetic enumeration protocol. The assembly line mechanism of biosynthesis of polyketides involves various chemical transformations like condensation, reductive steps, chain release involving hydrolysis or macro-ring formation, other complex cyclizations and various post-PKS and post-NRPS modifications. To develop a retro approach 25 such reactions were stored as generic reactions (Fig. 3, Supplementary File S2). Functional groups of products were also stored in a separate database in SMARTS language. The generic reactions and functional groups were generated based on sub structural changes that occur in a reaction (Supplementary methods in Supplementary File S1). Given a polyketide or nonribosomal peptide chemical structure, the retro-biosynthetic enumeration process first searches for a functional group using Obgrep tool of Open Babel.67 The Reactor module of ChemAxon (JChem 6.1.3, 2013, ChemAxon (http://www.chemaxon.com)) is used to transform the given metabolite into its precursor based on the corresponding generic reaction. This precursor metabolite becomes the new input and another round of functional group search and reaction enumeration is then processed. The process is continued until no other functional group is detected in the compound. In order to test the developed retro-biosynthetic approach chemical structures of 78 experimentally characterized secondary metabolites were downloaded from SBSPKS database (Supplementary File S2). This set consisted of 49 polyketides from modular PKS section of SBSPKS, 27 nonribosomal peptides from NRPS section and two compounds from hybrid PKS/NRPS section. For each of these 78 secondary metabolites complete biosynthetic pathways were available in published literature. Reactions for each compound were enumerated and the predicted steps were cross checked with known biosynthetic pathways for correctness. Supplementary File S2 lists the total number of reactions in the biosynthetic pathways of each compound, number of correctly predicted reactions, and sum of the incorrect and missing reactions. For a given compound the prediction was classified as “correct” if the number of correctly predicted reactions was 100%, “minor error” if correctly predicted reactions were within 80%–100%, “partially correct” if the number of correctly predicted reactions was within 50–80% and “Incorrect” if the number of correctly predicted reactions was less than 50%. Table 1 shows the summary of the results of retro-biosynthetic enumeration for 78 secondary metabolites. Out of these 78 secondary metabolites consisting of 51 polyketides/hybrid metabolites and 27 nonribosomal peptides, all the enzymatic reaction steps could be completely enumerated for 17 polyketides/hybrids and 12 nonribosomal peptides. An example of completely enumerated biosynthetic pathway is that of halstoctacosanolide (Fig. 4). Macrolactonization, oxidation, spontaneous cyclization and 18 steps of condensation and reduction were correctly predicted for halstoctacosanolide. Ten other compounds from the polyketide set were in the “minor error” category due to post-PKS modifications or conjugation of double bonds. For example in geldanamycin a post-PKS hydroxylation step changes a completely reduced extender unit (KS-AT-DH-ER-KR-ACP) to its hydroxylated form. The hydroxylated form is seen by the retro-biosynthesis algorithm as one synthesized by KS-AT-KR-ACP module. For 9 polyketides/hybrids and 10 nonribosomal peptides partially correct predictions could be made. One such example is monensin (Fig. 5). Although initial cyclization and post-PKS reactions were predicted correctly, the first condensation and reduction step was incorrectly predicted. The last module of monensin PKS adds a methyl malonyl-coA and completely reduces the keto group (C-26) using the KR, DH and ER domains. A hydroxylation step at the end adds a hydroxy group back to C-26 atom. Although the retro-biosynthesis approach correctly predicts condensation of a methyl malonyl-coA, presence of a hydroxyl group is mistaken as partial reduction by the PKS module. Hence, reduction by only a KS-AT-KR module is predicted. In addition, there was error in prediction of reaction of another module. Another example of partially enumerated pathway is the biosynthetic pathway for non-ribosomal peptide A40926 (Supplementary Fig. S1). The steps predicted correctly have been marked in blue and the missing or wrong predictions have been marked in red. As the cross-linking could not be predicted the algorithm is unable to locate a regular amino acid after the hydrolytic termination step and hence terminates. For the remaining 15 polyketides/hybrids and 5 nonribosomal peptides more than 50% of the reactions could not be enumerated, hence they were classified as incorrect predictions. This set also includes compounds like ambruticin, aureothin, chlorothricin, coronafacic acid and curacin, for which no reaction could be enumerated, mainly due to presence of unusual and complex cyclizations. In summary, out of 78 secondary metabolites correct or partially correct enumeration could be done for 58 compounds.
