

Abstract
In heart failure, the β-adrenergic receptors (βARs) become desensitized and uncoupled from heterotrimeric G proteins. This process is initiated by G protein-coupled receptor kinases (GRKs), some of which are upregulated in the failing heart making them desirable therapeutic targets. The serotonin reuptake inhibitor paroxetine was previously identified as a GRK2 inhibitor. Utilizing a structure based drug design approach we modified paroxetine to generate a small compound library. Included in this series is a highly potent and selective GRK2 inhibitor, 14as, with an IC50 of 30 nM against GRK2 and greater than 230-fold selectivity over other GRKs and kinases. Furthermore, 14as showed a 100-fold improvement in cardiomyocyte contractility assays over paroxetine and a plasma concentration higher than its IC50 for over seven hours. Three of these inhibitors, including 14as, were additionally crystallized in complex with GRK2 to give insights into the structural determinants of potency and selectivity of these inhibitors.
Keywords: G protein-coupled receptor kinases, G protein-coupled receptors, β-adrenergic receptors, protein crystallography, inhibitors, kinases, paroxetine, CCG215022, CCG224406, Takeda101, Takeda103A, GSK180736A
Graphical Abstract
G protein-coupled receptors (GPCRs) mediate cellular responses to many different kinds of extracellular stimuli1 and, with over 800 members, they are the largest class of receptors in the human genome. Given their many physiological roles, they are targeted by a large fraction of the pharmaceuticals on the market as well as illicit drugs.2 Upon agonist binding, GPCRs recruit and activate heterotrimeric G proteins, stimulating a variety of downstream intracellular signaling events including the recruitment of GPCR kinases (GRKs), which regulate GPCRs via phosphorylation of their cytoplasmic loops or tails.3, 4 In response, β-arrestins bind to the phosphorylated receptors to induce desensitization and ultimately internalization of the GPCRs.5, 6
There are seven known GRKs in humans, subdivided by homology into three sub-families: GRK1, GRK2, and GRK4. All belong to the protein kinase A, G, and C (AGC) family. The GRK1 subfamily consists of GRK1 and GRK7 and their expression is largely confined to the retina. The GRK2 subfamily consists of the ubiquitously expressed GRK2, and GRK3, which predominates in olfactory neurons. The GRK4 subfamily consists of GRK4, which is mainly expressed in the testes and kidneys, and GRK5 and 6, which are ubiquitously expressed, although GRK6 is more prevalent in the brain.7–10
In response to the failing heart, the sympathetic nervous system acts to increase the levels of circulating catecholamines such as norepinephrine and epinephrine.11–13 Upon binding of these hormones to β-adrenergic receptors (βARs) in cardiomyocytes, downstream signaling is initiated to improve the positive inotropic response in the heart.4, 14 In the failing heart, activation of βARs also leads to upregulation of GRK2 and GRK515, 16 which in turn leads to uncoupling of the βARs from G proteins,11, 16, 17 decreased βARs at the cellular membrane, and decreased cardiac output in response to hormonal stimulation.13, 18, 19 Decreasing the levels of GRK2, in particular through gene knockdown in the heart, has been shown to be beneficial in preventing heart failure by renormalizing levels of catecholamines and cell surface βARs, and improving cardiac function in mouse and swine models, making GRK2 an attractive target for heart failure therapy.19–21
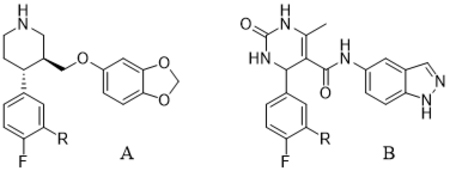
Efforts to selectively target GRK2 have generated several reported inhibitor series. Among these are a series of highly potent and selective compounds developed by Takeda.22 Two of these inhibitors, Takeda103A (1) and Takeda101 (2) (IC50 = 20 nM and 30 nM, respectively with >50-fold selectivity over other AGC kinases, Figure 1), were crystallized with the GRK2-Gβγ complex in order to ascertain their binding modes.23 They both stabilize an inactive conformation of GRK2, and their C and D rings pack into the polyphosphate and hydrophobic subsites of the active site, respectively, which contributes to their high potency and selectivity.23 Although these compounds never advanced to clinical trials, they served as proof of principle for development of new GRK2 inhibitors in our laboratory.24
Figure 1:
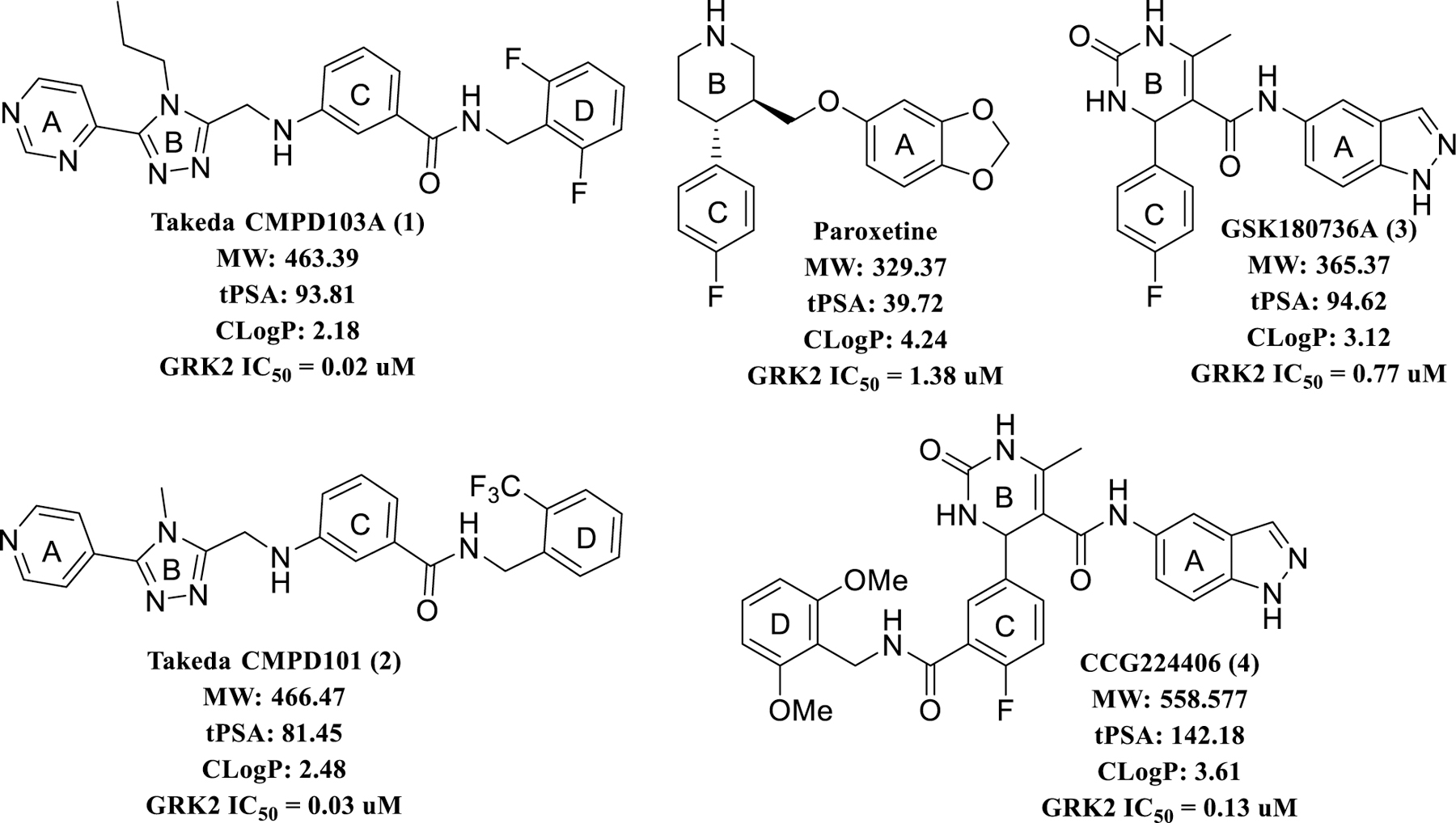
Known GRK2 inhibitors. The A, B, C, and D labels designate ring systems that bind in the adenine, ribose, pyrophosphate and hydrophobic subsites of the active site, respectively.
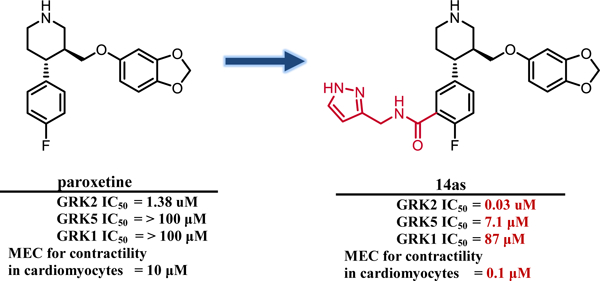
The FDA-approved serotonin-reuptake inhibitor, paroxetine hydrochloride, was identified as a modest GRK2 inhibitor with an IC50 of 1.4 µM (Figure 1).25 Further in vivo investigation in a mouse heart failure model revealed that paroxetine improves cardiac function post-myocardial infarction and renormalizes the levels of catecholamines and β-adrenergic receptor density in the heart, effects that persist for up to two weeks post-treatment.26 Because paroxetine is only a modestly potent and selective inhibitor of GRK2, but also an FDA-approved drug with excellent ADMET properties, it was an attractive lead compound for further optimization as a GRK2 selective agent.
In parallel, we identified another, more potent GRK2 inhibitor, GSK180736A (3) with an IC50 of 0.8 μM (Figure 1), a compound that is similar to paroxetine in both size and shape.27 This compound was originally developed as a Rho-associated coiled-coil containing kinase 1 (ROCK1) inhibitor (IC50 = 100 nM), but exhibited limited bioavailabilty.28 Through a hybrid-based drug design approach utilizing compounds 3 and 1 and their respective GRK2 crystal structures, we generated a small library of potent and selective GRK2 inhibitors. Some of these compounds such as 4 (CCG224406, Figure 1) were demonstrated to enhance myocardial contractility in mouse cardiomyocytes at a 20-fold lower concentration than paroxetine.24
Comparison of the binding poses of paroxetine and 3 in the active site of GRK2 (Figure 2A) shows that both compounds form interactions with the adenosine, ribose, and polyphosphate subsites, but neither occupies the hydrophobic subsite like the Takeda compounds (Figure 2B), leaving room to increase potency and selectivity through the addition of favorable moieties to their fluorophenyl rings. In the adenine subsite, the benzodioxole ring of paroxetine makes analogous hydrogen bonds to the backbone of atoms in the kinase domain hinge as the indazole ring of 3, with the exception that one of the paroxetine hydrogen bonds is a CH–O hydrogen bond. The B-rings of the two compounds both occupy the ribose subsite, where paroxetine makes a hydrogen bond with the carbonyl of Ala321 that is not seen with 3. The fluorophenyl rings of both molecules pack underneath the P-loop, superimposing nearly perfectly.25, 27 Based on these largely overlapping binding modes, we hypothesized that the structure activity relationships from our 3 hybrid campaign would be translatable, providing guidance for novel extensions to the paroxetine scaffold. Furthermore, because paroxetine is an FDA-approved drug we anticipated that the analogous paroxetine-series of compounds would exhibit superior pharmacokinetic properties. Paroxetine has nearly two-fold lower polar surface area, a lower molecular weight, and fewer hydrogen bond donors and acceptors than 3, physical properties that are all predictive of better absorption and permeability (Figure 1).29
Figure 2.
