

ABSTRACT
Mutations in the tRNA methyltransferase 1 (TRMT1) gene have been identified as the cause of certain forms of autosomal-recessive intellectual disability (ID). However, the molecular pathology underlying ID-associated TRMT1 mutations is unknown, since the biological role of the encoded TRMT1 protein remains to be determined. Here, we have elucidated the molecular targets and function of TRMT1 to uncover the cellular effects of ID-causing TRMT1 mutations. Using human cells that have been rendered deficient in TRMT1, we show that TRMT1 is responsible for catalyzing the dimethylguanosine (m2,2G) base modification in both nucleus- and mitochondrion-encoded tRNAs. TRMT1-deficient cells exhibit decreased proliferation rates, alterations in global protein synthesis, and perturbations in redox homeostasis, including increased endogenous ROS levels and hypersensitivity to oxidizing agents. Notably, ID-causing TRMT1 variants are unable to catalyze the formation of m2,2G due to defects in RNA binding and cannot rescue oxidative stress sensitivity. Our results uncover a biological role for TRMT1-catalyzed tRNA modification in redox metabolism and show that individuals with TRMT1-associated ID are likely to have major perturbations in cellular homeostasis due to the lack of m2,2G modifications.
KEYWORDS: RNA, TRMT1, Trm1, dimethylguanosine, intellectual disability, methylation, methyltransferase, modification, neurodevelopment, tRNA modification
INTRODUCTION
All cellular tRNAs undergo a multitude of posttranscriptional modifications catalyzed by RNA modification enzymes (1–4). There are over 100 chemically distinct tRNA modifications that range from addition of a single methyl group to complex molecular transformations involving multiple enzymes and cofactors. Certain tRNA modifications are required for the proper processing, folding, and stability of tRNA, while other modifications are crucial for tRNA function in protein synthesis by modulating translation efficiency and fidelity (5–11). Consequently, organisms lacking certain tRNA modifications exhibit major perturbations in tRNA biogenesis and protein expression with downstream effects on cellular growth, metabolism, and development (12–16). The critical role of tRNA modification is further underscored by the numerous mutations in tRNA modification enzymes that have been linked to human diseases and disorders, including neurodevelopmental, mitochondrial, and cancer-related pathologies (17–22).
One of the most common modifications in tRNA is the methylation of the nitrogenous base or sugar residue of tRNA by S-adenosyl-methionine (SAM)-dependent methyltransferases (23, 24). The yeast Saccharomyces cerevisiae tRNA methyltransferase 1 (Trm1p) represents one of the first tRNA methyltransferases discovered, with homologs identified in archaea and all known eukaryotes (25–32). Yeast Trm1p has been found to be imported into the nucleus and mitochondria, where it catalyzes the methylation of a specific guanosine residue at position 26 in numerous tRNAs to yield the N2,N2-dimethylguanosine (m2,2G) modification (33–40). While Trm1p is nonessential for yeast viability, Trm1-null strains exhibit temperature sensitivity and defects in tRNA stability that are exacerbated in combination with deletion of either the Trm4p tRNA methyltransferase or the La RNA binding protein (41, 42). These observations suggest that Trm1p-catalyzed m2,2G modifications act in concert with additional tRNA modifications and RNA chaperones to assist in the proper folding and stability of tRNA (43). Consistent with a function in tRNA structure, crystallographic and simulation studies have shown that the m2,2G modification plays a key role in preventing alternative tRNA conformations through the destabilization of certain base-pairing interactions (44–48). Recent studies have also shown that the m2,2G modification is dynamically regulated in response to environmental conditions such as nutrient availability and exposure to DNA-damaging agents, suggesting that the m2,2G modification plays a role in the cellular stress response (12, 49, 50).
Two potential mammalian orthologs of yeast Trm1p have been identified by sequence homology that are encoded by the TRMT1 and TRMT1L genes (32, 51–54). Intriguingly, genome-wide analyses have implicated frameshift mutations in human TRMT1 as a novel cause for autosomal-recessive intellectual disability (ID) (55, 56). Individuals with biallelic TRMT1 mutations display cognitive impairment, neurodevelopmental delays, and facial dysmorphism. TRMT1L has also been linked to neurological development, since Trmt1l-deficient mice exhibit altered motor coordination and aberrant behavior without any major anatomical changes (32). While TRMT1 and TRMT1L are predicted to catalyze the m2,2G RNA modification, methyltransferase activity has been demonstrated only for recombinant TRMT1 on a single tRNA substrate in vitro (57). Most critically, it remains to be shown whether either TRMT1 or TRMT1L plays a role in RNA modification in vivo and, if so, which RNA targets are subject to modification. Thus, the cellular role of the human Trm1p paralogs as well as the molecular consequences of ID-associated TRMT1 mutations remain enigmatic.
Here, we focus on TRMT1 to gain understanding into its cellular targets and functions along with the potential mechanism underlying ID-associated TRMT1 mutations. Using CRISPR gene knockout (KO), we show that TRMT1 is required for catalyzing the m2,2G modification in cytoplasmic and mitochondrial tRNAs of human cells. Significantly, we find that TRMT1-deficient cells exhibit decreased global protein translation, perturbations in cellular ROS levels, and hypersensitivity to oxidizing agents. Moreover, we demonstrate that ID-associated TRMT1 mutants exhibit defects in m2,2G formation and lack the ability to rescue cellular translation or cell survival in response to oxidative stress. Our results uncover a role for TRMT1-catalyzed tRNA modification in redox homeostasis and provide insight into the cellular effects caused by ID-associated TRMT1 mutations.
RESULTS
Human TRMT1 is a nucleus-encoded protein that is imported into mitochondria and the nucleus.
The human TRMT1 gene is predicted to encode a 659-amino-acid polypeptide containing a class I S-adenosyl-methionine (SAM) binding methyltransferase domain. In addition, sequence analysis revealed the presence of a C-terminal bipartite nuclear localization signal (NLS) embedded within a C3H1-type zinc finger motif, suggesting the nuclear import of TRMT1 (Fig. 1A; see also Fig. S1A in the supplemental material). Moreover, TRMT1 is also predicted to contain a mitochondrial targeting signal and mitochondrial protease cleavage sites at the amino terminus, suggesting the import of nucleus-encoded TRMT1 into mitochondria (Fig. 1A; Fig. S1B and C). In comparison, the predicted TRMT1 paralog, TRMT1L, also contains a putative bipartite nuclear localization signal at the C terminus but no predicted mitochondrial targeting signal (Fig. 1A; Fig. S1A).
FIG 1.

Subcellular localization of TRMT1 and TRMT1L. (A) Schematic of TRMT1 and TRMT1L with predicted domains and localization sequences. (B) TRMT1 is highly enriched in purified mitochondrial and nuclear fractions indicative of mitochondrial import and processing. IN, input; CM, cytoplasmic and mitochondrial fractions; C, cytoplasm; M, mitochondrial fraction; Mx5, 5-fold more mitochondrial equivalents loaded; NE, nuclear extract. Arrows indicate TRMT1 isoforms. *, nonspecific band detected by anti-TRMT1 antibody. Lamin B1, tubulin, and NADH:ubiquinone oxidoreductase core subunit S7 (NDUFS7) served as nuclear, cytoplasmic (Cyto), and mitochondrial (Mito) markers, respectively. (C and D) Endogenous TRMT1 and TRMT1-FLAG display nuclear and mitochondrial localization in HeLa cervical carcinoma cells. (E and F) TRMT1L is localized primarily to the nucleus and colocalizes with the nucleolar marker nucleophosmin. Mitochondria were identified using mitochondrion-targeted red fluorescent protein, and nuclear DNA was stained with DAPI.
To experimentally monitor the localization of the human Trm1 homologs, we performed subcellular fractionation on human HeLa cervical carcinoma cells followed by immunoblotting of the different fractions for TRMT1 or TRMT1L. Consistent with subcellular predictions, we detected the presence of TRMT1 in the nuclear and mitochondrial fractions of human cells (Fig. 1B). The TRMT1 found in the mitochondrial fraction was present as a lower-molecular-weight isoform that comigrated with a nonspecific protein band (Fig. 1B; arrows denote TRMT1, and the asterisk denotes the nonspecific band). The presence of a truncated form of TRMT1 in mitochondria is consistent with the predicted mitochondrial protease cleavage sites noted above and N-terminal processing of the mitochondrial targeting sequence, a common occurrence for many imported mitochondrial proteins. Using immunofluorescence in HeLa cells, we detected uniform TRMT1 staining in the nucleus as well as a filamentous TRMT1 signal in the cytoplasm that colocalized with mitochondria (Fig. 1C). In addition, a TRMT1 fusion protein with either a carboxyl-terminal FLAG or green fluorescent protein tag also displayed pan-nuclear and mitochondrial localization (Fig. 1D; Fig. S1D).
In contrast to TRMT1, endogenous TRMT1L exhibited discrete nucleolar localization without any cytoplasmic accumulation in mitochondria, as evidenced by subcellular fractionation and microscopy (Fig. 1B and E). In addition, we confirmed the nucleolar accumulation of TRMT1L through colocalization with the nucleolar marker nucleophosmin (Fig. 1F). The accumulation of TRMT1 in both the mitochondria and nucleus compared to the nucleus-only localization of TRMT1L is consistent with the sequence predictions noted above. Moreover, the differential subcellular localization patterns of human TRMT1 and TRMT1L suggest distinct targets and roles for each putative enzyme. Here, we focus on TRMT1 and its potential role in the methylation of nuclear and mitochondrial RNA transcripts.
TRMT1-deficient cells lack m2,2G modifications in tRNA.
To investigate the cellular role of TRMT1, we used CRISPR-mediated gene editing to abolish TRMT1 expression in 293T human embryonic kidney cells. We targeted the first exon of TRMT1 for CRISPR-induced mutagenesis using two different guide RNAs and generated single-cell clones for further analysis (Fig. 2A). As a wild-type (WT) control, we generated a cell line in which the AAVS1 gene locus was targeted for CRISPR mutagenesis (control-WT). The AAVS1 locus is a validated genomic region that can be disrupted without any known or discernible phenotypic effects in mammalian cells (58, 59). Genotyping of two independent TRMT1-knockout (KO) clones revealed the presence of indel frameshift mutations that are predicted to generate truncated polypeptides less than 70 amino acid residues in length (TRMT1-KO1 and -KO2) (Fig. 2A). Indeed, immunoblotting revealed the absence of detectable TRMT1 protein in both TRMT1-KO cell lines compared to the control-WT cell line (Fig. 2B). Moreover, loss of TRMT1 expression has no significant effect on TRMT1L levels (Fig. 2B, TRMT1L).
FIG 2.

TRMT1 is required for the formation of m2,2G in cellular tRNA. (A) CRISPR/Cas9 gene knockout (KO) strategy depicting sequence guide RNAs (sgRNAs) targeting exon 1 of the human TRMT1 gene. Indel mutations in the genomic sequence of the TRMT1-KO strains used in this study are noted below. (B) Immunoblot of TRMT1 and TRMT1L levels in the wild-type control (control-WT) and TRMT1 knockout cell lines (TRMT1-KO1 and -KO2). An asterisk denotes a nonspecific band detected by the TRMT1 antibody. (C) Molar percentage of m2,2G or m2G modification in tRNA isolated from control-WT or TRMT1-KO cell lines. (D) Fold change in tRNA modification levels of the indicated TRMT1-KO strain relative to the control-WT cell line. Quantification for panels C and D was based on 3 independent RNA samples from each cell line.
We next directly measured the levels of 20 different tRNA modifications in the human cell lines through quantitative mass spectrometry of modified ribonucleosides (60). Focusing on m2,2G and using absolute quantification of modified nucleosides, we found that the m2,2G modification percentage per tRNA molecule was ∼50% in the control-WT strain, which decreased to near background levels in both TRMT1-KO cell lines (Fig. 2C). Comparing the relative change between cell lines, both TRMT1-KO cell lines displayed a >100-fold decrease in m2,2G modification levels relative to the control-WT strain (Fig. 2D). Interestingly, no other modification displayed a statistically significant change between the control versus TRMT1-KO cell lines, including the similar methyl modification, N2-monomethylguanosine (m2G) (Fig. 2C and D). These results indicate that TRMT1 is required for the formation of nearly all detectable m2,2G modifications in the tRNA of human cells.