Fig. 2.

Schematic representation of retro-biosynthetic enumeration. Schematic diagram representing the main steps involved in the retro-biosynthetic enumeration of reactions leading to a given polyketide and nonribosomal peptide product.
Fig. 3.

Examples of generic reactions used for Retro-biosynthetic approach. All possible modules required for the biosynthesis of polyketides and nonribosomal peptides. The second column lists an example reaction catalyzed by each type of module and the generic reaction or reaction rule associated with these modules. Circles indicate change in functional group.
Table 1.
Results of retro-biosynthetic enumeration for secondary metabolites.
| Number of compounds | Correct predictions (100%) | Minor error (80%–100%) | Partially correct (50%–80%) | Incorrect predictions (<50%) | |
|---|---|---|---|---|---|
| Polyketides/hybrid | 51 | 17 | 10 | 9 | 15 |
| Nonribosomal peptides | 27 | 12 | 0 | 10 | 5 |
| Total | 78 | 29 | 10 | 19 | 20 |
Fig. 4.

An example of reaction enumeration. An example of complete reaction enumeration starting from the polyketide – halstoctacosanolide to its starting metabolites using the retro-biosynthetic approach.
Fig. 5.
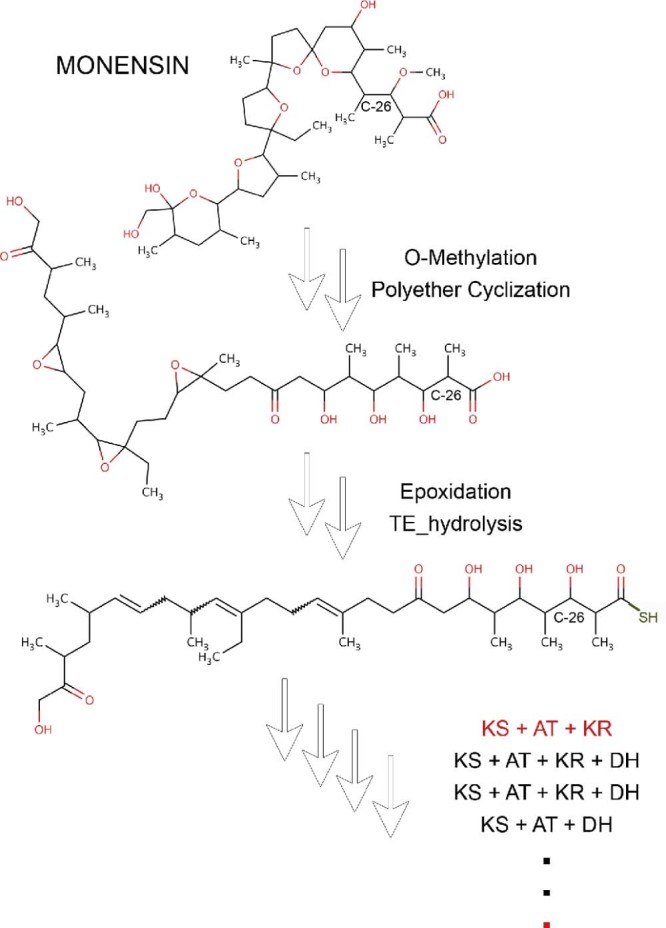
An example of incorrect reaction enumeration starting from the polyketide – monensin. The steps that were wrongly predicted have been highlighted in red.