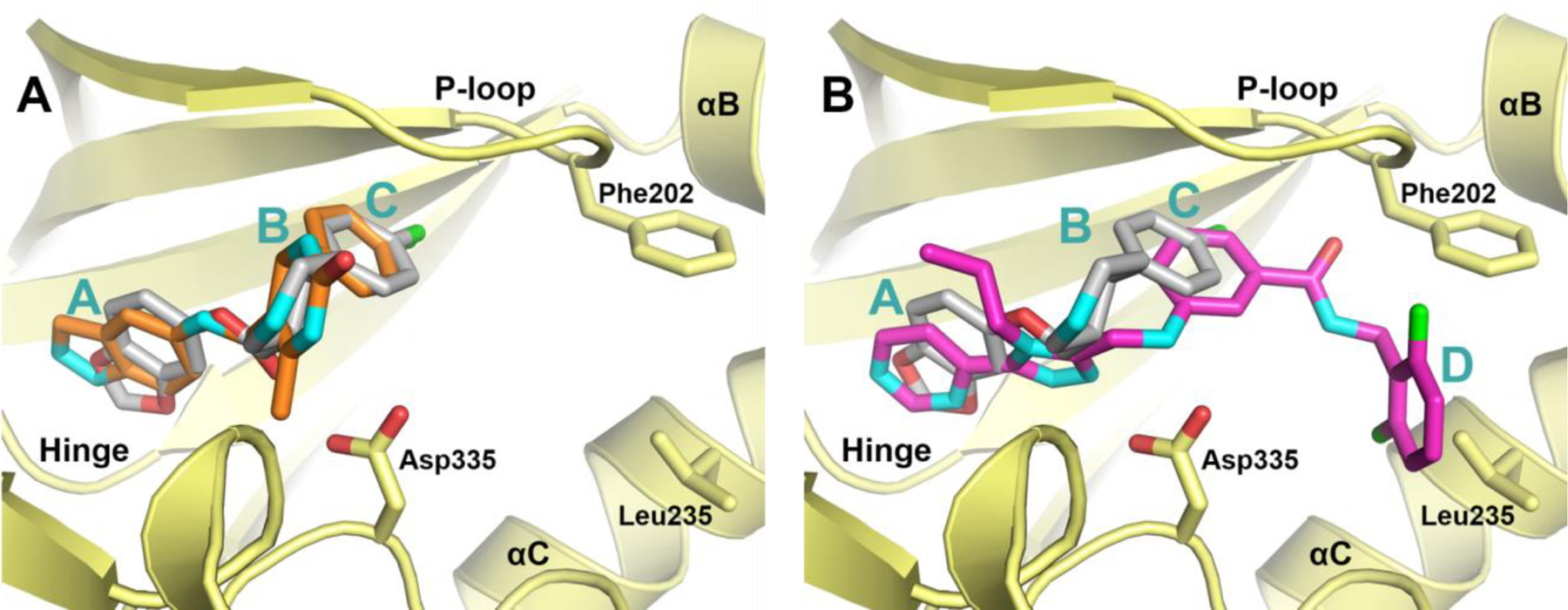
Comparison of lead compounds bound in the GRK2 active site suggest that extension of the paroxetine scaffold into the hydrophobic subsite is a route towards molecules with higher potency and selectivity. A) Overlay of paroxetine (gray carbons) and 3 (orange carbons) bound to the GRK2–Gβγ complex (PDB entries 3V5W and 4PNK, respectively). B) Overlay of paroxetine (gray carbons) and 1 (magenta carbons) bound to the GRK2–Gβγ complex (PDB entries 3V5W and 3PVW, respectively). The A, B, C, and D ring systems, which occupy the adenine, ribose, polyphosphate and hydrophobic subsites, respectively, are labeled for each compound, if present. Side chains of key residues that exhibit conformational changes upon inhibitor binding are shown as stick models.
We report herein the successful ligation of amide moieties to the fluorophenyl ring of paroxetine and generation of significantly more potent inhibitors of GRK2 (up to 50-fold) with high selectivity over GRK subfamilies and other AGC kinases (up to 230 fold over GRK1 and 5 as well as ROCK1 and PKA). Three representative compounds were crystallized revealing a new hydrogen bond with the amide linker and new polar contacts formed with the extensions in the hydrophobic subsite. Finally, we demonstrate that one of our optimized inhibitors (14as) enhances cardiomyocyte contractility with approximately 100-fold greater potency than paroxetine.
Results
Chemistry
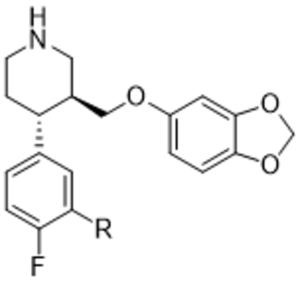
Initial hybrid analogues 14aa – 14ax were synthesized as described in Scheme 1. Synthesis commenced with commercially available ((3S,4R)-4-(4-fluorophenyl)-1-methylpiperidin-3-yl)methanol (5). Silyl protection of 5 using tert-butyl dimethyl silyl (TBS) chloride followed by N-demethylation with phenyl chloroformate (PCF) gave the phenyl carbamate 6.30 Base hydrolysis provided the free amine, which was then protected to give t-butyl carbamate 7. Sec-butyllithium mediated aryl lithiation of the position ortho to the fluorine, followed by trapping with carbon dioxide, yielded acid 8a.31 The acid was then methylated using trimethylsilyldiazomethane to give 9a. TBS deprotection of the alcohol followed by mesylation and nucleophilic displacement with sesamol afforded 11a. The methyl ester was then hydrolyzed under basic conditions to give the carboxylic acid 12a. Coupling of the free acid to various amines and final Boc deprotection yielded the carboxamides 14aa - ax.
Scheme 1:
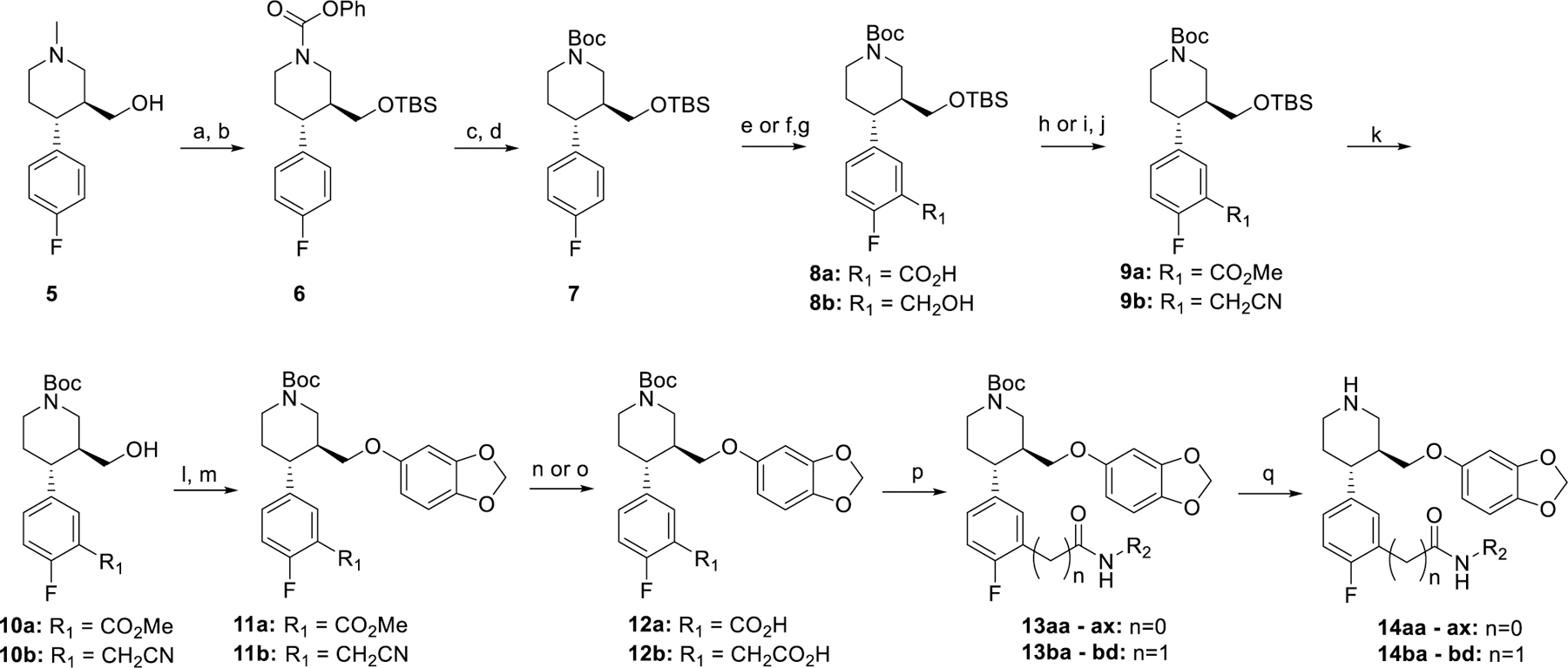
Synthesis of hybrid inhibitors 14aa – ax and 14ba - bd. Reagents and conditions. a) TBSCl, DIEA, imidazole, DCM, b) PCF, toluene, 110 ˚C then TEA, c) 8N NaOH, IPA, 80 ˚C, d) Boc2O, DIEA, DCM, e) TMEDA, sBuLi, THF, then CO2, f) TMEDA, sBuLi, THF, then DMF, g) NaBH4, THF, MeOH, h) TMS-diazomethane, MeOH/Toluene, i) Ms2O, DIEA, DCM, j) NaCN, DMSO, k) TBAF, THF, l) Ms2O, DIEA, DCM, m) NaH, 3,4-(methylenedioxy)phenol, DMF, 0 ˚C to 65 ˚C, n) 1N NaOH, MeOH, o) 50% NaOH, EtOH, 98 ˚C, p) DIEA, EDC, HOBt, R2NH2, q) HCl/dioxanes
Synthesis of homologated analogues 14ba – 14bd began with sec-butyllithium-mediated ortho lithiation of intermediate 7 followed by formylation with N, N-dimethylformamide to yield the carboxaldehyde.31 Subsequent reduction with sodium borohydride gave the alcohol 8b. Mesylation of the benzylic alcohol with methanesulfonic anhydride followed by nucleophilic substitution with sodium cyanide produced 9b. TBS deprotection of the remaining alcohol followed by mesylation and nucleophilic substitution with sesamol gave 11b. Base hydrolysis of the nitrile afforded carboxylic acid 12b. The acid was then coupled to various amines to give the carboxamides 13ba - bd which were then Boc-deprotected to provide the desired homologated compounds 14ba - bd.
Structure Activity Relationships.
All new compounds were assayed for in vitro activity against bovine GRK1, 2 and 5, as well as PKA and ROCK1 (Table 1). We envisioned that incorporation of an amide linker between the paroxetine scaffold and new hydrophobic subsite-binding substituents would allow for the formation of a hydrogen bond to the backbone nitrogen of Phe202 in the P-loop, as observed in our compound 3 derived series.23, 24 However, simple methyl amide (14aa) was two-fold less potent than paroxetine against GRK2, suggesting that any hydrogen bond that forms does not overcome desolvation and/or entropic penalties. Increasing the size of the amide substituent from a methyl to benzyl (14ab) was tolerated but not any more potent than paroxetine alone. Lengthening the amide substituent to phenethyl 14ac resulted in a slight loss in potency against GRK2, indicating that more lipophilicity on its own is not beneficial to improving potency. Moving to the 2,6–difluorobenzylamide (14ad), mimicking the D-ring of compound 1 and adding bulk, had no change in potency versus GRK2 and resulted in a decrease in selectivity against GRK5. Additionally, the added lipophilicity gave a modest increase in ROCK1 inhibition (34% at 10 µM). Further lengthening the substituent to the 2,6–difluorophenethylamide (14ae) resulted in an even further decrease in selectivity for GRK2 over GRK5 as it exhibited a 10-fold increase in potency for GRK5 and a 2-fold decrease in potency for GRK2. Moving to a hybrid of compound 2 we investigated the addition of a trifluoromethyl benzyl (14af) which resulted in a loss in potency for all three GRKs, suggesting a limit to the size of the substituent that could be tolerated.
Table 1.
Kinase inhibitory activity of paroxetine analogs
 | ||||||
|---|---|---|---|---|---|---|
| R | Compound | GRK2 IC50 (μM)a |
GRK1 IC50 (μM)a |
GRK5 IC50 (μM)a |
PKA IC50 (μM)a |
ROCK1* |
| H | Paroxetine | 1.38±1.00 | > 100 | > 100 | > 100 | 10% |
| 1 | 0.02±0.001 | 9.1±3.2 | 2.2±0.9 | ND | ND | |
| 2 | 0.03±0.006 | 52.1±26.3 | 9.2±3.0 | ND | ND | |
| 3 | 0.77±0.5 | > 100 | > 100 | 30±19 | 65% | |
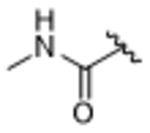 |
14aa | 2.1±0.72 | > 100 | > 100 | > 100 | 11% |
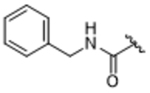 |
14ab | 0.77±0.17 | > 100 | 84.3±35.8 | > 100 | 17% |
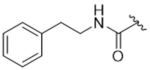 |
14ac | 2.68±2.11 | > 100 | > 100 | > 100 | 22% |
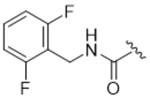 |
14ad | 1.53±0.49 | > 100 | 37.2±28.9 | > 100 | 34% |
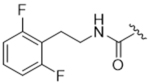 |
14ae | 2.03±0.33 | > 100 | 10.2±1.8 | > 100 | 0% |
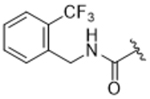 |
14af | 12.4±7.7 | > 100 | 75.7±39.9 | > 100 | 5% |
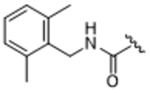 |
14ag | 2.17±0.79 | 24.5±19.6 | 42.3±16.3 | > 100 | 0% |
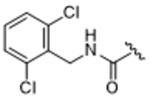 |
14ah | 1.76±1.43 | 25.8±16.1 | 33.2±2.48 | > 100 | 0% |
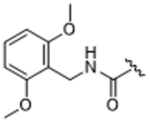 |
14ai | 2.04±1.02 | 32.0±14.0 | 38.4±16.1 | >100 | 13% |
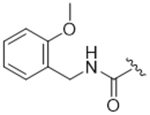 |
14aj | 37.7±9.14 | > 100 | > 100 | > 100 | 1% |
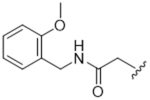 |
14ba | 2.37±2.27 | > 100 | > 100 | 25.8±25 | 18% |
 |
14ak | 0.61±0.12 | > 100 | 17.1±5.9 | > 100 | 6% |
 |
14al | 1.52±0.78 | > 100 | 76.3±15.3 | > 100 | 6% |
 |
14am | 2.04±0.74 | > 100 | > 100 | > 100 | 8% |
 |
14an | 3.28±1.8 | > 100 | > 100 | > 100 | 15% |
 |
14ao | 3.02±0.99 | > 100 | > 100 | > 100 | 18% |
 |
14ap | 3.24±1.0 | > 100 | > 100 | > 100 | 21% |
 |
14bb | 6.01±2.08 | 60.0±2.07 | > 100 | 48.2±35 | 0% |
 |
14bc | 5.9±3.2 | > 100 | > 100 | > 100 | 0% |
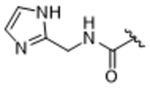 |
14aq | 0.75±0.32 | > 100 | 14.8±3.2 | > 100 | 18% |
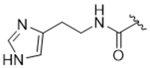 |
14ar | 0.6±0.21 | > 100 | > 100 | > 100 | 11% |
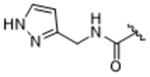 |
14as | 0.03±0.001 | 87.3±27.9 | 7.09±0.73 | > 100 | 9% |
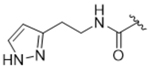 |
14at | 0.77±0.20 | > 100 | > 100 | > 100 | 18% |
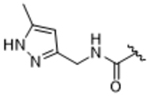 |
14au | 0.03±0.02 | 86.5±7.0 | 4.0±0.52 | > 100 | 12% |
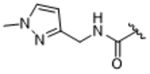 |
14av | 1.25±0.30 | > 100 | > 100 | > 100 | 17% |
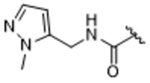 |
14aw | 2.11±0.52 | > 100 | > 100 | > 100 | 14% |
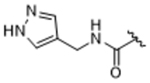 |
14ax | 0.39±0.11 | > 100 | > 100 | > 100 | 19% |
 |
14ay | 16.7±5.4 | > 100 | > 100 | > 100 | 13% |
 |
14bd | 0.63±0.33 | > 100 | > 100 | > 100 | 27% |
All IC50 measurements are an average of three separate experiments run in duplicate. Errors shown represent error of the mean.