TRMT1 catalyzes the formation of m2,2G in cytoplasmic and mitochondrial tRNAs.
To identify the tRNAs that are modified by TRMT1, we used the PHA (for positive hybridization in the absence of modification) assay (50). This Northern blot-based assay relies on differential probe hybridization to tRNA caused by the presence or absence of m2,2G, which impairs base pairing (47, 61, 62). Thus, a decrease in m2,2G modification leads to an increase in PHA probe signal that can be normalized against the probe signal from a different region of the same tRNA as an internal control (Fig. 3A). We tested a panel of PHA probes against nucleus-encoded tRNA isoacceptors that have been shown to contain an m2,2G modification at position 26 (63–65). As negative controls for differential hybridization, we probed for tRNAs that are known to lack m2,2G (tRNA-Gly-CCC and Val-CAC) (66–68). Using this assay, we find that the steady-state levels of all tested tRNAs remained similar between the WT and TRMT1-KO cells (Fig. 3B, T-loop signal). In contrast, we found that the PHA signal was increased for all predicted m2,2G-containing tRNAs in the TRMT1-KO cells, indicating the loss of the m2,2G modification (Fig. 3B, PHA). As expected, no change in PHA signal was detected for any of the control tRNAs that normally lack m2,2G at position 26 (Fig. 3B, tRNA-Gly-CCC and Val-CAC). These results demonstrate that TRMT1 is required for methylating at least 10 of the 12 tRNA isotypes containing G26. The remaining two tRNAs with G26, tRNA-Tyr and tRNA-Val, have been shown to be modified by TRMT1 in vitro or contain m2G at position 26, respectively (54, 69). Thus, TRMT1 is required for the formation of m2,2G in the majority of nucleus-encoded tRNAs.
FIG 3.

TRMT1 is necessary for the formation of m2,2G in cytoplasmic and mitochondrial tRNAs. (A) Assay of positive hybridization in the absence (PHA) of G26 modification to monitor m2,2G formation in tRNA. PHA probe spans position G26 between the D-AC stem-loops, while the T-loop probe was used for signal normalization. (B) Northern blot PHA assays with the indicated probes using RNA extracted from control-WT or TRMT1-KO cell lines. (C) Schematic of primer extension assay to monitor the presence of m2,2G at position 26 of tRNA. (D and E) Primer extensions with the indicated nucleus- or mitochondrion-encoded tRNA probes. RT, reverse transcriptase; Um, 2′-O-methyluridine; D, dihydrouridine; m1A, 1-methyladenosine; m1G, 1-methylguanosine; m2G, 2-methylguanosine; ψ, pseudouridine; *, nonspecific signal; >, labeled oligonucleotide used for primer extension.
To precisely determine whether TRMT1 catalyzes the methylation of G26 in tRNA, we used a primer extension assay that monitors RNA modification status at nucleotide resolution. In this assay, the presence of m2,2G leads to a block of reverse transcriptase (RT) at position 26 of tRNA, while a decrease in m2,2G allows for readthrough and an extended product up to a subsequent RT-blocking modification, such as dihydrouridine (D), 1-methyladenosine (m1A), or 1-methylguanosine (m1G) (Fig. 3C). As expected, we identified an RT block at the expected position of each tested tRNA in control-WT cells, indicative of the m2,2G modification (Fig. 3D, WT). In contrast, the RT block at position 26 of each tested m2,2G-containing tRNA was completely absent from both TRMT1-KO cell lines (Fig. 2D, KO1 and KO2). These results show that m2,2G formation at position 26 in tRNA is due to TRMT1-catalyzed modification.
The localization of TRMT1 in both the nucleus and mitochondria suggests a dual role for TRMT1 in the modification of nucleus- and mitochondrion (mt)-encoded tRNAs. In human cells, seven mitochondrial tRNAs contain G at position 26, with mt-tRNA-Ile being the only one known to contain m2,2G (68, 70). Testing all seven mt-tRNAs, we detected an increase in PHA signal for mt-tRNA-Ile in both TRMT1-KO cells, while the PHA status of all other mt-tRNAs remained the same (Fig. S2). Using primer extension as additional confirmation, we detected the presence of m2,2G at position 26 in mt-tRNA-Ile, which was absent from the TRMT1-KO cell lines (Fig. 3D, mt-Ile-UAU). These results indicate that TRMT1 catalyzes the formation of m2,2G in cytoplasmic tRNAs as well as mitochondrial tRNA-Ile.
Even though mt-tRNA-Ile is the only known mitochondrial tRNA in human cells to contain m2,2G, certain mitochondrion-encoded tRNAs can also contain m2G at position 26 (70–72). While we detected no change in PHA signal for any human mt-tRNA besides mt-tRNA-Ile, the hybridization-based PHA assay does not provide information on the presence or absence of m2G, since m2G can form canonical base-pairing interactions and maintain duplex stability (62, 73). However, the presence of m2G does inhibit RT-catalyzed primer extension leading to a pause signal (74–76). Thus, we used primer extension to test whether m2G was present in mt-tRNA-Ala and mt-tRNA-Arg, which both contain a G at position 26. We detected a primer extension pause at position 26 for both mitochondrial tRNAs from control-WT cells that was absent from the TRMT1-KO cell lines (Fig. 3E). Moreover, TRMT1 is specific for the methylation of G at position 26, since the m2G present at position 10 in mt-tRNA-Ala was maintained in TRMT1-KO cells (Fig. 3E, upper gel). While we detected no major change in m2G levels by mass spectrometry (Fig. 2C), this is likely due to the small fraction of TRMT1-catalyzed m2G present in only a subset of mitochondrial tRNAs compared to the abundant m2G at other tRNA positions. Altogether, these studies uncover TRMT1 as the human enzyme responsible for catalyzing the formation of m2,2G at position 26 in nucleus-encoded tRNAs as well as m2,2G and m2,G in certain mitochondrial tRNAs.
TRMT1 mutations that cause intellectual disability result in defective tRNA modification activity.
The TRMT1-deficient human cell lines generated in this study provide a system to investigate TRMT1 variants found in the human population through functional complementation. In this assay, we transiently transfected the TRMT1-KO1 cell line with expression constructs expressing wild-type TRMT1 or variants thereof, followed by primer extension assay to monitor m2,2G formation. We generated two known TRMT1 variants that are predicted to arise from point mutations in the TRMT1 gene of individuals diagnosed with ID (55, 56). The ID-associated TRMT1 variants encode truncated proteins lacking all of the C3H1 zinc finger motif while retaining all or a portion of the SAM-MT domain (Fig. 4A, Q219fs and Y445fs). In addition, we engineered a TRMT1 variant lacking the first 36 amino acid residues constituting the MTS sequence to investigate the mitochondrial role of TRMT1 (TRMT1ΔMTS). As a control, we also generated a TRMT1 variant containing a single point mutation of a conserved cysteine residue in the SAM catalytic site that has been shown to abrogate activity in tRNA methyltransferases (Fig. 4A, C348R) (54). Each TRMT1 variant was transiently expressed as untagged proteins with expression confirmed by immunoblotting. All TRMT1 variants exhibited levels of accumulation similar to that of WT-TRMT1 (Fig. 4B).
FIG 4.

Intellectual disability-causing mutations in TRMT1 abrogate tRNA modification activity and RNA binding. (A) Schematic of TRMT1 domains and variants. WT, wild type; ΔMTS, deletion of mitochondrial targeting signal; C348R, catalytic-inactive variant (an asterisk denotes point mutation); Q219fs and Y445fs, TRMT1 variants encoded by ID frameshift mutations in the TRMT1 gene. (B) Immunoblot of whole-cell extracts prepared from each human cell line transfected with the indicated constructs. (C) Primer extension assay to monitor the presence of m2,2G in tRNA from cell lines transfected with the indicated TRMT1 constructs. >, labeled oligonucleotide used for primer extension. (D) Immunoblot for TRMT1 of input (2%) or FLAG affinity purifications (6%) from 293T cells transfected to express the FLAG tag alone (vector) or FLAG-tagged TRMT1 variants. (E) Nucleic acid stain of RNAs extracted from the indicated input or purified samples after denaturing PAGE. The migration pattern of tRNAs, 5.8S rRNA, and 5S rRNA are denoted. (F) Northern blot hybridization analysis of TRMT1-associated RNAs with the indicated probes.
Through primer extension analysis of m2,2G-containing tRNAs, we found that expression of wild-type TRMT1 in TRMT1-KO cells is necessary and sufficient to restore the presence of m2,2G in all tested cytoplasmic and mitochondrial tRNAs (Fig. 4C, WT). In contrast to wild-type TRMT1, expression of TRMT1 containing a point mutation in the SAM-MT catalytic site was unable to restore m2,2G formation in any tRNA (Fig. 4C, C348R). While expression of TRMT1ΔMTS can restore formation of m2,2G in cytoplasmic tRNAs, this TRMT1 variant was severely impaired in rescuing m2,2G formation in mitochondrial tRNAs (Fig. 4C, TRMT1ΔMTS). This result provides functional evidence that the predicted MTS sequence in TRMT1 is an active mitochondrial import signal that is necessary for TRMT1 to catalyze mitochondrial tRNA modification. Strikingly, we find that neither ID-associated TRMT1 variant was able to restore m2,2G modification in cytoplasmic or mitochondrial tRNAs of TRMT1-KO cells (Fig. 4C, Q219fs or Y445fs). The lack of tRNA modification activity is not due to TRMT1 expression levels, since the ID-associated variants accumulated to levels similar to that of wild-type TRMT1 (Fig. 4B). Our results demonstrate that nearly all m2,2G modifications in tRNA are catalyzed by TRMT1 and that ID-associated TRMT1 variants are defective in tRNA modification activity.
To decipher the molecular basis by which ID-associated mutations disrupt TRMT1 activity, we examined the interaction between TRMT1 and tRNA substrates by RNA immunoprecipitation. We expressed a FLAG-tagged version of each TRMT1 variant in 293T HEK cells, followed by affinity purification and analysis of copurifying RNAs. Immunoblotting confirmed the expression and purification of each TRMT1 variant on anti-FLAG resin (Fig. 4D). While no RNAs were found in the control purification from vector-transfected cells, we detected RNAs of ∼70 nucleotides that copurified with WT-TRMT1 and TRMT1-ΔMTS (Fig. 4E). By Northern blotting, we identified the TRMT1-associated RNAs as m2,2G-containing tRNAs (Fig. 4F). Moreover, the TRMT1 interaction was specific to m2,2G-containing tRNAs, since tRNA-Glu-UUC lacking m2,2G as well as U6 snRNA displayed only background binding to TRMT1. In contrast to WT-TRMT1, neither of the ID-associated TRMT1 mutants copurified with any tRNA species (Fig. 4F, Q219fs or Y445fs). These results demonstrate that ID-associated TRMT1 variants are abrogated in their ability to modify tRNA due to RNA binding defects and indicate that individuals homozygous for ID-associated TRMT1 alleles will be deficient in m2,2G tRNA modifications.
Loss of TRMT1-catalyzed tRNA modifications perturbs cellular protein synthesis.
We next investigated the molecular role of TRMT1-catalyzed tRNA modifications to provide insight into the potential biological effects caused by ID-associated TRMT1 mutations. Due to the prevalence of tRNAs containing the m2,2G modification and its predicted role in modulating tRNA structure, we tested whether the presence or absence of m2,2G could influence the biological activity of tRNA in cellular translation. Global de novo protein synthesis in the control-WT and TRMT1-KO cell lines was monitored via the incorporation of puromycin into nascent polypeptides, followed by detection of puromycin-labeled proteins through immunoblotting (77). The puromycin-labeling method has been shown to be a sensitive measure of global protein synthesis rates in multiple cell types and conditions, including changes that affect the translation machinery (78, 79).