The database of secondary metabolite biosynthetic reactions can be improved to add complex cyclization steps and many other post-PKS and post-NRPS modifications catalyzed by tailoring enzymes. This will aid in widening the scope of this approach. The tool can be further developed to link the biosynthetic reactions to their respective genes. Genome mining could be used to identify PKSs in completely sequenced genomes and stored in a separate database. Therefore, after the reactions are enumerated and enzymes are identified, co-occurrence of these enzymes together in a gene cluster can be checked using the PKS sequence database. Tailoring enzymes usually co-occur in the genomic neighborhood of PKSs. Hence, neighboring genes of PKS should also be stored in the database. Therefore, the retro-biosynthetic approach can be a very useful resource for enumeration of secondary metabolite biosynthetic pathways and relating it to polyketide and nonribosomal peptide biosynthetic clusters by genome mining.
5. Discussion
The two major classes of natural products biosynthesized by various microbial, fungal and plant species are polyketides and nonribosomal peptides. Connecting these natural products and their gene clusters would not only broaden the understanding of their complex biosynthesis, but will also help in discovery of novel natural products and help in designing new natural product-based drugs. In silico tools for identification of new secondary metabolites have played an important role in successful experimental characterization of new polyketides and nonribosomal peptides. Most of these computational tools facilitate connecting “genes to metabolite”. These tools use various sequence and structure based bioinformatics approaches to predict the reaction catalyzed by each domain, its substrate and product. Occurrence of tailoring enzymes, complex cyclization patterns and iterative use of catalytic domains and order of catalytic reactions add to the complexity of the chemical structure of these metabolites. A retro-biosynthetic approach of identifying genes associated with the metabolite, i.e., connecting “metabolites to genes”, would overcome the hurdle of complexity of reactions. In this article, we have given a brief overview of a retro-biosynthetic approach to connect orphan polyketides and nonribosomal peptides to their biosynthetic gene clusters. This computational approach will be made available in the next update of SBSPKS web-server developed by our group. The predictive power of the aforementioned computational approaches can be enhanced by expanding the knowledge base with information about tailoring enzymes, cyclization patterns and iterative use of catalytic domains.
Both “Genes to Metabolites” and “Metabolites to Genes” approaches are based on understanding of the evolution of sequence/structural features of individual catalytic domains of PKS or NRPS biosynthetic pathways. Availability of large number of experimentally characterized modular PKS and NRPS clusters has opened up the opportunity for integrative analysis of the evolution of complete PKS or NRPS biosynthetic pathways by insertion, deletion and substitution of various catalytic domains. The PKS and NRPS gene clusters have evolved by insertion, deletion and substitution of various catalytic domains. Thus, it would be interesting to explore the possibility of correlating the combinatorial organization of domains in a genomic space and the diversity of the products in the chemical structure space. It is possible to develop new computational approaches, where different PKS and NRPS modules can be represented by unique identifiers and hence the gene cluster can be represented as a module string. The insertions, additions and deletions can be taken into account by aligning these module strings using modified version of standard alignment tools or dynamic programming. The best alignments can be picked and used to predict the probable metabolite synthesized by the biosynthetic cluster. It may be noted that such domain string approach is similar to the clusterBLAST method available in antiSMASH. However, domain string approach will be computationally faster in view of reduced representation of modules in terms of single identifiers. Hence, it can be used for quick comparison of newly identified clusters with experimentally characterized clusters present in various databases.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work is supported by grants to National Institute of Immunology, New Delhi from Department of Biotechnology (DBT), Government of India. DM also acknowledges financial support from DBT, India under BTIS project (BT/BI/03/009/2002) and Bioinformatics R&D grant (BT/PR13526/BID/07/311/2010). SK acknowledges the support from DBT, India in the form of BINC fellowship and SA was supported by senior research fellowship from CSIR, India during the course of this work.
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
Supplementary data to this article can be found online at doi:10.1016/j.synbio.2016.03.001.
Appendix. Supplementary material
The following is the supplementary data to this article:
Supplementary Methods and Supplementary Figure.
Reactions used, Test dataset and the results.
Information for downloading Source code for Retro-biosynthetic algorithm as a perl script.