Percent inhibition at 10 μM inhibitor concentration. ND, not determined.
We next tested structure activity relationships generated by our previously reported compound 3-based series, which revealed that bulkier substituents in the hydrophobic subsite of GRK2 can achieve both increased potency for GRK2 as well as increased selectivity over the other GRKs and related AGC kinases like ROCK and PKA.24 Three representative substituents from this endeavor are 2,6–dimethylbenzylamide, 14ag, 2,6–dichlorobenzylamide, 14ah, and 2,6–dimethoxybenzylamide, 14ai. Although the added bulk improved selectivity against ROCK1, all compounds displayed reduced selectivity for GRK2 over GRK1 and 5, and no improvement in potency, suggesting inadequate engagement of the hydrophobic subsite.
Less bulky but still lipophilic amide substituents were then investigated. In our previous SAR campaign, the most potent compound against GRK2 (IC50 = 60 nM) placed a 2-methoxybenzylamide substituent in the hydrophobic subsite.24 Surprisingly, translating the 2-methoxybenzylamide onto the paroxetine scaffold resulted in our least potent GRK2 inhibitor, 14aj (IC50 = 40 μM). Interestingly, homologating the 2-methoxybenzylamide by adding a methylene group between the amide and fluorophenyl ring (14ba) brought back some GRK2 potency (IC50 = 2.4 μM) but also picked up affinity for GRK5. These results, in addition to those above, revealed that although the binding poses and structures of 3 and paroxetine are similar, the SAR of their extended scaffolds are poorly translatable. One possible explanation is that the overall conformation of the GRK2 kinase domain in complex with compound 3 is more closed than in the GRK2·paroxetine complex due to different interactions formed by their hinge-binding moieties. This results in small but significant structural differences in the polyphosphate and hydrophobic subsites and the relative orientation of the small and large lobes that form the active site.25, 32
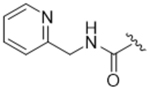
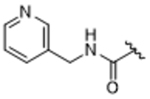
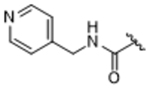
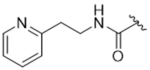
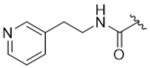
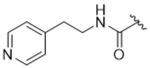
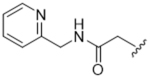
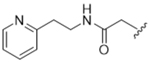
As bulky and lipophilic groups were not tolerated, we turned our attention to smaller more polar groups including a series of regioisomeric pyridyl methylamides. The 2-pyridine, 14ak, afforded a 2-fold increase in potency for GRK2 along with a 5-fold increase in GRK5 potency. Thus, for the first time we were able to modestly improve GRK potency. The corresponding 3- and 4-pyridyl analogs (14al and 14am) were less potent, suggesting that the added potency of 14ak was due to the position of the nitrogen rather than the aromatic ring alone. Lengthening the carbon linker between the amide and the pyridines (14an, 14ao, 14ap) or between the amide and the phenyl ring (14bb, 14bc) also reduced potency, providing evidence for the importance of the position of the pyridyl nitrogen relative to the fluorophenyl ring. From the overall SAR of these pyridine amides, it was clear that the ortho position of the nitrogen in 14ak is key for potency against both GRK2 and GRK5.
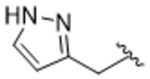
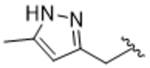
To more efficiently exploit this polar contact we next sought to design molecules that would have a stronger hydrogen bond acceptor in a similar position as the pyridine nitrogen of 14ak. The 2-imidazolylmethyl amide 14aq, as well as its homolog 14ar, showed potency comparable to 14ak. They also had increased potency for GRK5, presumably by making a hydrogen bond to Lys220 as we observed previously in the GRK5·215022 (17) complex.33 Replacing the imidazole amide with 3-pyrazolylmethyl amide (14as), resulted in a dramatic increase in potency for GRK2 (IC50 = 30 nM) while maintaining 230-fold selectivity over GRK5 and more than 2500-fold selectivity over GRK1, PKA, and ROCK1. Lengthening the amide linker to give 3-pyrazolylethyl amide, 14at, resulted in a 26-fold decrease in potency for GRK2, relative to 14as. Furthermore, methylation of either nitrogen of 14as (14av and 14aw) resulted in 40-fold and 70-fold decreases in GRK2 affinity, respectively, whereas placing a methyl group adjacent to the two nitrogens (14au) did not diminish potency. These results strongly suggest that both nitrogens of the pyrazole of 14as are necessary for its high GRK2 potency and are likely making either one or two polar interactions. Simple one-atom transposition of the two pyrazole nitrogens (14ax) resulted in a 10-fold loss in GRK2 potency relative to 14as, further confirming the importance of the location of the nitrogens. Homologation of the 4-pyrazole methyl amide of 14ax (14ay, 14bd) could not rescue the loss in potency.
Crystallographic Analysis
To confirm our design strategy and to guide our analogue design early in our campaign, we determined the 2.6 Å crystal structure of 14ak bound to the GRK2–Gβγ complex (Figure 3A). The complex crystallized in an unusual space group (P2) not previously observed for this assembly, suggesting unique conformational changes in the kinase domain. We additionally determined co-crystal structures of 14bd and 14as in the space group C2 at resolutions of 2.2 Å and 3.0 Å, respectively (Figure 3B and 3C). Due to the addition of their various amide linked D rings, there are slight variations in the fitting of their A, B and C rings among the molecules, although overall they bind similarly to paroxetine (PDB ID: 3V5W).25 The benzodioxole A rings of the three inhibitors form analogous hinge interactions, the piperidine B ring sits puckered in the ribose subsite where it forms a hydrogen bond with the backbone carbonyl of Ala321, and the fluorophenyl C ring packs under the P-loop in the polyphosphate subsite. As seen in our previously reported compound 3 derived inhibitors, the carbonyl of the amide linkers of all three inhibitors forms a hydrogen bond with the backbone nitrogen of Gly201. Beyond the amide linker of 14bd, the electron density for the pyrazole is relatively poor, likely because it extends out of the active site towards solvent. Because the D rings of 14ak and 14as do occupy the hydrophobic subsite, we interpret the divergent packing of 14bd as a consequence of the extra methylene preceding its amide group, which makes the substituent too long to pack into the hydrophobic subsite. Phe202 in the 14bd complex also adopts a distinct rotamer from the paroxetine, 14ak, and 14as complexes, which allows it to pack with the solvent-extended pyrazole of 14bd. Relative to 14bd, the amide nitrogen of 14ak and 14as forms an additional hydrogen bond with the side chain of Asp335, which is also a consequence of the fact that 14bd is homologated, moving this nitrogen out of reach.
Figure 3.
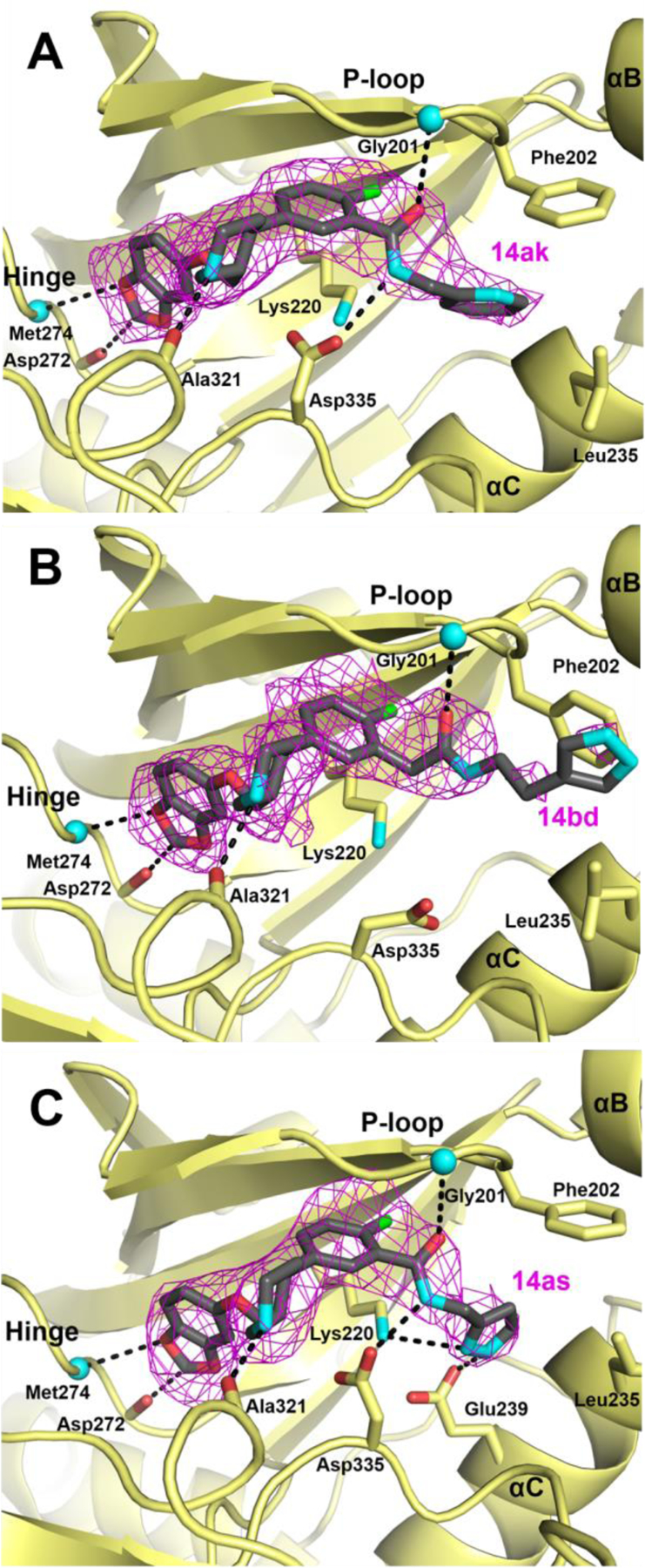
Binding modes of three different paroxetine adducts in the GRK2 active site reveal rules for productive engagement of the hydrophobic subsite. 3σ |Fo|-|Fc| omit maps of A) 14ak, B) 14bd, and C) 14as are shown as magenta wire cages superimposed on the fully refined co-crystal structures. Hydrogen bonds are represented as black dashed lines. All three compounds impose distinct conformational changes in the P-loop, Glu329 and Asp335. The homologated D ring of 14bd extends into solvent, consistent with its weak difference density.
The co-crystal structure of the 14bd crystal complex was the most analogous to the parent paroxetine complex in that it aligns nearly identically (r.m.s.d. deviation of 0.22 Å for the Cα atoms of residues 185–271 in the small lobe). This is consistent with our SAR in that 14bd has only a 2-fold increase in GRK2 potency relative to paroxetine but retains similar IC50 values for both GRK1 and GRK5. The increase in potency for GRK2 seems unlikely to be due to the additional hydrogen bond picked up by the amide linker because 14aa, which contains just an amide substituent, is two-fold less potent. Instead, it is likely due to additional buried surface area, a key feature for GRK2 inhibitors.24 Selectivity is likely similar to that of paroxetine because of the lack of contacts of its D-ring in the hydrophobic subsite.