By monitoring puromycin incorporation over time and normalization against total protein loaded, we found that both TRMT1-KO cell lines displayed a notably lower rate of puromycin incorporation than control-WT cell lines (Fig. 5A). The reduction in puromycin incorporation exhibited by TRMT1-KO cells could be detected starting at 30 min postincubation, with an eventual plateau as the labeling became saturated (Fig. 5B; Fig. S3). Most critically, reexpression of WT-TRMT1 but not the ID-associated TRMT1 variants could increase the levels of nascent polypeptides labeled by puromycin in the TRMT1-KO1 strain (Fig. 5C and D). Thus, the tRNA modification activity of TRMT1 is required for rescuing global protein synthesis in the TRMT1-KO cell lines. As a control, the formation of puromycin conjugates was diminished to near background levels if protein synthesis was inhibited by cycloheximide, indicating that translating ribosomes are required for puromycin incorporation into nascent polypeptide chains (Fig. S3). These results are consistent with a role for TRMT1-catalyzed tRNA modifications in maintaining basal levels of global translation and suggest that the steady-state accumulation of numerous proteins and their downstream functions are impacted in individuals with ID-causing TRMT1 mutations.
FIG 5.

TRMT1-catalyzed tRNA modifications are required for basal levels of protein synthesis. (A) Protein synthesis assay based upon translation-dependent puromycin (puro) incorporation. Endogenous nascent polypeptide chains were labeled with puromycin for the indicated times in control-WT or TRMT1-KO cell lines, followed by fractionation and immunoblot detection with antipuromycin antibodies. Representative blots are shown with total protein serving as a loading control. (B) Quantification of relative puromycin incorporation for TRMT1-KO cells compared to control-WT for each puromycin time point. The means and standard deviations represent the puromycin signal of each cell line relative to that of control-WT after normalization to total protein at each time point from three independent labeling experiments. (C) Rescue of protein synthesis by reexpression of TRMT1-WT but not ID-associated mutants. Immunoblot of puromycin-labeled polypeptides from cell lines transfected with the indicated TRMT1 expression constructs. (D) Quantification of relative de novo protein synthesis from panel C as measured by the accumulation of puromycin-labeled polypeptides from two independent labeling experiments after transfection. *, P < 0.05; **, P < 0.01. (E and F) Mitochondrial protein translation analysis. Control-WT and TRMT1-KO cells were incubated with [35S]methionine-cysteine in the presence of the cytoplasmic translation inhibitor emetine alone or with the mitochondrial translation inhibitor chloramphenicol (Cam). Whole-cell extracts were resolved by SDS-PAGE and stained with Coomassie (E) and 35S-labeled proteins were visualized by phosphorimaging (F), with putative assignments noted on the left. *, nonspecific labeling. (G) Quantification of the fold difference in translation of the indicated mitochondrion-encoded proteins relative to control-WT cells. Measurements represent the averages from three independent labeling wells.
In addition to cytoplasmic protein synthesis, we tested if mitochondrial translation was affected by TRMT1-catalyzed tRNA modifications. We used [35S]methionine-cysteine to metabolically label nascent mitochondrial polypeptides while inhibiting cytoplasmic translation using emetine (80). Based upon this assay, we detected an array of 35S-labeled products that matched the expected pattern of mitochondrion-encoded proteins (Fig. 5E, total protein, and F, 35S-labeled proteins). As a control, incubation of cells with both emetine and the mitochondrial translation inhibitor chloramphenicol blocked the formation of 35S-labeled mitochondrial proteins (Fig. 5F, Cam). All predicted mitochondrion-encoded proteins were observed except for MT-ND4 and ATP8, which could not be resolved on the gel due to their low molecular masses (<10 kDa). Mitochondrion-encoded proteins that displayed detectable signal were compared between cell lines either individually or paired depending on the resolution of the protein bands. No significant or consistent change in translation was detected for cytochrome b (CYB) or ATP synthase subunit 6 (ATP6) between the cell lines (Fig. 5G, CYB and ATP6). However, increased mitochondrial translation was detected for ND5/COI, ND4, ND2, ND1, and CO2/CO3 (Fig. 5G). Intriguingly, the mitochondrion-encoded proteins that displayed increased translation are all subunits of complex I or IV. The increased translation of particular mitochondrion-encoded proteins contrasts with the decreased cytoplasmic translation in the TRMT1-KO cells and suggests that TRMT1-catalyzed tRNA modifications play differential roles in the cytoplasm versus the mitochondria. Moreover, the increased mitochondrial translation exhibited by TRMT1-KO cells represents a unique phenotype that differs from the mitochondrial protein synthesis defects caused by deficiencies in other mitochondrial tRNA modifications (81–87).
TRMT1-catalyzed tRNA modifications are required for proper cell proliferation and redox homeostasis.
Previous studies have linked tRNA modification-dependent translation with cellular growth and survival in response to stress stimuli (11, 88–91). However, the role of TRMT1-catalyzed tRNA modifications in human cell physiology is unknown. While no overt differences in morphology or size were detected between any of the cell lines based upon microscopy or flow cytometry (Fig. 6A and B), we found that TRMT1-deficient cells displayed noticeably slower proliferation than the control-WT strain under standard growth conditions of serum and oxygen (Fig. 6C). The slow growth was not due to increased cell death in the TRMT1-KO cell lines, since viability remained >95% for each cell line across all time points (Fig. 6D). Based upon cell cycle analysis, we found that TRMT1-KO cell lines exhibited an increase in the percentage of cells in G2/M phase concomitant with a decrease in cells in G1/G0 phase relative to control-WT cells (Fig. 6E and F). These results suggest that loss of TRMT1 expression and tRNA modification leads to slower progression through G2/M phase and decreased cellular proliferation.
FIG 6.

TRMT1-deficient cell lines display decreased cellular proliferation and altered cell cycle dynamics without significant changes in cell morphology or size. (A) Phase-contrast images of control-WT versus TRMT1-KO cell lines. (B) Representative flow cytometry histogram plots of control-WT versus TRMT1-KO cell lines, with the x axis representing forward scatter (FSC), which is indicative of cell size. (C and D) Cell proliferation and viability plots of control-WT versus TRMT1-KO cell lines. Cell count and viability were measured by flow cytometry at the indicated time points after plating. Cell count and deviation represent measurements from three independent cultures. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (E and F) Cell cycle analysis of control-WT versus TRMT1-KO cell lines. (E) Representative flow cytometry histograms of cell cycle content are shown as overlays relative to control-WT. (F) Relative percentages of G0/G1, S, and G2/M-phase cells measured by flow cytometry analysis were quantified and plotted.
Since TRMT1 is required for the formation of m2,2G in mitochondrial tRNA-Ile, we investigated whether changes in mitochondrial physiology could account for the slow-growth phenotype of TRMT1-KO cells. However, no major differences in mitochondrial membrane potential or mitochondrial reactive oxygen species (ROS) were detected between any of the cell strains, even though TRMT1-KO cells lack m2,2G in mitochondrial tRNA-Ile and altered mitochondrial translation (Fig. S4A to D). The absence of any significant mitochondrial defects in TRMT1-KO cells is consistent with the clinical observation that individuals with ID-associated TRMT1 mutations exhibit cognitive deficits without the pathological features commonly associated with mitochondrial tRNA disorders, such as myopathy or stroke (84, 92–95).
While no major changes in mitochondrial function were detected, we were surprised to find that TRMT1-KO cells exhibited increased levels of total ROS, as determined by flow cytometry analysis with the oxidation-sensitive probe dihydroethidium (DHE) (Fig. 7A, compare red and green outlines of TRMT1-KO cells to gray outline of control-WT). In both populations of TRMT1-KO cell lines, the mean fluorescence intensity of DHE was increased by 20 to 30% relative to that of control-WT cells (Fig. 7B). As an independent measure of ROS levels, we also used the cytoplasmic ROS probe CellROX to quantify the number of cells with increased superoxide. In agreement with ROS measurements using DHE, we detected an increase in CellROX mean fluorescence intensity along with the number of cells with increased ROS for TRMT1-KO cells relative to control-WT cells (Fig. S4E and F). In addition, we confirmed that the increase in DHE signal of TRMT1-KO cell lines was due to ROS, since growth in the antioxidant N-acetylcysteine (NAC) was sufficient to decrease the DHE signal specifically in TRMT1-KO cells (Fig. 7B, +NAC). Most significantly, growth in NAC could partially alleviate the slow-growth phenotype of TRMT1-KO cells (Fig. 7C). These results uncover an unexpected role for TRMT1 in redox homeostasis and indicate that the growth defect observed in TRMT1-KO cells is due in part to elevated levels of oxidative stress.
FIG 7.

TRMT1-deficient cell lines exhibit defects in ROS homeostasis and oxidative stress survival. (A) Flow cytometry plots of cells stained with the superoxide detection probe dihydroethidium (DHE). Intensity values are shown on the x axis with cell counts on the y axis. (B) Quantification of relative ROS levels based on DHE signal of the indicated cell lines in the absence or presence of the antioxidant N-acetylcysteine (+NAC). (C) Cell proliferation curves of control-WT versus TRMT1-KO cell lines in the absence or presence of NAC. Cell count and deviations represent measurements from independent cultures. (D) Flow cytometry plots of DHE-stained cells after growth in the absence or presence of 100 μM menadione (+Men). (E) Relative levels of ROS were quantified as described for panel A. (F) Dose-response curve of control-WT or TRMT1-KO cell lines after exposure to tert-butyl-hydroperoxide (t-bu-OOH). (G) Viability of control-WT or TRMT1-KO cell lines after incubation with the indicated chemical agent. (H) Viability of control-WT or TRMT1-KO1 cell lines transfected with the indicated TRMT1 plasmid after treatment with t-bu-OOH. Error bars for ROS measurements and viability assays represent the standard deviations from two to four independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
We further investigated the function of TRMT1 in redox homeostasis by comparing ROS levels between control-WT and TRMT1-KO cell lines after treatment with menadione, a naphthoquinone vitamin K analog that induces the formation of endogenous ROS by futile redox cycling (96, 97). As expected, treatment of control-WT or TRMT1-KO cells with menadione led to a shift in cell population with elevated levels of ROS (Fig. 7D). Remarkably, the fold increase in ROS between untreated and treated TRMT1-KO cells was significantly higher than the fold increase detected between untreated and treated control-WT cells (Fig. 7D and E, compare menadione-treated populations of control-WT versus TRMT1-KO1 or -KO2). In other words, menadione further exacerbated the already elevated levels of ROS in the TRMT1-KO cell lines. Since ROS formation by menadione is dependent upon metabolism by one-electron reducing enzymes, these results suggest that TRMT1 modulates redox homeostasis through regulation of the levels or activity of reductive enzymes present in the cytosol or mitochondria.
To determine the cellular consequences of perturbations in TRMT1-dependent redox homeostasis, we tested whether TRMT1-KO cells exhibited any changes in sensitivity when challenged with additional forms of stress or damage. Consistent with a defect in the redox homeostasis, we found that TRMT1-KO cells displayed hypersensitivity to multiple oxidizing agents, including tert-butyl-hydroperoxide (t-bu-OOH), hydrogen peroxide (H2O2), and arsenic trioxide (As2O3) (Fig. 7F and G). Moreover, the sensitivity defect exhibited by TRMT1-KO cells was specific to oxidizing agents, since no significant difference in viability or cell growth was found for an alkylating agent (methylmethane sulfonate [MMS]) or the topoisomerase inhibitor etoposide. Intriguingly, previous studies have shown that loss of Trm1p in S. cerevisiae confers sensitivity to MMS but not H2O2, even though m2,2G levels increase in response to H2O2 (49, 98). Thus, these findings reveal differential contributions of the m2,2G modification in stress survival between yeast and human cells.
Focusing on the cellular response to t-bu-OOH due to its chemical stability and consistent lethal dose (99, 100), we found that TRMT1-KO cells exhibited a dose-dependent hypersensitivity to t-bu-OOH that resulted in loss of plasma membrane integrity and severely diminished viability (Fig. 7F and G; Fig. S5A and B). Importantly, the sensitivity to oxidative stress exhibited by TRMT1-KO cells could be partially rescued by transient reexpression of TRMT1 (Fig. 7H). Due to heterogeneous TRMT1 expression caused by incomplete transfection efficiency, oxidative stress survival was increased but not completely restored to the level of the control-WT cell line. In contrast to WT TRMT1, neither ID-associated TRMT1 variant displayed any rescue of the oxidative stress sensitivity phenotype in the TRMT1-KO cell strain despite being expressed to levels similar to that of WT-TRMT1 (Fig. 7H, Q219fs and Y445fs; Fig. S5C). Moreover, the partial rescue of t-bu-OOH sensitivity by TRMT1 reexpression is linked to TRMT1 catalytic activity, since transfection with the TRMT1-C348R catalytic-dead mutant had no detectable effect on t-bu-OOH sensitivity in the TRMT1-KO cell line (Fig. 7H, C348R). Altogether, these results implicate a key role for TRMT1 and TRMT1-catalyzed tRNA modifications in cell proliferation and ROS homeostasis that are abrogated by ID-associated mutations in TRMT1.