References
- 1.Cane D.E., Walsh C.T. The parallel and convergent universes of polyketide synthases and nonribosomal peptide synthetases. Chem Biol. 1999;6:R319–25. doi: 10.1016/s1074-5521(00)80001-0. [DOI] [PubMed] [Google Scholar]
- 2.McKenney J.M. Lovastatin: a new cholesterol-lowering agent. Clin Pharm. 1988;7:21–36. [PubMed] [Google Scholar]
- 3.Cheng Y.Q., Tang G.L., Shen B. Type I polyketide synthase requiring a discrete acyltransferase for polyketide biosynthesis. Proc Natl Acad Sci U S A. 2003;100:3149–3154. doi: 10.1073/pnas.0537286100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walsh C.T., Fischbach M.A. Natural products version 2.0: connecting genes to molecules. J Am Chem Soc. 2010;132:2469–2493. doi: 10.1021/ja909118a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hertweck C. The biosynthetic logic of polyketide diversity. Angew Chem Int Ed Engl. 2009;48:4688–4716. doi: 10.1002/anie.200806121. [DOI] [PubMed] [Google Scholar]
- 6.Hill A.M. The biosynthesis, molecular genetics and enzymology of the polyketide-derived metabolites. Nat Prod Rep. 2006;23:256–320. doi: 10.1039/b301028g. [DOI] [PubMed] [Google Scholar]
- 7.Schwarzer D., Finking R., Marahiel M.A. Nonribosomal peptides: from genes to products. Nat Prod Rep. 2003;20:275–287. doi: 10.1039/b111145k. [DOI] [PubMed] [Google Scholar]
- 8.Doekel S., Marahiel M.A. Biosynthesis of natural products on modular peptide synthetases. Metab Eng. 2001;3:64–77. doi: 10.1006/mben.2000.0170. [DOI] [PubMed] [Google Scholar]
- 9.Meier J.L., Burkart M.D. Chapter 9. Synthetic probes for polyketide and nonribosomal peptide biosynthetic enzymes. Methods Enzymol. 2009;458:219–254. doi: 10.1016/S0076-6879(09)04809-5. [DOI] [PubMed] [Google Scholar]
- 10.Weissman K.J., Leadlay P.F. Combinatorial biosynthesis of reduced polyketides. Nat Rev Microbiol. 2005;3:925–936. doi: 10.1038/nrmicro1287. [DOI] [PubMed] [Google Scholar]
- 11.Xu Y., Zhou T., Zhang S., Espinosa-Artiles P., Wang L., Zhang W. Diversity-oriented combinatorial biosynthesis of benzenediol lactone scaffolds by subunit shuffling of fungal polyketide synthases. Proc Natl Acad Sci U S A. 2014;111:12354–12359. doi: 10.1073/pnas.1406999111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Genilloud O. The re-emerging role of microbial natural products in antibiotic discovery. Antonie Van Leeuwenhoek. 2014;106:173–188. doi: 10.1007/s10482-014-0204-6. [DOI] [PubMed] [Google Scholar]
- 13.Helfrich E.J., Reiter S., Piel J. Recent advances in genome-based polyketide discovery. Curr Opin Biotechnol. 2014;29:107–115. doi: 10.1016/j.copbio.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 14.Du J., Shao Z., Zhao H. Engineering microbial factories for synthesis of value-added products. J Ind Microbiol Biotechnol. 2011;38:873–890. doi: 10.1007/s10295-011-0970-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bennur T., Ravi Kumar A., Zinjarde S.S., Javdekar V. Nocardiopsis species: a potential source of bioactive compounds. J Appl Microbiol. 2016;120:1–16. doi: 10.1111/jam.12950. [DOI] [PubMed] [Google Scholar]
- 16.Traxler M.F., Kolter R. Natural products in soil microbe interactions and evolution. Nat Prod Rep. 2015;32:956–970. doi: 10.1039/c5np00013k. [DOI] [PubMed] [Google Scholar]