Although the 2-pyridine of analogue 14ak packs snuggly into the hydrophobic subsite of GRK2, this seems to require displacement of the αB helix and adjoining loops away from the active site by up to 1.3 Å, consistent with a larger r.m.s.d. of 0.76 Å for the Cα atoms in the small lobe of the kinase compared to the paroxetine complex, as well as a unique hinge conformation that changes the relative orientation of the small and large lobes by ~15˚ relative to the 14ak and 14bd co-crystal structures. These changes are probably responsible for the unique crystal form that this complex adopts. Because the SAR showed that placement of the pyridine nitrogen at the 3 or 4 position was detrimental to potency, it had been surmised that a polar contact or hydrogen bond might exist between the nitrogen and the catalytic lysine as seen in our formerly reported GRK5·17 inhibitor complex.33 Here the pyridine nitrogen is too distant (4.5 Å) and not in the right orientation to form a hydrogen bond with the side chain of the catalytic lysine (GRK2-Lys220), as does the analogous pyridine D ring in the GRK5·17 co-crystal structure. Thus it is instead modeled facing the solvent, as in our GRK2-Gβγ·17 structure.24 The loss in GRK2 potency upon migration of the pyridine nitrogen to the 3 or 4 position is likely a consequence of the loss of lipophilicity at those positions, which can make non-polar contacts in the hydrophobic subsite. The increased potency exhibited by 14ak for both GRK2 and GRK5 relative to paroxetine is therefore likely due to additional van der Waals interactions formed by the D ring in the hydrophobic subsite.
The small lobe in the 14as co-crystal structure is more similar to the paroxetine complex (r.m.s.d. of 0.41 Å) than the 14ak complex (r.m.s.d. of 0.86 Å). As in 14ak, the non-homologated pyrazole moiety of 14as packs in the hydrophobic subsite but packs lower in the site than the pyridine of 14ak. The pyrazole nitrogens are within hydrogen bond distance of the side chains of both Glu239 and Lys220, with the closest being formed with Glu239. Therefore, it is most likely that the significant increase in potency for GRK2 results from not only the specific interactions picked up by the pyrazole nitrogens, but also because the pyrazole does not require a conformational change ( pushing out the αB helix) as required for the pyridine of 14ak. The carbon of the pyrazole adjacent to the two nitrogens packs against Leu235 in the αC helix, thus it is not clear how 14au (methylated) retains high potency, but the observed interactions explain why 14av and 14aw (N-methylated variants), 14ax and 14aq (pyrazole nitrogens moved around the ring) exhibit large decreases in potency, as does lengthening the linker between the amide and the pyrazole (14at).
Without a crystal structure of GRK5 in complex with one of our paroxetine-derived compounds, the ability of these pyrazole derivatives to retain such high selectivity over GRK5 remains unclear. We have observed that the hydrophobic subsite in GRK2 can accommodate bulkier D-rings than GRK5.24, 33, 34 Furthermore, of the compounds reported here only those with non-homologated amide linkages exhibit any potency for GRK5, suggesting that the hydrogen bond formed between the amide nitrogen and GRK2-Asp335 (GRK5-Asp329) is important for potency against GRK5. The non-homolgated methylene-linked pyrazoles, 14as and 14au, exhibited the highest potency for GRK5 (but retained >100 fold selectivity for GRK2), consistent with the idea that hydrogen bond formation by the amide linker in combination with favorable polar contacts is similarly important for GRK5 potency. Thus, it seems most likely that differences in the kinase domain hinges and, consequently, the overall conformation of the GRK2 and GRK5 kinase domains are responsible for the observed selectivity in the paroxetine-derived compounds. For example, if one overlays the small lobe of the GRK2·14as complex with that of the GRK5·17 complex, the glycine in the DFG loop of the large lobe of GRK5 would sterically collide with the pyrazole of 14as. The preferred conformation of the GRK5 hinge may also engender less optimal interactions with the benzodioxole of paroxetine series inhibitors.
Metabolic Stability
To guide the design and selection of analogs with favorable pharmacokinetic (PK) properties, we evaluated the stability of selected compounds to incubation with mouse liver microsomes (MLM) (Table 2). Our lead compound, paroxetine, had a t1/2 of 24 min in our MLM assay while 3 had a t1/2 of 20.6 min and 2 had a t1/2 of only 1.9 min. Appending lipophilic carboxamides onto 3, giving our previously reported GRK2 inhibitors (15, 16, 4), resulted in a substantial drop in stability with all three compounds having a t1/2 lower than 4 min.24 Conversely, addition of the same moieties onto the paroxetine scaffold (analogs 14ag, 14ah, 14ai) could be done without eroding metabolic stability and in the case of 14ag improving upon the t1/2 of paroxetine two-fold. Addition of a 2-pyridine carboxamide to the scaffold of compound 3 (17) was still detrimental but not as poorly tolerated as the lipophilic appendages. Surprisingly, on the paroxetine template, the polar carboxamides (14ak, 14as, 14a4) exhibited substantial metabolic instability, perhaps due to targeting of the heterocyclic nitrogens to the heme of cytochrome P450. These conflicting results between the paroxetine and 3 scaffolds suggest that the mechanism of metabolism differs substantially between the two series.
Table 2:
Comparison of MLM stability between paroxetine and GSK180736A analogs
 | |||
|---|---|---|---|
| R = | Name | Scaffold | t1/2 in MLM (min) |
| N/A | 2 | N/A | 1.91 |
| H | 3 | B | 20.6 |
| H | Paroxetine | A | 24.3 |
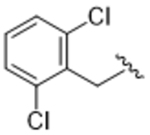 |
14ah | A | 36.4 |
| 15 | B | 3.37 | |
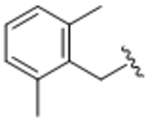 |
14ag | A | 45.9 |
| 16 | B | 3.46 | |
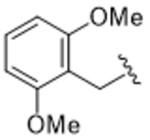 |
14ai | A | 24.9 |
| 4 | B | 3.82 | |
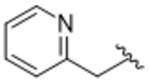 |
14ak | A | 3.15 |
| 17 | B | 11.32 | |
 |
14as | A | 7.0 |
 |
14au | A | 10.3 |
Contractility in Mouse Cardiomyocytes
As an underlying mechanism of heart failure is the inability of the heart to properly contract we selected compounds 14ak and 14as in addition to Takeda 2 for evaluation in an in vivo contractility assay. Following incubation with varying doses of the inhibitors to give a baseline contraction (Figure 3, grey bars) mouse cardiomyocytes were then stimulated with the βAR agonist isoproterenol. The resulting maximal increase in contraction was then measured (Figure 3, black bars).33 As inhibition of GRK2 should increase the number of activated βARs we would expect our inhibitors to enhance the maximal increase in comparison to a DMSO control. Previously, we showed that both paroxetine and 3 produce an increase in contractility. The minimum dose needed for paroxetine to produce a significant response was 10 μM, whereas 3 showed similar efficacy at 1 μM.24, 25 These results agree with the higher potency of 3 relative to paroxetine.
Evaluation of modestly potent 14ak in the cardiomyocyte contractility assay did not show a significant increase in contractility at 0.5 μM or 1 μM and thus showed efficacy similar to the lead paroxetine, consistent with its modest two-fold increase in GRK2 inhibition potency. The highly potent 14as, on the other hand, showed a significant increase in contractility at a concentration of only 0.1 μM, a 100-fold lower concentration than paroxetine, consistent with the approximately 50-fold increase in potency 14as has for GRK2 relative to paroxetine. Additionally, 14as showed a 5-fold improvement over Takeda lead 2, although both compounds had equal potency for GRK2 (30 nM), suggesting that 14as may have better cell permeability. In comparison to our previously reported compound 4, this paroxetine hybrid inhibitor also shows a 5-fold improvement in efficacy.24 Importantly, these results suggest that significant improvement in βAR-stimulated contractility in mouse cardiomyocytes can be achieved with potent GRK2-selective inhibition.
Preliminary Pharmacokinetic Study in Mice
Compounds 2 and 14as, were evaluated in an abbreviated in vivo pharmacokinetic study in mice (Table 3). The compounds were intraperitoneally injected into CD-1 mice at a dose of 10 mg/kg, plasma samples were collected at four different time points over seven hours, and drug levels in plasma were quantified. Due to the abbreviated nature of this study, PK parameters could not be rigorously calculated. Nevertheless, it is clear the half-life in vivo for 14as which can be estimated from the four time points to be approximately one hour is superior to that of 2 which is too low to quantify in this study. Of particular relevance to future in vivo efficacy studies in mice, 14as maintained plasma drug levels exceeding the GRK2 IC50 (30 nM) seven hours after single IP administration.
Table 3.
In Vivo Exposure Following IP Administration to Mice*
| plasma (µM) | AUC 0 – 7hobs
(hr*µM) |
||||
|---|---|---|---|---|---|
| Compound (dose) | 30 min | 2 h | 4 h | 7 h | |
| 2 (10 mg/kg) | 1.83 | 0.032 | 0.005 | 0.006 | 1.90 |
| 14as (10 mg/kg) | 2.71 | 1.44 | 0.27 | 0.043 | 5.97 |
CD-1 mice were injected intraperitoneally with a single indicated dose. The data shown are mean values from 3 mice at each time point.
Conclusion
Because paroxetine was identified as a modestly potent GRK2 inhibitor with good selectivity, we sought to improve potency for GRK2 while retaining selectivity. Our previous GRK2 inhibitor series based on 3 successfully utilized a hybrid approach to develop improved GRK2 inhibitors, and thus we hypothesized that a similar approach could be used with paroxetine. A small library of paroxetine-derived GRK2 inhibitors were then synthesized, most of which retained selectivity for GRK2. Although we discovered that the SAR of our previous series was not translatable to the paroxetine series, we did learn that smaller heterocycles were advantageous to increasing GRK2 potency. Addition of a 3-pyrazolylmethyl amide to the paroxetine scaffold (14as), resulted in a highly potent GRK2 inhibitor (IC50 = 30 nM) with high selectivity over GRK1, GRK5, PKA, and ROCK1.
Co-crystal structures of three of the synthesized paroxetine derivatives were determined, revealing additional hydrogen bonds with the added amide linker and the ability of GRK2 to adopt a more open conformation than in the 3-based series, perhaps due to how the benzodioxole group packs with the hinge. This probably underlies the high selectivity of these compounds. The hydrophobic subsite exhibits some conformational flexibility in GRK2, as evidenced by the 14ak complex compared to the other two structures, and excludes the D ring if it is too big to bind in the pocket or is unable to overcome a desolvation penalty (both of which probably apply to 14bd).
Our compounds showed improved contractility in mouse cardiomyocytes at concentrations as low as 100 nM (14as) which is 100-fold lower than paroxetine and 5-fold lower than the Takeda compound 2, suggesting that selective and potent inhibition of GRK2 is sufficient for cardiac stimulation in the failing heart (i.e. does not require inhibition of GRK5). Evaluation in a short pharmacokinetics study revealed that 14as was able to maintain plasma concentrations higher than its IC50 for over seven hours, indicative of its potential as an in vivo therapeutic. 14as has thus been selected for additional studies in murine models of heart failure in which paroxetine has previously been evaluated.26 Because 14as shows such improved potency in cardiomyocyte assays, we hypothesize that it will exhibit superior results in vivo relative to paroxetine, confirming the utility of this series of GRK2 inhibitors as optimized leads for heart failure therapeutics.
Experimentals
Chemistry.
All reagents were used without further purification as received from commercial sources unless noted otherwise. 1H NMR spectra were taken in DMSO-d6, MeOD, or CDCl3 at room temperature on Varian Inova 400 MHz or Varian Inova 500 MHz instruments. Reported chemical shifts for the 1H NMR spectra were recorded in parts per million (ppm) on the δ scale from an internal standard of residual tetramethylsilane (0 ppm). Mass spectrometry data was measured using a Waters Corporation LCT. HPLC was used to determine purity of biologically tested compounds on an Agilent 1100 series with an Agilent Zorbax Eclipse Plus−C18 column. A gradient of 10–90% acetonitrile/water over 6 min followed by 90% acetonitrile/water for 7 min was used with detection at 254 nm. All tested compounds had purity >95%. Solvent abbreviations used: MeOH (methanol), DCM (dichloromethane), EtOAc (ethyl acetate), hex (hexanes), DMSO (dimethylsulfoxide), DMF (dimethylformamide), H2O (water), THF (tetrahydrofuran). Reagent abbreviations used: HOBt (hydroxybenzotriazole), EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide), DIEA (diisopropylethylamine), MgSO4 (magnesium sulfate), NaHCO3 (sodium bicarbonate), Na2CO3 (sodium carbonate), NH4Cl (ammonium chloride), NaCl (sodium chloride), K2CO3 (potassium carbonate) and HCl (hydrogen chloride).