TRMT1 is required for oxidative stress survival in human neural cells.
The increased levels of ROS and hypersensitivity of TRMT1-KO cells to oxidative stress suggests that a deficiency in TRMT1-catalyzed tRNA modifications could perturb neural tissue physiology in humans with ID-associated TRMT1 mutations. Thus, we investigated TRMT1 function in the ReNcell VM human neural stem cell (NSC) system, which is an established neural progenitor cell line derived from the ventral mesencephalon region of human fetal brain (101). Due to the technical challenges of isolating single mutant clones from NSC populations, we used two different lentiviral RNA interference (RNAi) constructs targeting TRMT1 to generate polyclonal NSC lines depleted of TRMT1 (Fig. 8A, TRMT1-KD1 and -KD2). As a control, we generated an NSC cell line containing a nonsilencing RNAi vector targeting enhanced green fluorescent protein (EGFP) (Fig. 8A, EGFP-KD). Validating the role of TRMT1 in m2,2G formation shown above, depletion of TRMT1 in ReNcell NSCs decreases the extent of m2,2G modification in tRNA, as detected by the increase in primer extension readthrough product for tRNA-Ile-UAU, Met-CAU, and mt-Ile-GAU (Fig. 8B). Notably, we find that both NSC lines depleted of TRMT1 display increased sensitivity to t-bu-OOH compared to the control NSC line (Fig. 8C). Thus, even partial depletion of TRMT1 and m2,2G modification in tRNA is sufficient to alter the oxidative stress sensitivity of human neural cells. Collectively, our studies reveal a function for TRMT1 and TRMT1-catalyzed tRNA modifications in protein translation and ROS homeostasis to ensure proper cell proliferation and oxidative stress survival.
FIG 8.
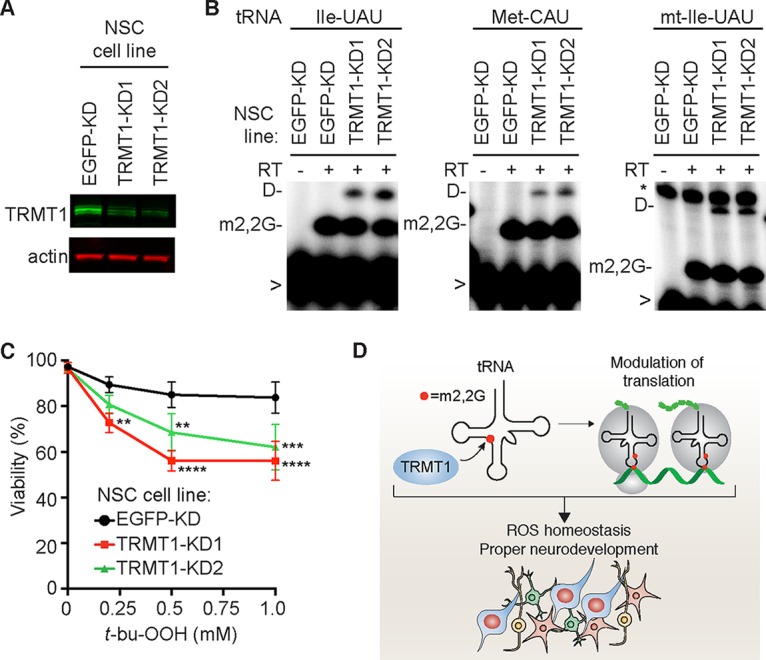
TRMT1 contributes to oxidative stress resistance in human neural stem cells (NSC). (A) Immunoblot analysis of TRMT1 levels in control or TRMT1-KD NSC lines. (B) Primer extension assay to monitor the presence of m2,2G in the tRNAs of the indicated NSC lines. RT, reverse transcriptase; D, dihydrouridine; >, labeled oligonucleotide for primer extension. (C) Viability of the indicated NSC line after treatment with t-bu-OOH. Error bars for ROS measurements and viability assays represent the standard deviations from at least two to six independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (D) Proposed function for TRMT1-catalyzed tRNA modifications in translation, ROS homeostasis, and neurodevelopment. See Discussion for details.
DISCUSSION
While mutations in tRNA modification enzymes have been identified as the cause of diverse neurodevelopmental disorders, the molecular basis for many of these pathologies remains unknown, including the etiology of TRMT1-associated ID. Here, we show that TRMT1 plays a critical role in global protein synthesis and ROS metabolism through a mechanism dependent upon the tRNA modification activity of TRMT1. Significantly, our studies suggest that a deficiency in redox homeostasis due to the loss of TRMT1-catalyzed tRNA modifications could perturb the growth and viability of neurons within individuals that are homozygous for ID-associated TRMT1 alleles (Fig. 7D). Consistent with this expectation, certain neurons in the brain are exquisitely sensitive to changes in the levels of reactive oxygen species, including neural stem cells that contribute to neurogenesis and cognition (102–105). Moreover, elevated levels of oxidative stress have been implicated in pathogenesis of neurological disorders such as Down syndrome and Parkinson's disease (106, 107). Finally, our results suggest that treatment with antioxidants could alleviate some of the cellular sensitivity to oxidative stress that contributes to neurodevelopmental delays, as demonstrated in vivo using mouse models of other neurological disorders (90, 108).
The loss of methyltransferase activity displayed by the ID-associated TRMT1 variants shown here suggests that individuals homozygous for such alleles will be deficient in m2,2G tRNA modifications. Based upon the proposed role for m2,2G modification in tRNA folding and the prevention of alternative conformations (45, 47, 48), the loss of m2,2G modification could have a significant effect on tRNA structure. In turn, the change in tRNA folding could affect the steady-state levels of tRNAs by perturbing the processing or stability of one or more tRNA species. In addition, the m2,2G modification could be required for subsequent RNA modifications occurring in the nucleus or cytoplasm. While we did not detect any significant changes in the accumulation or processing of tRNAs in TRMT1-deficient cells, there could be subtle differences in tRNA biogenesis that account for the phenotypic consequences in individuals with TRMT1 mutations. Moreover, different stress conditions could reveal differences in stability associated with tRNAs lacking m2,2G modification, as shown previously in yeast (41, 42). Thus, a global analysis of tRNA levels and modification status under different conditions will be the focus of future studies to uncover the potential tRNAs that are functionally affected by loss of TRMT1-catalyzed modification.
In addition to tRNA biogenesis, the m2,2G modification could modulate one or more aspects of translation by impacting molecular interactions between the tRNA and ribosome. Indeed, previous studies have suggested that the m2,2G modification plays a role in enhancing the activity of a nonsense suppressor tRNA in decoding the ochre stop codon in fission yeast (109). Intriguingly, our results suggest that in human cells, TRMT1-catalyzed m2,2G modifications are critical for ensuring the translational activity of a broad spectrum of tRNAs to ensure the maintenance of proper protein synthesis rates in the cytoplasm and mitochondria. By affecting translation, TRMT1-catalyzed tRNA modifications could have a significant effect on the levels of key proteins that are necessary for cellular growth, homeostasis, and stress response (Fig. 8D). In particular, experiments in yeast as well as the current study have implicated tRNA modifications in the cellular response to heat stress, nutrient starvation, and DNA damage (49, 50, 98, 110). Thus, the m2,2G modification in tRNA could be necessary for the proper translation of specific homeostatic or stress response proteins that are required for cellular stress survival, as shown previously for other tRNA modifications (49, 88, 90, 91, 111–113). TRMT1-catalyzed tRNA modifications could also play an indirect role in modulating translation by signaling to cellular stress response proteins to regulate protein synthesis.
In addition to the cellular stress response, tRNA modifications that modulate translation have been implicated in the regulation of stem cell pluripotency and organismal development (15, 89, 114, 115). Thus, loss-of-function mutations in human TRMT1 could perturb a number of cellular pathways that account for the spectrum of neurocognitive phenotypes exhibited by individuals with TRMT1-associated intellectual disability (55, 56). Future experiments will investigate the role of TRMT1-catalyzed m2,2G modifications in protein translation to identify cellular pathways that would be affected by TRMT1-catalyzed tRNA modification and their roles in cell physiology and development.
MATERIALS AND METHODS
Subcellular localization and fractionation.
Subcellular localization prediction was performed using the following online prediction algorithms: Mitofates (http://mitf.cbrc.jp/MitoFates/cgi-bin/top.cgi) (116), TPpred2 (http://tppred2.biocomp.unibo.it/tppred2) (117), and cNLS mapper (http://nls-mapper.iab.keio.ac.jp/cgi-bin/NLS_Mapper_form.cgi) (118). Results from each prediction program are shown in Fig. S1 in the supplemental material.
For immunofluorescence, mitochondria were visualized by infection of HeLa cells with baculovirus expressing red fluorescent protein targeted to mitochondria (CellLight Mitochondria-RFP, BacMam 2.0; Thermo Fisher). HeLa cells were then fixed with 4% formaldehyde on glass coverslips and probed with either mouse anti-TRMT1 (sc-373687; diluted 1:400; Santa Cruz Biotechnology), mouse anti-TRMT1L (H00081627-B01P; Abnova), or rabbit antinucleophosmin (35425; Cell Signaling) antibody diluted in 5% bovine serum albumin (BSA)–phosphate-buffered saline (PBS) overnight at 4°C. Following PBS washing of the coverslips, the primary antibody was detected using Alexa Fluor 488–goat anti-mouse IgG (L00031; Invitrogen) or goat anti-rabbit IgG with Alexa Fluor 594 (R37117; Thermo Fisher). For localization of GFP- or FLAG-tagged TRMT1, HeLa cells were seeded onto coverslips in a 24-well plate, followed by transfection with pcDNA3.1-TRMT1-EGFP or pcDNA3.1-TRMT1-FLAG using Lipofectamine 3000 (Thermo Fisher). For mitochondrion localization, cells were infected with baculovirus expressing red fluorescent protein targeted to mitochondria (CellLight Mitochondria-RFP, BacMam 2.0). Cells were fixed, probed as described above with mouse anti-FLAG secondary antibody (F1804; Sigma-Aldrich), stained with 4′,6-diamidino-2-phenylindole (DAPI) to visualize the nucleus, and mounted in Aqua-Poly/Mount (Polysciences Inc.), followed by imaging on a Zeiss AxioImager Z1 microscope.
Subcellular fractionation of HeLa cells was performed by differential centrifugation as previously described (119, 120). HeLa cells were collected by trypsinization, washed with PBS, resuspended in 3 ml cell homogenization medium (CHM; 10 mM Tris-HCl, pH 6.7, 10 mM KCl, 0.15 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride [PMSF], and 1 mM dithiothreitol [DTT]), and lysed with 40 strokes of a Dounce homogenizer. Homogenate was transferred to a 15-ml conical tube followed by addition of 1 ml of cell homogenization medium containing 1 M sucrose to achieve a final sucrose concentration of 0.25 M, and it was centrifuged (1,000 × g) for 5 min at 4°C in a swinging-bucket rotor to pellet nuclei. The cytoplasmic supernatant was collected and centrifuged (5,000 × g) at 4°C for 10 min in a fixed-angle rotor to collect mitochondria. The pellet containing mitochondria was washed with sucrose-Mg2+ medium (10 mM Tris-HCl, pH 6.7, 0.15 mM MgCl2, 0.25 M sucrose, 1 mM PMSF, 1 mM DTT), and the mitochondrial pellet was resuspended in radioisotope immunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 7.5, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 2 mM EDTA). Nuclei were resuspended in RIPA buffer containing 0.25 U/μl Benzonase (70664-3; EMD Millipore). Subcellular fractions were analyzed by immunoblotting (see below).
CRISPR gene editing and shRNA gene silencing.