- 17.Manivasagan P., Kang K.H., Sivakumar K., Li-Chan E.C., Oh H.M., Kim S.K. Marine actinobacteria: an important source of bioactive natural products. Environ Toxicol Pharmacol. 2014;38:172–188. doi: 10.1016/j.etap.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 18.Reen F.J., Romano S., Dobson A.D., O'Gara F. The sound of silence: activating silent biosynthetic gene clusters in marine microorganisms. Mar Drugs. 2015;13:4754–4783. doi: 10.3390/md13084754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiang Y.M., Chang S.L., Oakley B.R., Wang C.C. Recent advances in awakening silent biosynthetic gene clusters and linking orphan clusters to natural products in microorganisms. Curr Opin Chem Biol. 2011;15:137–143. doi: 10.1016/j.cbpa.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Medema M.H., Fischbach M.A. Computational approaches to natural product discovery. Nat Chem Biol. 2015;11:639–648. doi: 10.1038/nchembio.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bachmann B.O. Biosynthesis: is it time to go retro? Nat Chem Biol. 2010;6:390–393. doi: 10.1038/nchembio.377. [DOI] [PubMed] [Google Scholar]
- 22.Cacho R.A., Tang Y., Chooi Y.H. Next-generation sequencing approach for connecting secondary metabolites to biosynthetic gene clusters in fungi. Front Microbiol. 2014;5:774. doi: 10.3389/fmicb.2014.00774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng X.-M., Corey E.J. 1st ed. John Wiley & Sons.; New York: 1989. The logic of chemical synthesis. [Google Scholar]
- 24.Irschik H., Kopp M., Weissman K.J., Buntin K., Piel J., Muller R. Analysis of the sorangicin gene cluster reinforces the utility of a combined phylogenetic/retrobiosynthetic analysis for deciphering natural product assembly by trans-AT PKS. Chembiochem. 2010;11:1840–1849. doi: 10.1002/cbic.201000313. [DOI] [PubMed] [Google Scholar]
- 25.Milshteyn A., Schneider J.S., Brady S.F. Mining the metabiome: identifying novel natural products from microbial communities. Chem Biol. 2014;21:1211–1223. doi: 10.1016/j.chembiol.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deane C.D., Mitchell D.A. Lessons learned from the transformation of natural product discovery to a genome-driven endeavor. J Ind Microbiol Biotechnol. 2014;41:315–331. doi: 10.1007/s10295-013-1361-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weber T. In silico tools for the analysis of antibiotic biosynthetic pathways. Int J Med Microbiol. 2014;304:230–235. doi: 10.1016/j.ijmm.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Boddy C.N. Bioinformatics tools for genome mining of polyketide and non-ribosomal peptides. J Ind Microbiol Biotechnol. 2014;41:443–450. doi: 10.1007/s10295-013-1368-1. [DOI] [PubMed] [Google Scholar]
- 29.Conway K.R., Boddy C.N. ClusterMine360: a database of microbial PKS/NRPS biosynthesis. Nucleic Acids Res. 2013;41:D402–7. doi: 10.1093/nar/gks993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hadjithomas M., Chen I.M., Chu K., Ratner A., Palaniappan K., Szeto E. IMG-ABC: a knowledge base to fuel discovery of biosynthetic gene clusters and novel secondary metabolites. MBio. 2015;6:e00932. doi: 10.1128/mBio.00932-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Medema M.H., Kottmann R., Yilmaz P., Cummings M., Biggins J.B., Blin K. Minimum information about a biosynthetic gene cluster. Nat Chem Biol. 2015;11:625–631. doi: 10.1038/nchembio.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flissi A., Dufresne Y., Michalik J., Tonon L., Janot S., Noe L. Norine, the knowledgebase dedicated to non-ribosomal peptides, is now open to crowdsourcing. Nucleic Acids Res. 2015;44:D1113–8. doi: 10.1093/nar/gkv1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yadav G., Gokhale R.S., Mohanty D. Computational approach for prediction of domain organization and substrate specificity of modular polyketide synthases. J Mol Biol. 2003;328:335–363. doi: 10.1016/s0022-2836(03)00232-8. [DOI] [PubMed] [Google Scholar]