Phenyl(3S,4R)-3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluorophenyl)piperidine-1-carboxylate (6):
To a 250 mL round bottom flask was added ((3S,4R)-4-(4-fluorophenyl)-1-methylpiperidin-3-yl)methanol (4.0 g, 17.91 mmol) and dichloromethane (40 mL). Tert-butyldimethylsilyl chloride (4.05 g, 26.9 mmol) and imidazole (1.22 g, 17.9 mmol) were added to the reaction vessel producing a cloudy white mixture. Lastly, N,N-diisopropylethylamine (4.68 mL, 26.9 mmol) was added, giving a clear solution. The reaction was stirred overnight at room temperature. Methylene chloride was used to dilute the reaction followed by washing with brine (2x). The organic layer was dried over MgSO4, concentrated in vacuo, and purified using flash chromatography with a 20%−40% EtOAc/Hexane gradient to give phenyl (3S,4R)-3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluorophenyl)piperidine-1-carboxylate (5.51 g, 16.3 mmol, 91% yield). 1H NMR (500 MHz, Chloroform-d) δ 7.13 (dd, J = 8.5, 5.5 Hz, 2H), 6.96 (t, J = 8.7 Hz, 2H), 3.29 (dd, J = 10.3, 2.3 Hz, 1H), 3.15 (dd, J = 10.1, 6.0 Hz, 1H), 3.08 (dd, J = 7.6, 1.8 Hz, 1H), 2.92 (ddt, J = 11.2, 4.0, 2.4 Hz, 1H), 2.33 (s, 3H), 2.32 – 2.28 (m, 1H), 2.00 – 1.88 (m, 3H), 1.85 – 1.72 (m, 2H), 0.84 (s, 9H), −0.11 (d, J = 9.3 Hz, 6H). Phenyl (3S,4R)-3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluorophenyl)piperidine-1-carboxylate (0.47 g, 1.40 mmol) and toluene (8 mL) were added to a 50mL flask and refluxed at 110ºC. Phenyl chloroformate (0.32 mL, 2.53 mmol) was then added dropwise and the reaction was stirred another four hours at reflux. The reaction was then cooled to 60ºC and trimethylamine (0.20 mL, 1.44 mmol) was added to quench the reaction which was stirred another 40 minutes and then cooled to room temperature. The reaction was diluted with toluene and brine. The layers were separated and the toluene layer was washed 2x with brine, dried over MgSO4, and concentrated. Purified the resulting clear viscous liquid with flash chromatography (0 – 10% EtOAc/Hexanes) to give a clear syrup, phenyl (3S,4R)-3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluorophenyl)piperidine-1-carboxylate (0.467g, 75% yield). Took forward as is without further purification.
Tert-butyl (3S,4R)-3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluorophenyl)piperidine-1-carboxylate (7).
Compound 6 (6.03 g, 13.6 mmol) was added to a 250 mL round bottom flask and dissolved in isopropanol (45 mL). Sodium hydroxide (8 N, 15mL) was then added and the reaction was refluxed at 80ºC overnight. The reaction was diluted with water and toluene. The layers were separated and the organic layer was washed 2x with brine, dried over MgSO4, and concentrated. Purified using flash chromatography (0–20% MeOH/DCM) to give as a clear oil (3S,4R)-3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluorophenyl)piperidine (2.41 g, 55% yield). 1H NMR (DMSO – d6, 400 MHz) δ: 7.24–7.19 (m, 2H), 7.10 (t, J=8 Hz, 2H), 3.21 (dd, J = 10.2, 3.0 Hz, 1H), 3.12 (td, J = 11.2, 10.2, 7.7 Hz, 2H), 2.98 – 2.92 (m, 1H), 2.40 (td, J = 11.4, 4.6 Hz, 1H), 2.34 (dd, J = 12.0, 10.7 Hz, 1H), 2.18 (s, 1H), 1.70 (tdt, J = 11.0, 7.2, 3.5 Hz, 1H), 1.60 – 1.47 (m, 2H), 0.8 (s, 9H), −0.13 (d, J = 6.7 Hz, 6H). (3S,4R)-3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluorophenyl)piperidine (1.05 g, 3.25 mmol), boc anhydride (0.99 g, 4.54 mmol), N,N-diisopropylethylamine (0.85 mL, 4.88 mmol) and THF (30 mL) were added to a 250 mL round bottom flask and stirred under nitrogen overnight. Reaction was quenched with water and diluted with EtOAc. The organic layer was washed 2x with brine, dried with MgSO4, and concentrated to give a clear oil. The oil was further purified using flash chromatography (10% EtOAc/Hexanes) to give, as a clear syrup, tert-butyl (3S,4R)-3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluorophenyl)piperidine-1-carboxylate (1.35 g, 98% yield). 1H NMR (DMSO – d6, 400 MHz) δ: 7.26–7.19 (m, 2H), 7.09 (t, J=8 Hz, 2H), 4.26 (d, J=8.0 Hz, 1H), 4.01 (br s, 1H), 3.46–3.02 (m, 4H), 2.69 (s, 1H), 1.81–1.43 (m, 3H), 1.39 (s, 9H), 0.80 (s, 9H), −0.13 (s, 6H); HPLC purity: 95%; MS (ESI+) m/z: 424.4 (M+1), 446.4 (M+Na+).
5-((3S,4R)-1-(tert-butoxycarbonyl)-3-(((tert-butyldimethylsilyl)oxy)methyl)piperidin-4-yl)-2-fluorobenzoic acid (8a).
N1,N1,N2,N2-tetramethylethylene-1,2-diamine (0.549 g, 4.72 mmol) was added to THF (10 mL) in a 50 mL round bottom flask cooled to −78 °C under argon. Sec-buytllithium (3.63 mL, 4.72 mmol) was added and the reaction mixture was stirred for thirty minutes then (3S,4R)-tert-butyl 3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluorophenyl)piperidine-1-carboxylate (1.0 g, 2.36 mmol) in THF (10 mL) was added and the solution was stirred at −78 °C for 1 hour. Freshly broken CO2 was added and the mixture allowed to warm to room temperature. The reaction was extracted with ether 2x, washed with NaCl 2x, dried with MgSO4 and concentrated in vacuo. The resulting intermediate was used without further purification.
(3S,4R)-tert-butyl 3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluoro-3-(methoxycarbonyl)phenyl) piperidine-1-carboxylate (9a).
To a solution of 5-((3S,4R)-1-(tert-butoxycarbonyl)-3-(((tert-butyldimethylsilyl)oxy)methyl)piperidin-4-yl)-2-fluorobenzoic acid (1.08 g, 2.31 mmol) in 20% (V/V) Methanol/toluene (30 mL) was added drop wise TMS-diazomethane (1.27 mL, 2.54 mmol). Gas evolved and reaction is slightly yellow. The reaction was stirred for 30 minutes and was then quenched with acetic acid until gas no longer evolved and the yellow color was gone. Then the reaction was concentrated in vacuo. Dissolved in ethyl acetate and ether and washed 2x with brine, dried with MgSO4 and concentrated. The crude product was purified using flash chromatography with a 20%−30% EtOAc/Hexane gradient to give (3S,4R)-tert-butyl 3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluoro-3-(methoxycarbonyl)phenyl) piperidine-1-carboxylate (0.96 g crude, 1.99 mmol, 86% yield over two steps). 1H NMR (400 MHz, DMSO-d6) δ 7.69 (dd, J = 7.2, 2.3 Hz, 1H), 7.56 (dq, J = 7.2, 2.3 Hz, 1H), 7.29 (dd, J = 10.8, 8.5 Hz, 1H), 4.25 (d, J = 13.0 Hz, 1H), 4.03 (d, J = 8.2 Hz, 1H), 3.84 (s, 3H), 3.22 (s, 1H), 3.14 (dd, J = 10.5, 7.4 Hz, 1H), 2.72 (s, 1H), 2.58 (s, 2H), 1.76 (s, 1H), 1.67 (d, J = 12.5 Hz, 1H), 1.63 – 1.48 (m, 1H), 1.41 (s, 9H), 0.82 (s, 9H), −0.11 (d, J = 4.4 Hz, 6H).
(3S,4R)-tert-butyl 4-(4-fluoro-3-(methoxycarbonyl)phenyl)-3-(hydroxymethyl)piperidine-1-carboxylate (10a).
To a solution of (3S,4R)-tert-butyl 3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluoro-3-(methoxycarbonyl)phenyl) piperidine-1-carboxylate (1.68 g, 3.49 mmol) in THF (50 mL) was added acetic acid (0.60 mL, 10.46 mmol) followed by tetrabutylammonium fluoride (1.0 M, 10.46 mL, 10.46 mmol). The resulting mixture was stirred overnight at 60 °C. Diluted with ethyl acetate/ether and treated with saturated aq. NH4Cl. The layers were separated and the organic layer was washed with NaCl (2x), dried with MgSO4, and concentrated in vacuo. Purified using flash chromatography with a 10%−30% EtOAc/Hexane gradient to give (3S,4R)-tert-butyl 4-(4-fluoro-3-(methoxycarbonyl)phenyl)-3-(hydroxymethyl)piperidine-1-carboxylate as a clear oil (1.29 g, 3.51 mmol, 100% yield). 1H NMR (400 MHz, Chloroform-d) δ 7.75 (dd, J = 6.9, 2.4 Hz, 1H), 7.34 (ddd, J = 8.4, 4.5, 2.4 Hz, 1H), 7.08 (dd, J = 10.5, 8.5 Hz, 1H), 4.36 (d, J = 13.0 Hz, 1H), 4.21 (s, 1H), 3.92 (s, 3H), 3.43 (dt, J = 11.1, 3.8 Hz, 1H), 3.25 (dt, J = 11.3, 6.0 Hz, 1H), 2.85 – 2.68 (m, 2H), 2.61 (s, 1H), 1.83 (dq, J = 7.1, 3.9, 3.5 Hz, 1H), 1.77 (d, J = 13.1 Hz, 1H), 1.67 (qd, J = 12.3, 4.2 Hz, 1H), 1.48 (s, 9H), 1.20 (s, 1H).
(3S,4R)-tert-butyl 4-(4-fluoro-3-(methoxycarbonyl)phenyl)-3-(((methylsulfonyl)oxy)methyl)piperidine-1-carboxylate) (11aa).
To a 0 °C solution of (3S,4R)-tert-butyl 4-(4-fluoro-3-(methoxycarbonyl) phenyl)-3-(hydroxymethyl)piperidine-1-carboxylate (0.687 g, 1.87 mmol) in 25 mL DCM was added diisopropylethylamine (0.980 mL, 5.61 mmol) followed by methanesulfonylchloride (0.651 g, 3.74 mmol). The reaction was allowed to warm to room temperature and stirred overnight. The resulting product was washed with water (1x), then brine (1x), dried over MgSO4, and concentrated in vacuo to give a clear oil (0.833 g crude). The product was used in subsequent reactions without further purification.
(3S,4R)-tert-butyl 3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-(methoxycarbonyl)phenyl) piperidine-1-carboxylate (11ab).
To a 0 °C solution of benzo[d][1,3]dioxol-5-ol (0.543 g, 3.93 mmol) in 5 mL DMF was added 60% sodium hydride in mineral oil (0.164g, 4.11 mmol). The solution turned light pink and was stirred for five minutes. To the reaction was added (3S,4R)-tert-butyl 4-(4-fluoro-3-(methoxycarbonyl)phenyl)-3-(((methylsulfonyl)oxy)methyl)piperidine-1-carboxylate (0.833 g, 1.87 mmol) in 5 mL DMF. The reaction was heated to 70 °C for 1 hour then cooled to room temperature and treated with sat. NH4Cl solution. Extracted with ethyl acetate/ether (2x), washed with brine, dried with MgSO4, and concentrated in vacuo. The crude product was purified by flash chromatography using a gradient of 5% MeOH/DCM to give as a clear oil (3S,4R)-tert-butyl 3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-(methoxycarbonyl)phenyl)piperidine-1-carboxylate (0.452 g, 0.927 mmol, 50% yield over two steps). 1H NMR (400 MHz, Methanol-d4) δ 7.76 (dd, J = 6.9, 2.4 Hz, 1H), 7.49 (ddd, J = 8.5, 4.6, 2.5 Hz, 1H), 7.14 (dd, J = 10.7, 8.5 Hz, 1H), 6.61 (d, J = 8.5 Hz, 1H), 6.35 (d, J = 2.5 Hz, 1H), 6.16 (dd, J = 8.5, 2.5 Hz, 1H), 5.85 (q, J = 1.2 Hz, 2H), 4.40 (d, J = 13.3 Hz, 1H), 4.22 (d, J = 3.3 Hz, 1H), 3.87 (s, 3H), 3.61 (dd, J = 9.8, 2.8 Hz, 1H), 3.50 (dd, J = 9.9, 6.6 Hz, 1H), 2.82 (td, J = 11.7, 3.8 Hz, 3H), 2.11 – 1.97 (m, 1H), 1.86 – 1.77 (m, 1H), 1.76 – 1.64 (m, 1H), 1.49 (s, 9H).