Lentiviral CRISPR-Cas9 constructs were generated by cloning double-strand oligonucleotide inserts into pLentiCRISPR v2.0 (Addgene) (oligonucleotides TRMT1 gs F1, R1, F3, and R3 [Table S1]). Lentiviral RNAi constructs targeting either EGFP (RHS4459) or TRMT1 (TRCN0000038764 and TRCN0000038765) were obtained from Dharmacon-GE Lifesciences. Lentiviral short hairpin RNA (shRNA) or shRNA constructs were transfected with packaging plasmids (psPAX2 and pMD2.G; Addgene) into 293T cells for lentivirus production. The 293T cell line was subsequently infected with lentivirus in the presence of hexadimethrine bromide, followed by stable clone selection. The presence of CRISPR-induced mutations in the TRMT1 gene was detected by Surveyor assay (IDT) and confirmed by Sanger sequencing of cloned PCR products amplified from genomic DNA using primers TRMT1 gPCR F2 and TRMT1 gPCR R6 (Table S1).
Cell culture.
HeLa S3 cervical carcinoma, 293T human embryonic kidney, and derivative cell lines were cultured in Dulbecco's minimal essential medium (DMEM) containing 10% fetal bovine serum, 2 mM l-alanyl–l-glutamine (GlutaMax; Gibco), and 1% penicillin-streptomycin (here referred to as complete DMEM). ReNcell VM neural progenitor cell lines (EMD Millipore) were cultured on laminin-coated plates in ReNcell NSC maintenance medium supplemented with 20 ng/ml each of enhanced growth factor and fibroblast growth factor beta.
For cell proliferation measurements, cells were seeded in 6-well plates at 1 × 105 cells per well. At 24-h time intervals, cells were trypsinized and live cell counts were quantified using a Muse flow cytometer (EMD Millipore). For cell cycle analyses, media containing mitotic cells were harvested and combined with trypsinized cells, fixed with ethanol containing 100 μg/ml RNase, and stained with 20 μg/ml propidium iodide for 30 min. Cells were subsequently analyzed by flow cytometry on a Muse cytometer and quantified using FlowJo software.
For cell treatment and viability assays, 293T cells were exposed to H2O2, t-bu-OOH, or MMS (all from Sigma) at the indicated concentrations in DMEM supplemented with 2 mM l-alanyl–l-glutamine and 1% penicillin-streptomycin without fetal bovine serum (FBS) for 30 min, followed by replacement with complete medium containing serum. Etoposide and arsenic trioxide (As2O3) were left in the medium for the duration of the experiment until analysis. Treatment with menadione (100 μM) was for 2 h in complete medium. For ReNcell treatment, t-bu-OOH was diluted into complete ReNcell maintenance medium containing growth factors and harvested for viability assays using Accutase cell detachment solution (EMD Millipore). For measurement of mitochondrial membrane potential, cells were stained with TMRE using the Muse mitopotential assay kit (EMD Millipore). As a negative control for mitochondrial potential, a duplicate sample of cells was treated with carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) for 5 min immediately before measurement. For analysis of ROS levels by DHE (A00142; AnaSpec), cells were harvested by trypsinization, resuspended in PBS, and stained with 3.75 μM DHE in PBS. Alternatively, cells were incubated with either 5 μM CellROX orange or 1 μM MitoSOX (ThermoFisher) in complete DMEM for 30 min, washed once with PBS, trypsinized, and harvested. For NAC treatment, cells were incubated with 1 mM NAC for 24 to 96 h before ROS measurement and/or cell counting.
For TRMT1 reexpression experiments, empty vector or untagged TRMT1 expression constructs were transiently transfected into the control-WT or TRMT1-KO1 cell lines using Lipofectamine 3000 (Thermo Fisher). Twenty-four hours after transfection, cells were split into 6-well plates for treatment with t-bu-OOH as indicated. Cell viability was measured 24 h posttreatment, while one well was harvested for protein extracts and immunoblotting.
Protein synthesis assay.
For de novo protein synthesis detection using puromycin, each cell line was seeded at 5 × 105 cells per well of a 6-well plate and allowed to attach for 24 h. Cells were treated with 5 μM puromycin in complete DMEM for the indicated times, harvested by trypsinization, washed with PBS, and resuspended in 100 μl of RIPA buffer. Following a 5-min incubation on ice, cell lysates were centrifuged at 14,000 × g at 4°C for 15 min, and the supernatant was collected for analysis by immunoblotting.
For monitoring mitochondrial protein synthesis, cell lines were seeded as described above and pretreated with 200 μM emetine for 10 min in cysteine and methionine-free DMEM (21013024; Thermo Fisher) containing 10% dialyzed FBS (Thermo Fisher) and 2 mM l-alanyl–l-glutamine (GlutaMAX; Gibco). After the initial emetine incubation, 200 μCi of [35S]methionine-cysteine protein labeling mix (NEG772; PerkinElmer) was added to the cells and further incubated for 1 h. Cells were harvested directly from the plate in PBS and resuspended in a buffer containing 50 mM Tris-HCl, pH 8.3, 1% SDS, and 0.25 U/μl Benzonase (70664-3; EMD Millipore). After a 15-min incubation on ice, lysed cells were centrifuged at 21,000 × g at 4°C for 5 min, and the supernatant was collected for analysis on a 12% Tris-glycine SDS-PAGE. The gel was stained with SimplyBlue Coomassie (Thermo Scientific), dried, exposed on a phosphor screen (GE Healthcare), and scanned on a Bio-Rad personal molecular imager followed by analysis using NIH ImageJ software.
RNA analysis.
RNA from input and purified samples was extracted using TRIzol (Invitrogen, Thermo Fisher) and fractionated on 7 M urea–1.0× Tris-borate-EDTA (TBE)–10% denaturing polyacrylamide gels, followed by Sybr gold nucleic acid staining (Invitrogen). RNA was visualized by 300 nM UV transillumination and subsequently transferred to a Hybond N+ membrane (GE Healthcare Life Sciences) for probe hybridization with radiolabeled oligonucleotides (Table S1). Blots were stripped by incubation at 75°C with stripping buffer (0.15 M NaCl, 0.015 M Na-citrate, 0.1% SDS) and repeated at least twice until there was no detectable signal.
For primer extension analysis, total RNA (∼3.3 μg) was preannealed with 0.625 pmol of 5′-32P-labeled oligonucleotide and 1.4 μl of 5× hybridization buffer (250 mM Tris, pH 8.5, and 300 mM NaCl) in a total volume of 7 μl. The mixture was heated at 95°C for 3 min, slowly cooled to 41°C, and combined with 7 μl of extension mix consisting of 0.12 μl avian myeloblastosis virus (AMV) reverse transcriptase (Promega), 2.8 μl 5× AMV buffer, 0.56 μl of 1 mM deoxynucleoside triphosphates (dNTPs) (40 μM final concentration), and 3.52 μl water. Following primer extension at 41°C for 1 h, samples were mixed with 2× formamide loading dye, heated at 95°C for 3 min, and run on a 7 M urea–1.0× TBE–15% denaturing polyacrylamide gel. Gels were exposed on a phosphor screen (GE Healthcare) and scanned on a Bio-Rad personal molecular imager, followed by analysis using NIH ImageJ software.
Plasmid constructs.
The coding region for human TRMT1 was PCR amplified from cDNA clone HsCD00323018 (PlasmID Repository, Harvard Medical School) for cloning into pcDNA3.1 (Thermo Fisher) for expression as either an untagged, EGFP-tagged, or triple FLAG-tagged protein as previously described (119, 121). TRMT1 variants were constructed by Gibson cloning and verified by Sanger sequencing. In addition, all TRMT1 constructs used for reexpression in TRMT1-KO cell lines contained silent mutations to prevent CRISPR-mediated editing of the transfected constructs. The mutagenesis oligonucleotides for generating the silent mutations in TRMT1 are listed in Table S1.
Affinity purification of TRMT1-FLAG.
Transient transfection and cellular extract production were performed as previously described (111). Whole-cell extract from transiently transfected cells cell lines (1 mg of total protein) was rotated with 10 μl of anti-DYKDDDDK magnetic beads (TaKaRa BioUSA, Clontech) for 2 h at 4°C in lysis buffer (20 mM HEPES at pH 7.9, 2 mM MgCl2, 0.2 mM EGTA, 10% glycerol, 1 mM DTT, 0.1 mM PMSF, 0.1% NP-40) with 150 mM NaCl. Resin was washed three times using the same buffer, followed by RNA extraction or protein analysis (see below).
Protein analysis.
Cellular extracts and purified protein samples were fractionated on NuPAGE Bis-Tris polyacrylamide gels (Thermo Scientific), followed by transfer to Immobilon FL polyvinylidene difluoride (PVDF) membrane (Millipore) for immunoblotting. Total protein on the blot was detected using REVERT total protein stain (LI-COR Biosciences). Antibodies were used against the following proteins: TRMT1 (L00029; Santa Cruz Biotechnology), NDUFS7 (L00007; Proteintech), FLAG epitope tag (L00018; Sigma), actin (L00003; EMD Millipore), glyceraldehyde-3-phosphate dehydrogenase (2118; Cell Signaling), puromycin (MABE342; Millipore), and Nrf2 (12721; Cell Signaling). Primary antibodies were detected using IRDye 800CW goat anti-mouse IgG (925-32210; Thermo Fisher) and IRDye 680RD goat anti-mouse IgG (926-68070; LI-COR Biosciences). Immunoblots were scanned using direct infrared fluorescence via the Odyssey system (LI-COR Biosciences).
Liquid chromatography-mass spectrometry (LC-MS) analysis of tRNA modifications. (i) Chemicals.
All chemicals and reagents were obtained at the highest purity available and were used without further purification unless stated. Benzonase, bacterial alkaline phosphatase, butylated hydroxytoluene, acetonitrile, and buffer salts were purchased from Sigma-Aldrich (Steinheim, Germany). Coformycin was obtained from the National Cancer Institute. Snake venom phosphodiesterase I was purchased from VWR (Darmstadt, Germany). Water purified through a Milli-Q system was used throughout our studies.
(ii) RNA fractionation.
Total RNA was loaded on a size-exclusion column (3 μm, 300 Å, 7.8 by 300 mm; Agilent Bio SEC-3; Agilent, Waldbronn, Gemany) and RNA fractions eluted with 100 mM ammonium acetate at pH 7 as the mobile phase. tRNA was separated from rRNA and small RNAs, vacuum concentrated, and reconstituted in water. tRNA concentrations were determined by UV spectroscopy at 260 nm.
(iii) Nucleoside preparation.
One microgram of purified tRNA from each sample (biological triplicates of each strain) was digested using a mixture of Benzonase (21.5 U), bacterial alkaline phosphatase (0.5 U), and phosphodiesterase I (0.21 U) in a final reaction volume of 50 μl. The reaction mixture was supplemented with MgCl2 and Tris-HCl (pH 8.0), each at a concentration of 0.1 mM. The nucleobase deaminase inhibitor coformycin was added at a concentration of 10 μg/ml, and butylated hydroxytoluene (an antioxidant) was added at a concentration of 0.5 mM. The digestion was allowed to proceed for 2 h at 37°C and was stopped upon removal of the enzymes by microfiltration with 10-kDa spin filters (VWR). [15N]deoxyadenosine ([15N]dA) was added to a final concentration of 5 nM as an internal standard to account for mass spectrometric detection fluctuations.
(iv) LC-tandem MS analysis of RNA nucleosides.
Ribonucleosides were separated, using a Synergy Fusion RP (2.5-μm particle size, 100-Å pore size, 100-mm length, 2-mm inner diameter) from Phenomenex (Torrance, CA), on an Agilent 1290 series high-performance liquid chromatography (HPLC) system equipped with a diode array detector (DAD). Mobile phase A was 5 mM ammonium acetate adjusted to pH 5.3 with glacial acetic acid, and mobile phase B was pure acetonitrile. Gradient elution started with 100% A for 1 min and then increased to 10% B after 10 min, 40% after 14 min, and regeneration of starting conditions with 100% A for 4 additional minutes. The flow rate was 0.35 ml/min, and the column temperature was 35°C. The effluent from the column was directed through the DAD before entering the Agilent 6490 triple-quadrupole mass spectrometer in dynamic multiple reaction monitoring (MRM) mode. The MS was operated in positive ion mode with the following parameters: electrospray ionization (ESI-MS; Agilent Jetstream); fragmentor voltage (set in a tune file), 250 V; cell accelerator voltage, 2 V; N2-gas temperature, 150°C; N2-gas flow, 15 liters/min; nebulizer, 30 lb/in2; sheath gas (N2) temperature, 275°C; sheath gas flow, 11 liters/min; capillary voltage, 2,500 V; nozzle voltage, 500 V.