- 34.Yadav G., Gokhale R.S., Mohanty D. Towards prediction of metabolic products of polyketide synthases: an in silico analysis. PLoS Comput Biol. 2009;5:e1000351. doi: 10.1371/journal.pcbi.1000351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ansari M.Z., Yadav G., Gokhale R.S., Mohanty D. NRPS-PKS: a knowledge-based resource for analysis of NRPS/PKS megasynthases. Nucleic Acids Res. 2004;32:W405–13. doi: 10.1093/nar/gkh359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anand S., Prasad M.V., Yadav G., Kumar N., Shehara J., Ansari M.Z. SBSPKS: structure based sequence analysis of polyketide synthases. Nucleic Acids Res. 2010;38:W487–96. doi: 10.1093/nar/gkq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tae H., Kong E.B., Park K. ASMPKS: an analysis system for modular polyketide synthases. BMC Bioinformatics. 2007;8:327. doi: 10.1186/1471-2105-8-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Starcevic A., Zucko J., Simunkovic J., Long P.F., Cullum J., Hranueli D. ClustScan: an integrated program package for the semi-automatic annotation of modular biosynthetic gene clusters and in silico prediction of novel chemical structures. Nucleic Acids Res. 2008;36:6882–6892. doi: 10.1093/nar/gkn685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li M.H., Ung P.M., Zajkowski J., Garneau-Tsodikova S., Sherman D.H. Automated genome mining for natural products. BMC Bioinformatics. 2009;10:185. doi: 10.1186/1471-2105-10-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rottig M., Medema M.H., Blin K., Weber T., Rausch C., Kohlbacher O. NRPSpredictor2 – a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 2011;39:W362–7. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bachmann B.O., Ravel J. Chapter 8. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol. 2009;458:181–217. doi: 10.1016/S0076-6879(09)04808-3. [DOI] [PubMed] [Google Scholar]
- 42.Kim J., Yi G.S. PKMiner: a database for exploring type II polyketide synthases. BMC Microbiol. 2012;12:169. doi: 10.1186/1471-2180-12-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khaldi N., Seifuddin F.T., Turner G., Haft D., Nierman W.C., Wolfe K.H. SMURF: genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol. 2010;47:736–741. doi: 10.1016/j.fgb.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Charlop-Powers Z., Milshteyn A., Brady S.F. Metagenomic small molecule discovery methods. Curr Opin Microbiol. 2014;19:70–75. doi: 10.1016/j.mib.2014.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reddy B.V., Milshteyn A., Charlop-Powers Z., Brady S.F. eSNaPD: a versatile, web-based bioinformatics platform for surveying and mining natural product biosynthetic diversity from metagenomes. Chem Biol. 2014;21:1023–1033. doi: 10.1016/j.chembiol.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ziemert N., Podell S., Penn K., Badger J.H., Allen E., Jensen P.R. The natural product domain seeker NaPDoS: a phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS ONE. 2012;7:e34064. doi: 10.1371/journal.pone.0034064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Medema M.H., Blin K., Cimermancic P., de Jager V., Zakrzewski P., Fischbach M.A. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011;39:W339–46. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weber T., Blin K., Duddela S., Krug D., Kim H.U., Bruccoleri R. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237–43. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cimermancic P., Medema M.H., Claesen J., Kurita K., Wieland Brown L.C., Mavrommatis K. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell. 2014;158:412–421. doi: 10.1016/j.cell.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finn R.D., Bateman A., Clements J., Coggill P., Eberhardt R.Y., Eddy S.R. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Skinnider M.A., Dejong C.A., Rees P.N., Johnston C.W., Li H., Webster A.L. Genomes to natural products PRediction Informatics for Secondary Metabolomes (PRISM) Nucleic Acids Res. 2015;43:9645–9662. doi: 10.1093/nar/gkv1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Minowa Y., Araki M., Kanehisa M. Comprehensive analysis of distinctive polyketide and nonribosomal peptide structural motifs encoded in microbial genomes. J Mol Biol. 2007;368:1500–1517. doi: 10.1016/j.jmb.2007.02.099. [DOI] [PubMed] [Google Scholar]