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-1-(tert-butoxycarbonyl)piperidin-4-yl)-2-fluorobenzoic acid (12a).
To a round bottom flask equipped with a stir bar was added (3S,4R)-tert-butyl 3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-(methoxycarbonyl)phenyl)piperidine-1-carboxylate (0.297g, 0.608 mmol), 1M NaOH (1.83 mL, 1.825 mmol), H2O (5 mL), and Methanol (12 mL). The reaction was stirred overnight at room temperature. Ether was added to the reaction and the resulting layers were separated. To the aqueous layer, 10% citric acid was added to give a pH of 4. The aqueous layer was then extracted 2x with ethyl acetate. The ethyl acetate layers were then combined and washed 1x with NaCl, dried with MgSO4, and concentrated to give, with no further purification, as an amorphous solid 5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-1-(tert-butoxycarbonyl)piperidin-4-yl)-2-fluorobenzoic acid (0.226g, 0.477 mmol, 79% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.77 (dd, J = 7.0, 2.4 Hz, 1H), 7.47 (ddd, J = 8.6, 4.5, 2.5 Hz, 1H), 7.13 (dd, J = 10.7, 8.5 Hz, 1H), 6.61 (d, J = 8.5 Hz, 1H), 6.36 (d, J = 2.4 Hz, 1H), 6.16 (dd, J = 8.5, 2.5 Hz, 1H), 5.84 (s, 2H), 4.41 (d, J = 13.4 Hz, 1H), 4.20 (d, J = 13.3 Hz, 1H), 3.61 (dd, J = 9.8, 3.0 Hz, 1H), 3.52 (dd, J = 9.9, 6.7 Hz, 1H), 2.87 (s, 1H), 2.81 (td, J = 11.7, 3.9 Hz, 2H), 2.05 (ddd, J = 15.0, 7.1, 4.0 Hz, 1H), 1.81 (dd, J = 13.5, 3.7 Hz, 1H), 1.70 (qd, J = 12.7, 4.6 Hz, 1H), 1.49 (s, 9H).
Tert-butyl (3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-(methylcarbamoyl)phenyl)piperidine-1-carboxylate. (13aa).
5-((3S,4R)-3-((benzo[d][1,3] dioxol-5-yloxy)methyl)-1-(tert-butoxycarbonyl)piperidin-4-yl)-2-fluorobenzoic acid (0.08 g, 0.169 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (0.073 g, 0.338 mmol), N,N-diisopropylethylamine (0.131 mL, 0.676 mmol), hydroxybenzotriazole (0.051g, 0.338 mmol), and 2M methylamine in THF (0.169 mL, 0.338 mmol) were added to THF (5 mL) in a 15 mL round bottom flask and stirred overnight at room temperature. The resulting solution was diluted with ethyl acetate and saturated sodium bicarbonate, and then the layers were separated. The organic layer was then washed with brine (2x), dried with MgSO4, and concentrated in vacuo. The crude product was purified using flash chromatography using 30% - 90% EtOAc/Hexanes to give as an amorphous solid tert-butyl (3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-(methylcarbamoyl)phenyl)piperidine-1-carboxylate (0.070 g, 0.144 mmol, 85% yield). 1H NMR (400 MHz, Chloroform-d) δ 7.94 (dd, J = 7.6, 2.4 Hz, 1H), 7.30 – 7.22 (m, 1H), 7.04 (dd, J = 11.8, 8.4 Hz, 1H), 6.78 – 6.65 (m, 1H), 6.62 (d, J = 8.4 Hz, 1H), 6.33 (t, J = 1.8 Hz, 1H), 6.12 (dt, J = 8.6, 1.8 Hz, 1H), 5.88 (d, J = 1.3 Hz, 2H), 4.44 (s, 1H), 4.24 (s, 2H), 3.58 (dd, J = 9.4, 2.8 Hz, 1H), 3.44 (dd, J = 9.4, 6.7 Hz, 1H), 3.03 (d, J = 4.0 Hz, 3H), 2.76 (q, J = 12.5, 11.8 Hz, 3H), 2.15 – 2.05 (m, 1H), 1.74 (dq, J = 17.3, 13.5, 13.0 Hz, 2H), 1.50 (d, J = 1.3 Hz, 9H).
Tert-butyl (3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(3-(benzylcarbamoyl)-4-fluorophenyl)piperidine-1-carboxylate (13ab).
Compound 13ab was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with benzylamine (59% yield). 1H NMR (500 MHz, Chloroform-d) δ 7.97 (dd, J = 7.5, 2.5 Hz, 1H), 7.38 – 7.34 (m, 3H), 7.29 (ddt, J = 8.7, 6.4, 3.4 Hz, 2H), 7.03 (ddd, J = 14.9, 10.0, 4.2 Hz, 2H), 6.62 (d, J = 8.4 Hz, 1H), 6.34 (d, J = 2.5 Hz, 1H), 6.13 (dd, J = 8.5, 2.5 Hz, 1H), 5.87 (s, 2H), 4.68 (d, J = 5.6 Hz, 2H), 4.45 (s, 1H), 4.25 (s, 1H), 3.59 (dd, J = 9.4, 2.8 Hz, 1H), 3.45 (dd, J = 9.5, 6.5 Hz, 1H), 2.77 (dt, J = 23.6, 11.9 Hz, 3H), 2.10 (ddq, J = 11.5, 7.7, 4.0 Hz, 1H), 1.80 (d, J = 13.2 Hz, 1H), 1.76 – 1.68 (m, 1H), 1.50 (s, 9H).
Tert-butyl (3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-(phenethylcarbamoyl)phenyl)piperidine-1-carboxylate (13ac).
Compound 13ac was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with 2-phenylethylamine (77% yield). 1H NMR (500 MHz, Chloroform-d) δ 7.93 (dd, J = 7.5, 2.4 Hz, 1H), 7.37 – 7.30 (m, 2H), 7.29 – 7.23 (m, 4H), 7.01 (dd, J = 11.6, 8.4 Hz, 1H), 6.80 – 6.71 (m, 1H), 6.63 (d, J = 8.4 Hz, 1H), 6.35 (d, J = 2.5 Hz, 1H), 6.14 (dd, J = 8.5, 2.5 Hz, 1H), 5.88 (s, 2H), 4.46 (s, 1H), 4.25 (s, 1H), 3.74 (q, J = 5.9 Hz, 2H), 3.59 (dd, J = 9.5, 2.9 Hz, 1H), 3.45 (dd, J = 9.5, 6.6 Hz, 1H), 2.94 (t, J = 7.0 Hz, 2H), 2.87 – 2.69 (m, 3H), 2.10 (tdt, J = 10.4, 7.0, 3.0 Hz, 1H), 1.83 – 1.67 (m, 2H), 1.51 (s, 9H).
(3S,4R)-tert-butyl −3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(3-((2,6-difluorobenzyl)carbamoyl)-4-fluorophenyl)piperidine-1-carboxylate. (13ad)
Compound 13ad was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with 2,6-difluorobenzylamine (24% yield). 1H NMR (CDCl3, 400 MHz) δ 7.95 (dd, J = 7.5, 2.4 Hz, 1H), 7.35 – 7.20 (m, 2H), 7.16–7.13 (m, 1H), 7.01 (dd, J = 11.8, 8.4 Hz, 1H), 6.94 – 6.75 (m, 2H), 6.60 (d, J = 8.5 Hz, 1H), 6.32 (d, J = 2.4 Hz, 1H), 6.11 (dd, J = 8.5, 2.5 Hz, 1H), 5.87 (s, 2H), 4.46 (br s, 1H), 4.19 (br s, 1H), 3.56 (dd, J = 9.5, 2.8 Hz, 1H), 3.42 (dd, J = 9.4, 6.6 Hz, 1H), 2.80 – 2.69 (m, 3H), 2.07 (m, 2H), 1.74– 1.69 (m, 3H), 1.49 (s, 9H).
(3S,4R)-tert-butyl −3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(3-((2,6-difluorophenethyl)carbamoyl)-4-fluorophenyl)piperidine-1-carboxylate (13ae).
Compound 13ae was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with 2-(2,6-difluorophenyl)ethan-1-amine (87% yield). 1H NMR (CDCl3, 400 MHz) δ 7.90 (dd, J = 7.5, 2.4 Hz, 1H), 7.24 – 7.07 (m, 2H), 7.00 (dd, J = 11.6, 8.4 Hz, 1H), 6.95 – 6.65 (m, 4H), 6.60 (d, J = 8.5 Hz, 1H), 6.32 (d, J = 2.4 Hz, 1H), 6.11 (dd, J = 8.5, 2.5 Hz, 1H), 5.86 (s, 2H), 4.41 (br s, 1H), 4.22 (br s, 1H), 3.70 (q, J = 6.9 Hz, 1H), 3.56 (m, 1H), 3.49 – 3.28 (m, 1H), 3.02 (t, J = 6.8 Hz, 2H), 2.79 – 2.68 (m, 2H), 2.10–2.04 (m, 1H), 1.87 – 1.65 (m, 2H), 1.48 (s, 9H).
tert-butyl (3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-((2-(trifluoromethyl)benzyl)carbamoyl)phenyl)piperidine-1-carboxylate (13af).
Compound 13af was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with (2-(trifluoromethyl)phenyl)methanamine (28% yield). Yellow oil. 1H NMR (400 MHz, Chloroform-d) δ 7.91 (m, 1H), 7.65 (t, J = 6.2 Hz, 2H), 7.52 (t, J = 7.6 Hz, 1H), 7.38 (t, J = 7.7 Hz, 1H), 7.25–7.23 (m, 3H), 7.12 – 6.92 (m, 2H), 6.20 (br s, 1H), 5.87 (m, 2H), 5.09–4.96 (m, 2H), 4.83 (d, J = 5.9 Hz, 2H), 4.26 (br s, 1H), 3.64 (m, 2H), 2.71–2.61 (m, 2H), 1.75 (m, 1H), 1.49 (s, 9H).
(3S,4R)-tert-butyl −3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(3-((2,6-dimethylbenzyl)carbamoyl)-4-fluorophenyl)piperidine-1-carboxylate (13ag).
Compound 13ag was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with 2,6-dimethylbenzylamine (75% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.48 (dd, J = 6.8, 2.3 Hz, 1H), 7.35 (ddd, J = 8.5, 4.9, 2.4 Hz, 1H), 7.13 – 6.98 (m, 4H), 6.59 (d, J = 8.5 Hz, 1H), 6.34 (d, J = 2.5 Hz, 1H), 6.14 (dd, J = 8.5, 2.5 Hz, 1H), 5.83 (q, J = 1.2 Hz, 2H), 4.60 (s, 2H), 4.40 (d, J = 13.3 Hz, 1H), 4.18 (dt, J = 13.5, 2.4 Hz, 1H), 3.58 (dd, J = 9.8, 2.9 Hz, 1H), 3.49 (dd, J = 9.8, 6.9 Hz, 1H), 2.89 (s, 1H), 2.77 (td, J = 11.7, 3.9 Hz, 2H), 2.38 (s, 6H), 2.04 (dtq, J = 11.0, 7.0, 3.5 Hz, 1H), 1.82 – 1.61 (m, 2H), 1.48 (s, 8H).
(3S,4R)-tert-butyl 3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(3-((2,6-dichlorobenzyl)carbamoyl)-4-fluorophenyl)piperidine-1-carboxylate (13ah).
Compound 13ah was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with 2,6-dichlorobenzylamine (41% yield). 1H NMR (CD3OD, 400 MHz) δ 7.52 (dd, J = 6.9, 2.4 Hz, 1H), 7.45–7.33 (m, 3H), 7.29 (dd, J = 8.7, 7.4 Hz, 1H), 7.10 (dd, J = 10.5, 8.5 Hz, 1H), 6.59 (d, J = 8.4 Hz, 1H), 6.34 (d, J = 2.4 Hz, 1H), 6.14 (dd, J = 8.5, 2.5 Hz, 1H), 5.84 (q, J = 1.2 Hz, 2H), 4.86 (s, 2H), 4.40 (d, J = 13.2 Hz, 1H), 4.23–4.14 (m, 1H), 3.59 (dd, J = 9.8, 3.0 Hz, 1H), 3.49 (dd, J = 9.9, 6.9 Hz, 1H), 2.98–2.72 (m, 3H), 2.10–1.99 (m, 1H), 1.82–1.74 (m, 1H), 1.68 (qd, J = 12.6, 4.5 Hz, 1H), 1.49 (s, 9H).
(3S,4R)-tert-butyl 3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(3-((2,6-dimethoxybenzyl)carbamoyl)-4-fluorophenyl)piperidine-1-carboxylate (13ai).