The mass transitions for each modified nucleoside are found in Table S2.
Data analysis.
Using Agilent's qualitative data analysis software, the UV peak areas at 260 nm of the four canonical ribonucleosides, cytidine, uridine, guanosine, and adenosine, were integrated and summed for each sample. Peaks corresponding to detection of MRM transitions for the modified nucleosides were first normalized to the peak area of the internal standard ([15N]deoxyadenosine) to account for intersample detection fluctuations, followed by a second normalization to the summed peak areas of the canonical nucleosides to account for the amount of injected RNA. The adjusted values from the knockout mutant samples were divided by the wild-type sample value to yield the fold change in modification level for each modified nucleosides.
Absolute quantification of m2,2G and m2G.
Calibration solutions of m2,2G, m2G, and the canonical nucleosides, cytidine, uridine, guanosine, and adenosine, were prepared with their respective synthetic standards (Sigma-Aldrich) and mixed to a starting concentration of 2.2 pmol/μl for the modified nucleosides and 2.2 nmol/μl for the canonical nucleosides. The resulting solution was serially diluted 1:10 to a final concentration of 2.2 amol/μl for the modified nucleosides and 2.2 fmol/μl for the canonicals. A 1/10 volume of the [15N]dA synthetic standard was added to each calibration solution. Ten microliters of each calibration solution (range for m2,2G, 20 amol to 2 pmol; range for canonicals, 20 fmol to 20 nmol) was subjected to LC-MS analysis before sample analysis using the same method as that used for the samples.
[15N]dA intersample fluctuation was 2.4%. The limit of detection (LOD; defined as S/N of >3) and limit of quantification (LOQ; defined as S/N of >10) of m2G both were found to be 200 amol. For m2,2G, the LOD was 200 amol and the LOQ was 2 fmol. The MS peak areas of the modified nucleosides was plotted over the amount of injected material. The slope of the linear regression was used to determine the amount of m2G and m2,2G in the analyzed samples.
For the canonicals, the peak areas of the UV chromatogram at 260 nm was used for linear regression. The resulting response factors were used to determine the amount of injected canonicals in picomoles per sample. The amount of RNA was then determined by using the average tRNA composition of 20 cytidines, 10 uridines, 20 guanosines, and 15 adenosines per tRNA. In a last step, the amounts of modified nucleosides (in femtomoles) were divided by the amount of tRNA injected (in femtomoles) to yield the modification percentage per tRNA molecule.
Quantification and statistical analysis.
All statistical analyses were performed using GraphPad Prism 7.0. Immunoblots for de novo protein synthesis assays using puromycin were quantified using Image Studio (LI-COR Biosciences). For mass spectrometry, the experiment was carried out on 3 independent RNA samples isolated from 3 independent flasks of each cell line. All results represent the means ± standard deviations. The numbers of times experiments were repeated were the following: Fig. 1B, 3; Fig. 1C to F, 1 to 3, depending on the antibody; Fig. 2B, >3; Fig. 3B, 2 to 4, depending on the probe; Fig. 3D and E, 2 to 6, depending on the probe; Fig. 4B and C, 3; Fig. 4D to F, 2; Fig. 5A and B, 3; Fig. 5C and D, 2; Fig. 5E to G, 2; Fig. 6A and B, 3; Fig. 6C and D, 2; Fig. 6E and F, 3; Fig. 7A and B, 6; Fig. 7C, 2; Fig. 7D and E, 3; Fig. 7F, 2; Fig. 7G, 3 to 5, depending on the treatment; Fig. 7H, 2; Fig. 8A and B, 2; Fig. 8C, 3.
Supplementary Material
ACKNOWLEDGMENTS
We thank Elaine Sia, Sina Ghaemmaghami, and Eric Phizicky for comments on the manuscript and Jillian Ramos, Christopher Prevost, and Rebecca Galer for technical assistance.
This work was supported by a Liebig Fellowship from Fonds of Chemical Industries and a LMU Junior Research Fund to S.M.K., as well as a University of Rochester Furth Fund Award and NSF CAREER award 1552126 to D.F.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00214-17.
REFERENCES
- 1.Jackman JE, Alfonzo JD. 2013. Transfer RNA modifications: nature's combinatorial chemistry playground. Wiley Interdiscip Rev RNA 4:35–48. doi: 10.1002/wrna.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.El Yacoubi B, Bailly M, de Crecy-Lagard V. 2012. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu Rev Genet 46:69–95. doi: 10.1146/annurev-genet-110711-155641. [DOI] [PubMed] [Google Scholar]
- 3.Phizicky E, Hopper A. 2010. tRNA biology charges to the front. Genes Dev 24:1832–1860. doi: 10.1101/gad.1956510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Motorin Y, Helm M. 2011. RNA nucleotide methylation. Wiley Interdiscip Rev RNA 2:611–631. doi: 10.1002/wrna.79. [DOI] [PubMed] [Google Scholar]
- 5.Kadaba S, Krueger A, Trice T, Krecic AM, Hinnebusch AG, Anderson J. 2004. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev 18:1227–1240. doi: 10.1101/gad.1183804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alexandrov A, Chernyakov I, Gu W, Hiley SL, Hughes TR, Grayhack EJ, Phizicky EM. 2006. Rapid tRNA decay can result from lack of nonessential modifications. Mol Cell 21:87–96. doi: 10.1016/j.molcel.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 7.Lamichhane TN, Blewett NH, Crawford AK, Cherkasova VA, Iben JR, Begley TJ, Farabaugh PJ, Maraia RJ. 2013. Lack of tRNA modification isopentenyl-A37 alters mRNA decoding and causes metabolic deficiencies in fission yeast. Mol Cell Biol 33:2918–2929. doi: 10.1128/MCB.00278-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manickam N, Joshi K, Bhatt MJ, Farabaugh PJ. 2016. Effects of tRNA modification on translational accuracy depend on intrinsic codon-anticodon strength. Nucleic Acids Res 44:1871–1881. doi: 10.1093/nar/gkv1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaborske JM, DuMont VL, Wallace EW, Pan T, Aquadro CF, Drummond DA. 2014. A nutrient-driven tRNA modification alters translational fidelity and genome-wide protein coding across an animal genus. PLoS Biol 12:e1002015. doi: 10.1371/journal.pbio.1002015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zinshteyn B, Gilbert WV. 2013. Loss of a conserved tRNA anticodon modification perturbs cellular signaling. PLoS Genet 9:e1003675. doi: 10.1371/journal.pgen.1003675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nedialkova DD, Leidel SA. 2015. Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 161:1606–1618. doi: 10.1016/j.cell.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arimbasseri AG, Maraia RJ. 2016. RNA polymerase III advances: structural and tRNA functional views. Trends Biochem Sci 41:546–559. doi: 10.1016/j.tibs.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dedon P, Begley T. 2014. A system of RNA modifications and biased codon use controls cellular stress response at the level of translation. Chem Res Toxicol 27:330–337. doi: 10.1021/tx400438d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang HY, Hopper AK. 2016. Multiple layers of stress-induced regulation in tRNA biology. Life (Basel) 6:E16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frye M, Blanco S. 2016. Post-transcriptional modifications in development and stem cells. Development 143:3871–3881. doi: 10.1242/dev.136556. [DOI] [PubMed] [Google Scholar]
- 16.Pan T. 2013. Adaptive translation as a mechanism of stress response and adaptation. Annu Rev Genet 47:121–137. doi: 10.1146/annurev-genet-111212-133522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki T, Nagao A, Suzuki T. 2011. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu Rev Genet 45:299–329. doi: 10.1146/annurev-genet-110410-132531. [DOI] [PubMed] [Google Scholar]
- 18.Kirchner S, Ignatova Z. 2015. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat Rev Genet 16:98–112. doi: 10.1038/nrg3861. [DOI] [PubMed] [Google Scholar]
- 19.Sarin LP, Leidel SA. 2014. Modify or die? RNA modification defects in metazoans. RNA Biol 11:1555–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Popis MC, Blanco S, Frye M. 2016. Posttranscriptional methylation of transfer and ribosomal RNA in stress response pathways, cell differentiation, and cancer. Curr Opin Oncol 28:65–71. doi: 10.1097/CCO.0000000000000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grewal SS. 2015. Why should cancer biologists care about tRNAs? tRNA synthesis, mRNA translation and the control of growth. Biochim Biophys Acta 1849:898–907. doi: 10.1016/j.bbagrm.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 22.Powell CA, Nicholls TJ, Minczuk M. 2015. Nuclear-encoded factors involved in post-transcriptional processing and modification of mitochondrial tRNAs in human disease. Front Genet 6:79. doi: 10.3389/fgene.2015.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swinehart WE, Jackman JE. 2015. Diversity in mechanism and function of tRNA methyltransferases. RNA Biol 12:398–411. doi: 10.1080/15476286.2015.1008358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hori H. 2014. Methylated nucleosides in tRNA and tRNA methyltransferases. Front Genet 5:144. doi: 10.3389/fgene.2014.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phillips JH, Kjellin-Straby K. 1967. Studies on microbial ribonucleic acid. IV. Two mutants of Saccharomyces cerevisiae lacking N-2-dimethylguanine in soluble ribonucleic acid. J Mol Biol 26:509–518. [DOI] [PubMed] [Google Scholar]
- 26.Hopper AK, Furukawa AH, Pham HD, Martin NC. 1982. Defects in modification of cytoplasmic and mitochondrial transfer RNAs are caused by single nuclear mutations. Cell 28:543–550. doi: 10.1016/0092-8674(82)90209-4. [DOI] [PubMed] [Google Scholar]
- 27.Ellis SR, Morales MJ, Li JM, Hopper AK, Martin NC. 1986. Isolation and characterization of the TRM1 locus, a gene essential for the N2,N2-dimethylguanosine modification of both mitochondrial and cytoplasmic tRNA in Saccharomyces cerevisiae. J Biol Chem 261:9703–9709. [PubMed] [Google Scholar]
- 28.Liu J, Zhou GQ, Straby KB. 1999. Caenorhabditis elegans ZC376.5 encodes a tRNA (m2/2G(26))dimethyltransferance in which (246)arginine is important for the enzyme activity. Gene 226:73–81. doi: 10.1016/S0378-1119(98)00550-2. [DOI] [PubMed] [Google Scholar]
- 29.Constantinesco F, Benachenhou N, Motorin Y, Grosjean H. 1998. The tRNA(guanine-26,N2-N2) methyltransferase (Trm1) from the hyperthermophilic archaeon Pyrococcus furiosus: cloning, sequencing of the gene and its expression in Escherichia coli. Nucleic Acids Res 26:3753–3761. doi: 10.1093/nar/26.16.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bujnicki JM, Leach RA, Debski J, Rychlewski L. 2002. Bioinformatic analyses of the tRNA: (guanine 26, N2,N2)-dimethyltransferase (Trm1) family. J Mol Microbiol Biotechnol 4:405–415. [PubMed] [Google Scholar]
- 31.Takeda H, Hori H, Endo Y. 2002. Identification of Aquifex aeolicus tRNA (m2(2G26)) methyltransferase gene. Nucleic Acids Res 2002:229–230. doi: 10.1093/nass/2.1.229. [DOI] [PubMed] [Google Scholar]
- 32.Vauti F, Goller T, Beine R, Becker L, Klopstock T, Hölter S, Wurst W, Fuchs H, Gailus-Durner V, de Angelis M, Arnold H-H. 2007. The mouse Trm1-like gene is expressed in neural tissues and plays a role in motor coordination and exploratory behaviour. Gene 389:174–185. doi: 10.1016/j.gene.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 33.Smith JD, Dunn DB. 1959. The occurrence of methylated guanines in ribonucleic acids from several sources. Biochem J 72:294–301. doi: 10.1042/bj0720294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kjellin-Straby K, Boman HG. 1965. Studies on microbial RNA, 3. Formation of submethylated sRNA in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 53:1346–1352. doi: 10.1073/pnas.53.6.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hall RH. 1965. A general procedure for the isolation of “minor” nucleosides from ribonucleic acid hydrolysates. Biochemistry 4:661–670. doi: 10.1021/bi00880a008. [DOI] [PubMed] [Google Scholar]