- 53.Medema M.H., Paalvast Y., Nguyen D.D., Melnik A., Dorrestein P.C., Takano E. Pep2Path: automated mass spectrometry-guided genome mining of peptidic natural products. PLoS Comput Biol. 2014;10:e1003822. doi: 10.1371/journal.pcbi.1003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Horbach R., Graf A., Weihmann F., Antelo L., Mathea S., Liermann J.C. Sfp-type 4′-phosphopantetheinyl transferase is indispensable for fungal pathogenicity. Plant Cell. 2009;21:3379–3396. doi: 10.1105/tpc.108.064188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vats A., Singh A.K., Mukherjee R., Chopra T., Ravindran M.S., Mohanty D. Retrobiosynthetic approach delineates the biosynthetic pathway and the structure of the acyl chain of mycobacterial glycopeptidolipids. J Biol Chem. 2012;287:30677–30687. doi: 10.1074/jbc.M112.384966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prather K.L., Martin C.H. De novo biosynthetic pathways: rational design of microbial chemical factories. Curr Opin Biotechnol. 2008;19:468–474. doi: 10.1016/j.copbio.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 57.McClymont K., Soyer O.S. Metabolic tinker: an online tool for guiding the design of synthetic metabolic pathways. Nucleic Acids Res. 2013;41:e113. doi: 10.1093/nar/gkt234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Soh K.C., Hatzimanikatis V. DREAMS of metabolism. Trends Biotechnol. 2010;28:501–508. doi: 10.1016/j.tibtech.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 59.Chou C.H., Chang W.C., Chiu C.M., Huang C.C., Huang H.D. FMM: a web server for metabolic pathway reconstruction and comparative analysis. Nucleic Acids Res. 2009;37:W129–34. doi: 10.1093/nar/gkp264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fenner K., Gao J., Kramer S., Ellis L., Wackett L. Data-driven extraction of relative reasoning rules to limit combinatorial explosion in biodegradation pathway prediction. Bioinformatics. 2008;24:2079–2085. doi: 10.1093/bioinformatics/btn378. [DOI] [PubMed] [Google Scholar]
- 61.Moriya Y., Shigemizu D., Hattori M., Tokimatsu T., Kotera M., Goto S. PathPred: an enzyme-catalyzed metabolic pathway prediction server. Nucleic Acids Res. 2010;38:W138–43. doi: 10.1093/nar/gkq318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao J., Ellis L.B., Wackett L.P. The University of Minnesota Pathway Prediction System: multi-level prediction and visualization. Nucleic Acids Res. 2011;39:W406–11. doi: 10.1093/nar/gkr200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martin C.H., Nielsen D.R., Solomon K.V., Prather K.L. Synthetic metabolism: engineering biology at the protein and pathway scales. Chem Biol. 2009;16:277–286. doi: 10.1016/j.chembiol.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 64.Kanehisa M. KEGG bioinformatics resource for plant genomics and metabolomics. Methods Mol Biol. 2016;1374:55–70. doi: 10.1007/978-1-4939-3167-5_3. [DOI] [PubMed] [Google Scholar]
- 65.Kanehisa M. The KEGG database. Novartis Found Symp. 2002;247:91–101. discussion 101–103, 119–128, 244-152. [PubMed] [Google Scholar]
- 66.Kotera M., Hattori M., Oh H.M., Yamamoto R., Komeno T., Yabuzaki J. RPAIR: a reactant-pair database representing chemical changes in enzymatic reactions. Genome Inform. 2004;15:P062. [Google Scholar]
- 67.O'Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open Babel: an open chemical toolbox. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods and Supplementary Figure.
Reactions used, Test dataset and the results.
Information for downloading Source code for Retro-biosynthetic algorithm as a perl script.