Compound 13ai was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with 2,6-dimethoxybenzylamine (71% yield). 1H NMR (400 MHz, DMSO-d6) δ 12.86 (s, 1H), 7.99 (q, J = 4.8 Hz, 1H), 7.86 (s, 1H), 7.51 (dd, J = 7.0, 2.3 Hz, 1H), 7.44 – 7.36 (m, 2H), 7.25 (t, J = 8.3 Hz, 1H), 7.16 (dd, J = 10.6, 8.5 Hz, 1H), 6.92 – 6.86 (m, 2H), 6.66 (d, J = 8.4 Hz, 2H), 4.48 (d, J = 5.0 Hz, 2H), 4.36 (s, 1H), 4.09 – 4.04 (m, 1H), 3.78 (s, 6H), 3.64 – 3.56 (m, 2H), 2.82 (s, 0H), 2.71 (qd, J = 11.9, 2.8 Hz, 1H), 2.11 (dt, J = 8.9, 4.4 Hz, 1H), 1.71 (d, J = 11.3 Hz, 1H), 1.61 (dd, J = 13.8, 9.5 Hz, 1H), 1.42 (s, 9H).
(3S,4R)-tert-butyl-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-((2-methoxybenzyl)carbamoyl)phenyl)piperidine-1-carboxylate (13aj).
Compound 13aj was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with 2-methoxy benzylamine (71% yield). 1H NMR (CDCl3, 400 MHz) δ 8.71 (s, 1H), 8.05 – 7.82 (m, 1H), 7.43 (dd, J = 13.1, 6.0 Hz, 1H), 7.33 (dd, J = 7.4, 1.7 Hz, 1H), 7.29 – 7.13 (m, 2H), 7.00 (t, J = 10.0 Hz, 1H), 6.96 – 6.78 (m, 2H), 6.19 (s, 1H), 5.98 – 5.75 (m, 2H), 5.58 (dd, J = 5.9, 2.0 Hz, 1H), 4.97 (s, 2H), 4.65 (d, J = 5.7 Hz, 2H), 3.93 – 3.78 (m, 3H), 3.74 – 3.53 (m, 2H), 2.70 (d, J = 45.6 Hz, 3H), 1.67 – 1.53 (m, 1H), 1.36 – 0.97 (m, 9H).
(3S,4R)-tert-butyl 3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-((pyridin-2-ylmethyl)carbamoyl)phenyl)piperidine-1-carboxylate (13ak).
Compound 13ak was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with pyridin-2-ylmethanamine (74% yield). 1H NMR (400 MHz, Chloroform-d) δ 8.56 (ddd, J = 5.0, 1.8, 1.0 Hz, 1H), 8.04 – 7.91 (m, 2H), 7.67 (td, J = 7.7, 1.7 Hz, 1H), 7.32 (d, J = 7.9 Hz, 1H), 7.30 – 7.26 (m, 1H), 7.20 (ddd, J = 7.6, 4.9, 1.1 Hz, 1H), 7.05 (dd, J = 11.5, 8.4 Hz, 1H), 6.60 (d, J = 8.5 Hz, 1H), 6.33 (d, J = 2.4 Hz, 1H), 6.11 (dd, J = 8.5, 2.4 Hz, 1H), 5.85 (s, 2H), 4.78 (d, J = 4.8 Hz, 2H), 4.44 (s, 1H), 4.23 (s, 1H), 3.58 (dd, J = 9.6, 2.8 Hz, 1H), 3.44 (dd, J = 9.5, 6.6 Hz, 1H), 2.90 – 2.68 (m, 3H), 2.16 – 2.04 (m, 1H), 1.83 – 1.65 (m, 2H), 1.48 (s, 9H).
Tert-butyl (3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-((pyridin-3-ylmethyl)carbamoyl)phenyl)piperidine-1-carboxylate (13al).
Compound 13al was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with 3-picoylamine (94% yield). 1H NMR (500 MHz, Chloroform-d) δ 8.62 (d, J = 2.3 Hz, 1H), 8.55 (dd, J = 4.7, 1.6 Hz, 1H), 7.96 (dd, J = 7.5, 2.4 Hz, 1H), 7.71 (d, J = 7.7 Hz, 1H), 7.29 (td, J = 7.9, 4.9 Hz, 2H), 7.12 – 7.01 (m, 2H), 6.62 (d, J = 8.4 Hz, 1H), 6.34 (d, J = 2.4 Hz, 1H), 6.13 (dd, J = 8.4, 2.5 Hz, 1H), 5.87 (s, 2H), 4.69 (d, J = 5.9 Hz, 2H), 4.44 (s, 1H), 4.24 (s, 1H), 3.59 (dd, J = 9.4, 2.8 Hz, 1H), 3.45 (dd, J = 9.5, 6.5 Hz, 1H), 2.78 (dt, J = 22.3, 13.0 Hz, 3H), 2.14 – 2.06 (m, 1H), 1.84 – 1.69 (m, 2H), 1.50 (s, 9H).
Tert-butyl (3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-((pyridin-4-ylmethyl)carbamoyl)phenyl)piperidine-1-carboxylate (13am).
Compound 13am was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with 4-(aminomethyl)pyridine (94% yield). 1H NMR (500 MHz, Chloroform-d) δ 8.60 – 8.53 (m, 2H), 7.96 (dd, J = 7.5, 2.4 Hz, 1H), 7.32 (ddd, J = 7.9, 4.8, 2.4 Hz, 1H), 7.28 – 7.23 (m, 2H), 7.21 (dt, J = 12.7, 6.0 Hz, 1H), 7.07 (dd, J = 11.7, 8.5 Hz, 1H), 6.62 (d, J = 8.4 Hz, 1H), 6.34 (d, J = 2.4 Hz, 1H), 6.13 (dd, J = 8.5, 2.5 Hz, 1H), 5.87 (s, 2H), 4.68 (d, J = 6.0 Hz, 2H), 4.44 (s, 1H), 4.25 (s, 1H), 3.59 (dd, J = 9.5, 2.8 Hz, 1H), 3.45 (dd, J = 9.5, 6.4 Hz, 1H), 2.87 – 2.70 (m, 3H), 2.09 (dtt, J = 10.4, 6.9, 3.4 Hz, 1H), 1.85 – 1.67 (m, 2H), 1.50 (s, 9H).
Tert-butyl (3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-((2-(pyridin-2-yl)ethyl)carbamoyl)phenyl)piperidine-1-carboxylate (13an).
Compound 13an was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with 2-(2-aminoethyl)pyridine (66% yield). 1H NMR (500 MHz, Chloroform-d) δ 8.56 (dt, J = 5.1, 1.2 Hz, 1H), 7.91 (dd, J = 7.4, 2.4 Hz, 1H), 7.70 – 7.63 (m, 1H), 7.62 (td, J = 7.6, 1.8 Hz, 1H), 7.25 – 7.22 (m, 1H), 7.19 (d, J = 7.8 Hz, 1H), 7.16 (ddd, J = 7.7, 4.9, 1.1 Hz, 1H), 7.00 (dd, J = 11.5, 8.4 Hz, 1H), 6.61 (d, J = 8.5 Hz, 1H), 6.33 (d, J = 2.4 Hz, 1H), 6.12 (dd, J = 8.5, 2.5 Hz, 1H), 5.87 (s, 2H), 4.44 (s, 1H), 4.24 (s, 1H), 3.90 (q, J = 5.9 Hz, 2H), 3.58 (dd, J = 9.5, 2.9 Hz, 1H), 3.44 (dd, J = 9.4, 6.7 Hz, 1H), 3.10 (t, J = 6.4 Hz, 2H), 2.82 – 2.68 (m, 3H), 2.09 (dtd, J = 11.0, 7.5, 3.4 Hz, 1H), 1.80 – 1.66 (m, 2H), 1.50 (s, 9H).
Tert-butyl (3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-((2-(pyridin-3-yl)ethyl)carbamoyl)phenyl)piperidine-1-carboxylate (13ao).
Compound 13ao was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with 3-(2-aminoethyl)pyridine (82% yield). 1H NMR (500 MHz, Chloroform-d) δ 8.52 – 8.48 (m, 2H), 7.92 (dd, J = 7.5, 2.4 Hz, 1H), 7.58 (dt, J = 7.8, 2.0 Hz, 1H), 7.30 – 7.27 (m, 1H), 7.02 (dd, J = 11.7, 8.4 Hz, 1H), 6.76 (dt, J = 12.4, 5.8 Hz, 1H), 6.62 (d, J = 8.5 Hz, 1H), 6.34 (d, J = 2.4 Hz, 1H), 6.13 (dd, J = 8.5, 2.5 Hz, 1H), 5.87 (s, 2H), 4.44 (s, 1H), 4.24 (s, 1H), 3.73 (q, J = 6.8 Hz, 2H), 3.58 (dd, J = 9.5, 2.9 Hz, 1H), 3.44 (dd, J = 9.5, 6.5 Hz, 1H), 2.95 (t, J = 7.1 Hz, 2H), 2.88 – 2.69 (m, 3H), 2.09 (dtt, J = 10.8, 7.3, 3.5 Hz, 1H), 1.83 – 1.67 (m, 2H), 1.50 (s, 9H).
Tert-butyl (3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-((2-(pyridin-4-yl)ethyl)carbamoyl)phenyl)piperidine-1-carboxylate (13ap).
Compound 13ap was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with 4-(2-aminoethyl)pyridine (79% yield). 1H NMR (500 MHz, Chloroform-d) δ 8.56 – 8.52 (m, 2H), 7.92 (dd, J = 7.5, 2.4 Hz, 1H), 7.31 – 7.25 (m, 4H), 7.18 – 7.15 (m, 2H), 7.02 (dd, J = 11.7, 8.5 Hz, 1H), 6.76 (dt, J = 12.4, 5.8 Hz, 1H), 6.62 (d, J = 8.5 Hz, 1H), 6.34 (d, J = 2.5 Hz, 1H), 6.13 (dd, J = 8.5, 2.5 Hz, 1H), 5.87 (s, 2H), 4.44 (s, 1H), 4.24 (s, 1H), 3.75 (q, J = 6.8 Hz, 2H), 3.58 (dd, J = 9.5, 2.9 Hz, 1H), 3.49 (d, J = 4.9 Hz, 1H), 3.44 (dd, J = 9.5, 6.5 Hz, 1H), 2.95 (t, J = 7.0 Hz, 2H), 2.86 – 2.69 (m, 3H), 2.08 (dtd, J = 13.1, 6.8, 5.9, 3.2 Hz, 1H), 1.79 (d, J = 13.7 Hz, 1H), 1.76 – 1.66 (m, 1H), 1.50 (s, 9H).
Tert-butyl (3S,4R)-4-(3-(((1H-imidazol-2-yl)methyl)carbamoyl)-4-fluorophenyl)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidine-1-carboxylate (13aq).
Compound 13aq was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with 1H-imidazole-2-methanamine dihydrochloride (66% yield). 1H NMR (400 MHz, Chloroform-d) δ 7.93 (dd, J = 7.4, 2.4 Hz, 1H), 7.64 (dd, J = 12.7, 6.1 Hz, 1H), 7.30 (ddd, J = 7.9, 4.8, 2.4 Hz, 1H), 7.05 (dd, J = 11.7, 8.5 Hz, 1H), 6.98 (s, 2H), 6.61 (d, J = 8.4 Hz, 1H), 6.34 (d, J = 2.5 Hz, 1H), 6.12 (dd, J = 8.5, 2.5 Hz, 1H), 5.88 (d, J = 1.2 Hz, 2H), 4.66 (d, J = 5.8 Hz, 2H), 4.45 (s, 1H), 4.25 (s, 1H), 3.59 (dd, J = 9.6, 2.8 Hz, 1H), 3.43 (dd, J = 9.5, 6.3 Hz, 1H), 2.88 – 2.71 (m, 3H), 2.15 – 2.00 (m, 1H), 1.87 – 1.60 (m, 2H), 1.50 (s, 9H).
Tert-butyl (3S,4R)-4-(3-((2-(1H-imidazol-4-yl)ethyl)carbamoyl)-4-fluorophenyl)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidine-1-carboxylate (13ar).
Compound 13ar was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with histamine (86% yield). 1H NMR (400 MHz, Chloroform-d) δ 7.87 (dd, J = 7.4, 2.4 Hz, 1H), 7.59 (s, 1H), 7.35 – 7.28 (m, 1H), 7.24 (dq, J = 7.2, 2.4 Hz, 1H), 7.01 (dd, J = 11.4, 8.5 Hz, 1H), 6.85 (s, 1H), 6.61 (d, J = 8.4 Hz, 1H), 6.33 (d, J = 2.4 Hz, 1H), 6.12 (dd, J = 8.5, 2.5 Hz, 1H), 5.87 (s, 2H), 4.44 (s, 1H), 4.24 (s, 1H), 3.76 (q, J = 6.3 Hz, 2H), 3.57 (dd, J = 9.5, 2.8 Hz, 1H), 3.43 (dd, J = 9.5, 6.6 Hz, 1H), 2.92 (t, J = 6.5 Hz, 2H), 2.88 – 2.66 (m, 3H), 2.14 – 2.03 (m, 1H), 1.82 – 1.64 (m, 2H), 1.50 (s, 9H).
Tert-butyl (3S,4R)-4-(3-(((1H-pyrazol-5-yl)methyl)carbamoyl)-4-fluorophenyl)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidine-1-carboxylate (13as).