- 36.Martin NC, Hopper AK. 1994. How single genes provide tRNA processing enzymes to mitochondria, nuclei and the cytosol. Biochimie 76:1161–1167. doi: 10.1016/0300-9084(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 37.Rose AM, Joyce PB, Hopper AK, Martin NC. 1992. Separate information required for nuclear and subnuclear localization: additional complexity in localizing an enzyme shared by mitochondria and nuclei. Mol Cell Biol 12:5652–5658. doi: 10.1128/MCB.12.12.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li JM, Hopper AK, Martin NC. 1989. N2,N2-dimethylguanosine-specific tRNA methyltransferase contains both nuclear and mitochondrial targeting signals in Saccharomyces cerevisiae. J Cell Biol 109:1411–1419. doi: 10.1083/jcb.109.4.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ellis SR, Hopper AK, Martin NC. 1987. Amino-terminal extension generated from an upstream AUG codon is not required for mitochondrial import of yeast N2,N2-dimethylguanosine-specific tRNA methyltransferase. Proc Natl Acad Sci U S A 84:5172–5176. doi: 10.1073/pnas.84.15.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ellis SR, Hopper AK, Martin NC. 1989. Amino-terminal extension generated from an upstream AUG codon increases the efficiency of mitochondrial import of yeast N2,N2-dimethylguanosine-specific tRNA methyltransferases. Mol Cell Biol 9:1611–1620. doi: 10.1128/MCB.9.4.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Copela LA, Chakshusmathi G, Sherrer RL, Wolin SL. 2006. The La protein functions redundantly with tRNA modification enzymes to ensure tRNA structural stability. RNA 12:644–654. doi: 10.1261/rna.2307206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dewe JM, Whipple JM, Chernyakov I, Jaramillo LN, Phizicky EM. 2012. The yeast rapid tRNA decay pathway competes with elongation factor 1A for substrate tRNAs and acts on tRNAs lacking one or more of several modifications. RNA 18:1886–1896. doi: 10.1261/rna.033654.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Helm M. 2006. Post-transcriptional nucleotide modification and alternative folding of RNA. Nucleic Acids Res 34:721–733. doi: 10.1093/nar/gkj471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bavi RS, Sambhare SB, Sonawane KD. 2013. MD simulation studies to investigate iso-energetic conformational behaviour of modified nucleosides m(2)G and m(2)2G present in tRNA. Comput Struct Biotechnol J 5:e201302015. doi: 10.5936/csbj.201302015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steinberg S, Cedergren R. 1995. A correlation between N2-dimethylguanosine presence and alternate tRNA conformers. RNA 1:886–891. [PMC free article] [PubMed] [Google Scholar]
- 46.Bavi RS, Kamble AD, Kumbhar NM, Kumbhar BV, Sonawane KD. 2011. Conformational preferences of modified nucleoside N(2)-methylguanosine (m(2)G) and its derivative N(2), N(2)-dimethylguanosine (m(2)(2)G) occur at 26th position (hinge region) in tRNA. Cell Biochem Biophys 61:507–521. doi: 10.1007/s12013-011-9233-1. [DOI] [PubMed] [Google Scholar]
- 47.Pallan PS, Kreutz C, Bosio S, Micura R, Egli M. 2008. Effects of N2,N2-dimethylguanosine on RNA structure and stability: crystal structure of an RNA duplex with tandem m2 2G:A pairs. RNA 14:2125–2135. doi: 10.1261/rna.1078508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Urbonavicius J, Armengaud J, Grosjean H. 2006. Identity elements required for enzymatic formation of N2,N2-dimethylguanosine from N2-monomethylated derivative and its possible role in avoiding alternative conformations in archaeal tRNA. J Mol Biol 357:387–399. doi: 10.1016/j.jmb.2005.12.087. [DOI] [PubMed] [Google Scholar]
- 49.Chan CT, Dyavaiah M, DeMott MS, Taghizadeh K, Dedon PC, Begley TJ. 2010. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet 6:e1001247. doi: 10.1371/journal.pgen.1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arimbasseri AG, Blewett NH, Iben JR, Lamichhane TN, Cherkasova V, Hafner M, Maraia RJ. 2015. RNA polymerase III output is functionally linked to tRNA dimethyl-G26 modification. PLoS Genet 11:e1005671. doi: 10.1371/journal.pgen.1005671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petrossian TC, Clarke SG. 2011. Uncovering the human methyltransferasome. Mol Cell Proteomics 10:M110.000976. doi: 10.1074/mcp.M110.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Towns W, Begley T. 2012. Transfer RNA methytransferases and their corresponding modifications in budding yeast and humans: activities, predications, and potential roles in human health. DNA Cell Biol 31:434–454. doi: 10.1089/dna.2011.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buckland RA, Maule JC, Sealey PG. 1996. A cluster of transfer RNA genes (TRM1, TRR3, and TRAN) on the short arm of human chromosome 6. Genomics 35:164–171. doi: 10.1006/geno.1996.0335. [DOI] [PubMed] [Google Scholar]
- 54.Liu J, Straby KB. 2000. The human tRNA(m(2)(2)G(26))dimethyltransferase: functional expression and characterization of a cloned hTRM1 gene. Nucleic Acids Res 28:3445–3451. doi: 10.1093/nar/28.18.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini S, Chen W, Hosseini M, Behjati F, Haas S, Jamali P, Zecha A, Mohseni M, Püttmann L, Vahid L, Jensen C, Moheb L, Bienek M, Larti F, Mueller I, Weissmann R, Darvish H, Wrogemann K, Hadavi V, Lipkowitz B, Esmaeeli-Nieh S, Wieczorek D, Kariminejad R, Firouzabadi S, Cohen M, Fattahi Z, Rost I, Mojahedi F, Hertzberg C, Dehghan A, Rajab A, Banavandi M, Hoffer J, Falah M, Musante L, Kalscheuer V, Ullmann R, Kuss A, Tzschach A, Kahrizi K, Ropers H. 2011. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- 56.Davarniya B, Hu H, Kahrizi K, Musante L, Fattahi Z, Hosseini M, Maqsoud F, Farajollahi R, Wienker TF, Ropers HH, Najmabadi H. 2015. The role of a novel TRMT1 gene mutation and rare GRM1 gene defect in intellectual disability in two Azeri families. PLoS One 10:e0129631. doi: 10.1371/journal.pone.0129631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu J, Liu J, Straby KB. 1998. Point and deletion mutations eliminate one or both methyl group transfers catalysed by the yeast TRM1 encoded tRNA (m22G26)dimethyltransferase. Nucleic Acids Res 26:5102–5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Papapetrou EP, Lee G, Malani N, Setty M, Riviere I, Tirunagari LM, Kadota K, Roth SL, Giardina P, Viale A, Leslie C, Bushman FD, Studer L, Sadelain M. 2011. Genomic safe harbors permit high beta-globin transgene expression in thalassemia induced pluripotent stem cells. Nat Biotechnol 29:73–78. doi: 10.1038/nbt.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sadelain M, Papapetrou EP, Bushman FD. 2011. Safe harbours for the integration of new DNA in the human genome. Nat Rev Cancer 12:51–58. [DOI] [PubMed] [Google Scholar]
- 60.Cai WM, Chionh YH, Hia F, Gu C, Kellner S, McBee ME, Ng CS, Pang YL, Prestwich EG, Lim KS, Babu IR, Begley TJ, Dedon PC. 2015. A platform for discovery and quantification of modified ribonucleosides in RNA: application to stress-induced reprogramming of tRNA modifications. Methods Enzymol 560:29–71. doi: 10.1016/bs.mie.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hiley SL, Jackman J, Babak T, Trochesset M, Morris QD, Phizicky E, Hughes TR. 2005. Detection and discovery of RNA modifications using microarrays. Nucleic Acids Res 33:e2. doi: 10.1093/nar/gni002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chawla M, Oliva R, Bujnicki JM, Cavallo L. 2015. An atlas of RNA base pairs involving modified nucleobases with optimal geometries and accurate energies. Nucleic Acids Res 43:9573. doi: 10.1093/nar/gkv925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Machnicka M, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother K, Helm M, Bujnicki J, Grosjean H. 2013. MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res 41:7. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, Vendeix FA, Fabris D, Agris PF. 2011. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res 39:D195–D201. doi: 10.1093/nar/gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chan PP, Lowe TM. 2009. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res 37:D93–D97. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gupta RC, Roe BA, Randerath K. 1980. Sequence of human glycine transfer ribonucleic acid (anticodon CCC). Determination by a newly developed thin-layer readout sequencing technique and comparison with other glycine transfer ribonucleic acids. Biochemistry 19:1699–1705. [DOI] [PubMed] [Google Scholar]
- 67.Piper PW. 1975. The primary structure of the major cytoplasmic valine tRNA of mouse myeloma cells. Eur J Biochem 51:295–304. doi: 10.1111/j.1432-1033.1975.tb03929.x. [DOI] [PubMed] [Google Scholar]
- 68.Clark WC, Evans ME, Dominissini D, Zheng G, Pan T. 2016. tRNA base methylation identification and quantification via high-throughput sequencing. RNA 22:1771–1784. doi: 10.1261/rna.056531.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen EY, Roe BA. 1977. Sequence studies on human placenta tRNAVal: comparison with the mouse myeloma tRNAVal. Biochem Biophys Res Commun 78:631–640. doi: 10.1016/0006-291X(77)90226-1. [DOI] [PubMed] [Google Scholar]
- 70.Suzuki T, Suzuki T. 2014. A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Res 42:7346–7357. doi: 10.1093/nar/gku390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sibler AP, Dirheimer G, Martin RP. 1986. Codon reading patterns in Saccharomyces cerevisiae mitochondria based on sequences of mitochondrial tRNAs. FEBS Lett 194:131–138. doi: 10.1016/0014-5793(86)80064-3. [DOI] [PubMed] [Google Scholar]
- 72.Watanabe Y, Tsurui H, Ueda T, Furushima R, Takamiya S, Kita K, Nishikawa K, Watanabe K. 1994. Primary and higher order structures of nematode (Ascaris suum) mitochondrial tRNAs lacking either the T or D stem. J Biol Chem 269:22902–22906. [PubMed] [Google Scholar]
- 73.Rife JP, Cheng CS, Moore PB, Strobel SA. 1998. N 2-methylguanosine is iso-energetic with guanosine in RNA duplexes and GNRA tetraloops. Nucleic Acids Res 26:3640–3644. doi: 10.1093/nar/26.16.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lesnyak DV, Osipiuk J, Skarina T, Sergiev PV, Bogdanov AA, Edwards A, Savchenko A, Joachimiak A, Dontsova OA. 2007. Methyltransferase that modifies guanine 966 of the 16 S rRNA: functional identification and tertiary structure. J Biol Chem 282:5880–5887. doi: 10.1074/jbc.M608214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Motorin Y, Muller S, Behm-Ansmant I, Branlant C. 2007. Identification of modified residues in RNAs by reverse transcription-based methods. Methods Enzymol 425:21–53. doi: 10.1016/S0076-6879(07)25002-5. [DOI] [PubMed] [Google Scholar]
- 76.Youvan DC, Hearst JE. 1979. Reverse transcriptase pauses at N2-methylguanine during in vitro transcription of Escherichia coli 16S ribosomal RNA. Proc Natl Acad Sci U S A 76:3751–3754. doi: 10.1073/pnas.76.8.3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schmidt EK, Clavarino G, Ceppi M, Pierre P. 2009. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 6:275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- 78.Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H, Klann E. 2013. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 493:411–415. doi: 10.1038/nature11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lipton JO, Yuan ED, Boyle LM, Ebrahimi-Fakhari D, Kwiatkowski E, Nathan A, Guttler T, Davis F, Asara JM, Sahin M. 2015. The circadian protein BMAL1 regulates translation in response to S6K1-mediated phosphorylation. Cell 161:1138–1151. doi: 10.1016/j.cell.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sasarman F, Shoubridge EA. 2012. Radioactive labeling of mitochondrial translation products in cultured cells. Methods Mol Biol 837:207–217. doi: 10.1007/978-1-61779-504-6_14. [DOI] [PubMed] [Google Scholar]
- 81.Yasukawa T, Suzuki T, Ueda T, Ohta S, Watanabe K. 2000. Modification defect at anticodon wobble nucleotide of mitochondrial tRNAs(Leu)(UUR) with pathogenic mutations of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. J Biol Chem 275:4251–4257. doi: 10.1074/jbc.275.6.4251. [DOI] [PubMed] [Google Scholar]
- 82.Yasukawa T, Suzuki T, Ishii N, Ohta S, Watanabe K. 2001. Wobble modification defect in tRNA disturbs codon-anticodon interaction in a mitochondrial disease. EMBO J 20:4794–4802. doi: 10.1093/emboj/20.17.4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Suzuki T, Suzuki T, Wada T, Saigo K, Watanabe K. 2002. Taurine as a constituent of mitochondrial tRNAs: new insights into the functions of taurine and human mitochondrial diseases. EMBO J 21:6581–6589. doi: 10.1093/emboj/cdf656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wei FY, Zhou B, Suzuki T, Miyata K, Ujihara Y, Horiguchi H, Takahashi N, Xie P, Michiue H, Fujimura A, Kaitsuka T, Matsui H, Koga Y, Mohri S, Suzuki T, Oike Y, Tomizawa K. 2015. Cdk5rap1-mediated 2-methylthio modification of mitochondrial tRNAs governs protein translation and contributes to myopathy in mice and humans. Cell Metab 21:428–442. doi: 10.1016/j.cmet.2015.01.019. [DOI] [PubMed] [Google Scholar]
- 85.Patton JR, Bykhovskaya Y, Mengesha E, Bertolotto C, Fischel-Ghodsian N. 2005. Mitochondrial myopathy and sideroblastic anemia (MLASA): missense mutation in the pseudouridine synthase 1 (PUS1) gene is associated with the loss of tRNA pseudouridylation. J Biol Chem 280:19823–19828. doi: 10.1074/jbc.M500216200. [DOI] [PubMed] [Google Scholar]
- 86.Fernandez-Vizarra E, Berardinelli A, Valente L, Tiranti V, Zeviani M. 2007. Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA). J Med Genet 44:173–180. doi: 10.1136/jmg.2006.045252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nakano S, Suzuki T, Kawarada L, Iwata H, Asano K, Suzuki T. 2016. NSUN3 methylase initiates 5-formylcytidine biogenesis in human mitochondrial tRNA(Met). Nat Chem Biol 12:546–551. doi: 10.1038/nchembio.2099. [DOI] [PubMed] [Google Scholar]
- 88.Fernandez-Vazquez J, Vargas-Perez I, Sanso M, Buhne K, Carmona M, Paulo E, Hermand D, Rodriguez-Gabriel M, Ayte J, Leidel S, Hidalgo E. 2013. Modification of tRNA(Lys) UUU by elongator is essential for efficient translation of stress mRNAs. PLoS Genet 9:e1003647. doi: 10.1371/journal.pgen.1003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blanco S, Bandiera R, Popis M, Hussain S, Lombard P, Aleksic J, Sajini A, Tanna H, Cortes-Garrido R, Gkatza N, Dietmann S, Frye M. 2016. Stem cell function and stress response are controlled by protein synthesis. Nature 534:335–340. doi: 10.1038/nature18282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Blanco S, Dietmann S, Flores JV, Hussain S, Kutter C, Humphreys P, Lukk M, Lombard P, Treps L, Popis M, Kellner S, Holter SM, Garrett L, Wurst W, Becker L, Klopstock T, Fuchs H, Gailus-Durner V, Hrabe de Angelis M, Karadottir RT, Helm M, Ule J, Gleeson JG, Odom DT, Frye M. 2014. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J 33:2020–2039. doi: 10.15252/embj.201489282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chan CT, Pang YL, Deng W, Babu IR, Dyavaiah M, Begley TJ, Dedon PC. 2012. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat Commun 3:937. doi: 10.1038/ncomms1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.El-Hattab AW, Adesina AM, Jones J, Scaglia F. 2015. MELAS syndrome: clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab 116:4–12. doi: 10.1016/j.ymgme.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 93.Yarham JW, Lamichhane TN, Pyle A, Mattijssen S, Baruffini E, Bruni F, Donnini C, Vassilev A, He L, Blakely EL, Griffin H, Santibanez-Koref M, Bindoff LA, Ferrero I, Chinnery PF, McFarland R, Maraia RJ, Taylor RW. 2014. Defective i6A37 modification of mitochondrial and cytosolic tRNAs results from pathogenic mutations in TRIT1 and its substrate tRNA. PLoS Genet 10:e1004424. doi: 10.1371/journal.pgen.1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sasarman F, Antonicka H, Shoubridge EA. 2008. The A3243G tRNALeu(UUR) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially suppressed by overexpression of EFTu and EFG2. Hum Mol Genet 17:3697–3707. doi: 10.1093/hmg/ddn265. [DOI] [PubMed] [Google Scholar]
- 95.Ling J, Roy H, Qin D, Rubio MA, Alfonzo JD, Fredrick K, Ibba M. 2007. Pathogenic mechanism of a human mitochondrial tRNAPhe mutation associated with myoclonic epilepsy with ragged red fibers syndrome. Proc Natl Acad Sci U S A 104:15299–15304. doi: 10.1073/pnas.0704441104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Criddle DN, Gillies S, Baumgartner-Wilson HK, Jaffar M, Chinje EC, Passmore S, Chvanov M, Barrow S, Gerasimenko OV, Tepikin AV, Sutton R, Petersen OH. 2006. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J Biol Chem 281:40485–40492. doi: 10.1074/jbc.M607704200. [DOI] [PubMed] [Google Scholar]
- 97.Klotz LO, Hou X, Jacob C. 2014. 1,4-Naphthoquinones: from oxidative damage to cellular and inter-cellular signaling. Molecules 19:14902–14918. doi: 10.3390/molecules190914902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pang YL, Abo R, Levine SS, Dedon PC. 2014. Diverse cell stresses induce unique patterns of tRNA up- and down-regulation: tRNA-seq for quantifying changes in tRNA copy number. Nucleic Acids Res 42:e170. doi: 10.1093/nar/gku945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nieminen AL, Saylor AK, Tesfai SA, Herman B, Lemasters JJ. 1995. Contribution of the mitochondrial permeability transition to lethal injury after exposure of hepatocytes to t-butylhydroperoxide. Biochem J 307(Part 1):99–106. doi: 10.1042/bj3070099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Alia M, Ramos S, Mateos R, Bravo L, Goya L. 2005. Response of the antioxidant defense system to tert-butyl hydroperoxide and hydrogen peroxide in a human hepatoma cell line (HepG2). J Biochem Mol Toxicol 19:119–128. doi: 10.1002/jbt.20061. [DOI] [PubMed] [Google Scholar]
- 101.Donato R, Miljan E, Hines S, Aouabdi S, Pollock K, Patel S, Edwards F, Sinden J. 2007. Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci 8:36. doi: 10.1186/1471-2202-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Baulch JE, Craver BM, Tran KK, Yu L, Chmielewski N, Allen BD, Limoli CL. 2015. Persistent oxidative stress in human neural stem cells exposed to low fluences of charged particles. Redox Biol 5:24–32. doi: 10.1016/j.redox.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tseng BP, Giedzinski E, Izadi A, Suarez T, Lan ML, Tran KK, Acharya MM, Nelson GA, Raber J, Parihar VK, Limoli CL. 2014. Functional consequences of radiation-induced oxidative stress in cultured neural stem cells and the brain exposed to charged particle irradiation. Antioxid Redox Signal 20:1410–1422. doi: 10.1089/ars.2012.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yuan TF, Gu S, Shan C, Marchado S, Arias-Carrion O. 2015. Oxidative stress and adult neurogenesis. Stem Cell Rev 11:706–709. doi: 10.1007/s12015-015-9603-y. [DOI] [PubMed] [Google Scholar]
- 105.Wang X, Michaelis EK. 2010. Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci 2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Filograna R, Beltramini M, Bubacco L, Bisaglia M. 2016. Anti-oxidants in Parkinson's disease therapy: a critical point of view. Curr Neuropharmacol 14:260–271. doi: 10.2174/1570159X13666151030102718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pagano G, Castello G. 2012. Oxidative stress and mitochondrial dysfunction in Down syndrome. Adv Exp Med Biol 724:291–299. doi: 10.1007/978-1-4614-0653-2_22. [DOI] [PubMed] [Google Scholar]
- 108.Bavarsad Shahripour R, Harrigan MR, Alexandrov AV. 2014. N-acetylcysteine (NAC) in neurological disorders: mechanisms of action and therapeutic opportunities. Brain Behav 4:108–122. doi: 10.1002/brb3.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Niederberger C, Graub R, Costa A, Desgres J, Schweingruber ME. 1999. The tRNA N2,N2-dimethylguanosine-26 methyltransferase encoded by gene trm1 increases efficiency of suppression of an ochre codon in Schizosaccharomyces pombe. FEBS Lett 464:67–70. doi: 10.1016/S0014-5793(99)01679-8. [DOI] [PubMed] [Google Scholar]
- 110.Hillenmeyer ME, Fung E, Wildenhain J, Pierce SE, Hoon S, Lee W, Proctor M, St Onge RP, Tyers M, Koller D, Altman RB, Davis RW, Nislow C, Giaever G. 2008. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320:362–365. doi: 10.1126/science.1150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fu D, Brophy JA, Chan CT, Atmore KA, Begley U, Paules RS, Dedon PC, Begley TJ, Samson LD. 2010. Human AlkB homolog ABH8 is a tRNA methyltransferase required for wobble uridine modification and DNA damage survival. Mol Cell Biol 30:2449–2459. doi: 10.1128/MCB.01604-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Begley U, Dyavaiah M, Patil A, Rooney JP, DiRenzo D, Young CM, Conklin DS, Zitomer RS, Begley TJ. 2007. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol Cell 28:860–870. doi: 10.1016/j.molcel.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Damon JR, Pincus D, Ploegh HL. 2015. tRNA thiolation links translation to stress responses in Saccharomyces cerevisiae. Mol Biol Cell 26:270–282. doi: 10.1091/mbc.E14-06-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tuorto F, Herbst F, Alerasool N, Bender S, Popp O, Federico G, Reitter S, Liebers R, Stoecklin G, Grone HJ, Dittmar G, Glimm H, Lyko F. 2015. The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J 34:2350–2362. doi: 10.15252/embj.201591382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rai K, Chidester S, Zavala CV, Manos EJ, James SR, Karpf AR, Jones DA, Cairns BR. 2007. Dnmt2 functions in the cytoplasm to promote liver, brain, and retina development in zebrafish. Genes Dev 21:261–266. doi: 10.1101/gad.1472907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fukasawa Y, Tsuji J, Fu SC, Tomii K, Horton P, Imai K. 2015. MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol Cell Proteomics 14:1113–1126. doi: 10.1074/mcp.M114.043083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Savojardo C, Martelli PL, Fariselli P, Casadio R. 2014. TPpred2: improving the prediction of mitochondrial targeting peptide cleavage sites by exploiting sequence motifs. Bioinformatics 30:2973–2974. doi: 10.1093/bioinformatics/btu411. [DOI] [PubMed] [Google Scholar]
- 118.Kosugi S, Hasebe M, Tomita M, Yanagawa H. 2009. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci U S A 106:10171–10176. doi: 10.1073/pnas.0900604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fu D, Jordan JJ, Samson LD. 2013. Human ALKBH7 is required for alkylation and oxidation-induced programmed necrosis. Genes Dev 27:1089–1100. doi: 10.1101/gad.215533.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Graham JM. 2001. Isolation of mitochondria from tissues and cells by differential centrifugation. Curr Protoc Cell Biol 4:3.3:3.3.1–3.3.3.15. [DOI] [PubMed] [Google Scholar]
- 121.Fu D, Samson LD, Hubscher U, van Loon B. 2015. The interaction between ALKBH2 DNA repair enzyme and PCNA is direct, mediated by the hydrophobic pocket of PCNA and perturbed in naturally occurring ALKBH2 variants. DNA Repair (Amst) 35:13–18. doi: 10.1016/j.dnarep.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.