Compound 13as was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with 2H-pyrazol-3-ylmethylamine hydrochloride (69% yield). 1H NMR (400 MHz, Chloroform-d) δ 10.71 (bs, IH) 7.95 (dd, J = 7.5, 2.4 Hz, 1H), 7.52 (d, J = 2.1 Hz, 1H), 7.37 (dt, J = 11.8, 5.5 Hz, 1H), 7.28 (d, J = 5.7 Hz, 1H), 7.03 (dd, J = 11.6, 8.4 Hz, 1H), 6.61 (d, J = 8.5 Hz, 1H), 6.34 (d, J = 2.5 Hz, 1H), 6.28 (d, J = 2.1 Hz, 1H), 6.12 (dd, J = 8.5, 2.5 Hz, 1H), 5.87 (q, J = 1.4 Hz, 2H), 4.73 – 4.65 (m, 2H), 4.45 (s, 1H), 4.25 (s, 1H), 3.58 (dd, J = 9.5, 2.8 Hz, 1H), 3.43 (dd, J = 9.5, 6.5 Hz, 1H), 2.85 – 2.68 (m, 3H), 2.09 (td, J = 7.7, 7.0, 3.8 Hz, 1H), 1.73 (dq, J = 18.5, 14.0 Hz, 2H), 1.50 (s, 9H).
Tert-butyl (3S,4R)-4-(3-((2-(1H-pyrazol-3-yl)ethyl)carbamoyl)-4-fluorophenyl)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidine-1-carboxylate (13at)
Compound 13at was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with 2-(1H-pyrazol-3-yl)ethan-1-amine (64% yield). 1H NMR (500 MHz, Chloroform-d) δ 10.78 (s, 1H), 7.90 (dd, J = 7.5, 2.4 Hz, 1H), 7.52 (d, J = 2.2 Hz, 1H), 7.25 (ddd, J = 8.4, 4.8, 2.5 Hz, 1H), 7.17 (dt, J = 11.7, 5.7 Hz, 1H), 7.00 (dd, J = 11.5, 8.4 Hz, 1H), 6.61 (d, J = 8.5 Hz, 1H), 6.33 (d, J = 2.5 Hz, 1H), 6.17 (d, J = 2.2 Hz, 1H), 6.12 (dd, J = 8.5, 2.5 Hz, 1H), 5.89 – 5.85 (m, 4H), 4.44 (s, 1H), 4.24 (s, 1H), 3.79 (q, J = 6.3, 5.9 Hz, 2H), 3.58 (dd, J = 9.5, 2.9 Hz, 1H), 3.43 (dd, J = 9.5, 6.6 Hz, 1H), 3.01 (t, J = 6.6 Hz, 2H), 2.85 – 2.68 (m, 3H), 2.14 – 2.02 (m, 1H), 1.74 (dtd, J = 24.7, 13.0, 3.9 Hz, 2H), 1.50 (s, 9H), 8.04 – 7.99 (m, 1H).
Tert-butyl (3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-(((5-methyl-1H-pyrazol-3-yl)methyl)carbamoyl)phenyl)piperidine-1-carboxylate (13au).
Compound 13au was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with (5-methyl-1H-pyrazol-3-yl)methanamine (91% yield). 1H NMR (400 MHz, Chloroform-d) δ 9.71 (d, J = 105.6 Hz, 1H), 7.96 (dd, J = 7.4, 2.4 Hz, 1H), 7.33 – 7.27 (m, 2H), 7.03 (dd, J = 11.7, 8.5 Hz, 1H), 6.62 (d, J = 8.4 Hz, 1H), 6.34 (d, J = 2.5 Hz, 1H), 6.12 (dd, J = 8.5, 2.5 Hz, 1H), 6.03 (s, 1H), 5.88 (s, 2H), 4.62 (d, J = 5.3 Hz, 2H), 4.45 (s, 1H), 4.25 (s, 1H), 3.58 (dd, J = 9.5, 2.8 Hz, 1H), 3.44 (dd, J = 9.5, 6.6 Hz, 1H), 2.76 (dt, J = 19.9, 10.7 Hz, 3H), 2.29 (s, 3H), 2.09 (dp, J = 7.5, 4.2 Hz, 1H), 1.84 – 1.62 (m, 2H), 1.50 (s, 8H).
Tert-butyl (3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-(((1-methyl-1H-pyrazol-3-yl)methyl)carbamoyl)phenyl)piperidine-1-carboxylate (13av).
Compound 13av was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with (1-methyl-1H-pyrazol-3-yl)methanamine (78% yield). 1H NMR (500 MHz, Chloroform-d) δ 7.96 (dd, J = 7.5, 2.5 Hz, 1H), 7.30 (d, J = 2.2 Hz, 1H), 7.30 – 7.18 (m, 2H), 7.02 (dd, J = 11.6, 8.4 Hz, 1H), 6.61 (d, J = 8.4 Hz, 1H), 6.34 (d, J = 2.5 Hz, 1H), 6.22 (d, J = 2.2 Hz, 1H), 6.12 (dd, J = 8.5, 2.5 Hz, 1H), 5.87 (s, 2H), 4.66 (dd, J = 5.3, 1.2 Hz, 2H), 4.45 (s, 1H), 4.24 (s, 1H), 3.87 (s, 3H), 3.58 (dd, J = 9.5, 2.9 Hz, 1H), 3.44 (dd, J = 9.5, 6.6 Hz, 1H), 2.85 – 2.68 (m, 3H), 2.09 (dtd, J = 11.4, 7.0, 2.9 Hz, 1H), 1.87 – 1.66 (m, 1H), 1.50 (s, 9H).
Tert-butyl (3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-(((1-methyl-1H-pyrazol-5-yl)methyl)carbamoyl)phenyl)piperidine-1-carboxylate (13aw)
Compound 13aw was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with (1-methyl-1H-pyrazol-5-yl)methanamine (67% yield). 1H NMR (500 MHz, Chloroform-d) δ 7.94 (dd, J = 7.5, 2.4 Hz, 1H), 7.42 (d, J = 1.8 Hz, 1H), 7.30 (ddd, J = 7.8, 4.8, 2.4 Hz, 1H), 7.05 (dd, J = 11.6, 8.4 Hz, 1H), 6.91 (dt, J = 12.2, 5.5 Hz, 1H), 6.62 (d, J = 8.7 Hz, 1H), 6.33 (dd, J = 2.5, 0.8 Hz, 1H), 6.23 (d, J = 1.8 Hz, 1H), 6.13 (dd, J = 8.5, 1.7 Hz, 1H), 5.87 (s, 2H), 4.71 (d, J = 5.4 Hz, 2H), 4.44 (s, 1H), 4.23 (d, J = 19.4 Hz, 1H), 3.89 (d, J = 0.8 Hz, 3H), 3.59 (dd, J = 9.5, 2.8 Hz, 1H), 3.44 (dd, J = 9.5, 6.4 Hz, 1H), 2.85 – 2.71 (m, 3H), 2.09 (dtq, J = 10.6, 6.5, 3.4 Hz, 1H), 1.76 (dddd, J = 20.3, 17.1, 12.8, 3.8 Hz, 3H), 1.50 (d, J = 0.8 Hz, 9H).
Tert-butyl (3S,4R)-4-(3-(((1H-pyrazol-4-yl)methyl)carbamoyl)-4-fluorophenyl)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidine-1-carboxylate (13ax)
Compound 13ax was synthesized as described for intermediate 13aa from intermediate 12a replacing methylamine with (1H-pyrazol-4-yl)methanamine (41% yield). 1H NMR (500 MHz, Chloroform-d) δ 7.94 (dd, J = 7.5, 2.4 Hz, 1H), 7.63 – 7.61 (m, 2H), 7.28 – 7.25 (m, 1H), 7.02 (dd, J = 11.6, 8.4 Hz, 1H), 6.94 (dt, J = 12.0, 5.5 Hz, 1H), 6.61 (d, J = 8.4 Hz, 1H), 6.33 (d, J = 2.5 Hz, 1H), 6.12 (dd, J = 8.5, 2.5 Hz, 1H), 5.87 (s, 2H), 4.57 (d, J = 4.7 Hz, 1H), 4.44 (s, 1H), 4.24 (s, 1H), 3.58 (dd, J = 9.5, 2.8 Hz, 1H), 3.44 (dd, J = 9.5, 6.5 Hz, 1H), 2.85 – 2.70 (m, 3H), 2.13 – 2.04 (m, 1H), 1.83 – 1.65 (m, 2H), 1.50 (s, 9H).
(3S,4R)-tert-butyl 4-(3-((2-(1H-pyrazol-4-yl)ethyl)carbamoyl)-4-fluorophenyl)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidine-1-carboxylate (13ay).
Compound 13ay was synthesized as described for intermediate 13aa from intermediate 12a replacing pyridin-2-ylmethanamine with 2-(1H-pyrazol-4-yl)ethan-1-amine (36% yield). 1H NMR (500 MHz, Methanol-d4) δ 7.55 (dd, J = 6.9, 2.4 Hz, 1H), 7.50 (s, 2H), 7.39 (ddd, J = 8.5, 4.8, 2.4 Hz, 1H), 7.13 (dd, J = 10.6, 8.5 Hz, 1H), 6.61 (d, J = 8.5 Hz, 1H), 6.36 (d, J = 2.5 Hz, 1H), 6.16 (dd, J = 8.5, 2.5 Hz, 1H), 5.84 (s, 2H), 4.42 (d, J = 13.2 Hz, 1H), 4.25 – 4.16 (m, 1H), 3.60 (dd, J = 9.8, 3.0 Hz, 1H), 3.57 – 3.51 (m, 3H), 2.86 (bs, 2H), 2.83 – 2.78 (m, 3H), 2.06 (dtq, J = 11.0, 7.1, 3.8 Hz, 1H), 1.84 – 1.77 (m, 1H), 1.71 (qd, J = 12.6, 4.4 Hz, 1H), 1.49 (s, 9H).
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluoro-N-methylbenzamide (14aa).
Tert-butyl (3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-(methylcarbamoyl)phenyl)piperidine-1-carboxylate (13aa) (0.07 g, 0.144 mmol) was dissolved in 1,4-dioxanes (2 mL) followed by 4M HCl/1,4-dioxanes (2 mL) and stirred at room temperature for two hours. The reaction was concentrated in vacuo, then purified using 0–20% MeOH (3 M ammonia)/dichloromethane to give 5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluoro-N-methylbenzamide as a white solid (0.025 g, 0.064 mmol, 44% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.61 (dd, J = 7.0, 2.3 Hz, 1H), 7.40 (d, J = 9.1 Hz, 1H), 7.13 (dd, J = 10.7, 8.5 Hz, 1H), 6.60 (d, J = 8.5 Hz, 1H), 6.33 (d, J = 2.5 Hz, 1H), 6.14 (dd, J = 8.5, 2.5 Hz, 1H), 5.84 (s, 2H), 3.70 – 3.64 (m, 1H), 3.56 (dt, J = 9.0, 3.9 Hz, 2H), 3.49 (dd, J = 9.7, 6.5 Hz, 1H), 3.39 – 3.32 (m, 1H), 2.91 (s, 3H), 2.76 (dd, J = 11.9, 5.0 Hz, 2H), 2.13 (bs, 1H), 1.85 – 1.73 (m, 2H). MS (ESI+) m/z: 387.2 (M+1). HPLC purity: 95%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-N-benzyl-2-fluorobenzamide (CCG258202) (14ab).
Prepared from Intermediate 13ab as described for compound 14aa (76% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.61 (dd, J = 6.9, 2.4 Hz, 1H), 7.41 (ddd, J = 8.5, 4.8, 2.4 Hz, 1H), 7.37 – 7.28 (m, 4H), 7.27 – 7.22 (m, 1H), 7.15 (dd, J = 10.6, 8.5 Hz, 1H), 6.59 (d, J = 8.5 Hz, 1H), 6.35 (d, J = 2.5 Hz, 1H), 6.14 (dd, J = 8.5, 2.5 Hz, 1H), 5.83 (d, J = 1.3 Hz, 2H), 4.57 (s, 2H), 3.59 (dd, J = 9.7, 3.0 Hz, 1H), 3.50 (dd, J = 9.7, 6.5 Hz, 1H), 3.42 (dd, J = 12.5, 3.8 Hz, 1H), 3.23 (d, J = 12.5 Hz, 1H), 2.90 – 2.76 (m, 3H), 2.19 (dtd, J = 16.0, 8.1, 4.0 Hz, 1H), 1.89 – 1.82 (m, 2H). MS (ESI+) m/z: 463.2 (M+1). HPLC purity: 95%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluoro-N-phenethylbenzamide (CCG258201) (14ac).
Prepared from Intermediate 13ac as described for compound 14aa (76% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.54 (dd, J = 7.0, 2.4 Hz, 1H), 7.39 (ddd, J = 8.5, 4.8, 2.4 Hz, 1H), 7.30 – 7.23 (m, 4H), 7.22 – 7.16 (m, 1H), 7.11 (dd, J = 10.6, 8.5 Hz, 1H), 6.60 (d, J = 8.5 Hz, 1H), 6.34 (d, J = 2.4 Hz, 1H), 6.14 (dd, J = 8.5, 2.5 Hz, 1H), 5.82 (s, 2H), 3.62 – 3.54 (m, 3H), 3.47 (dd, J = 9.6, 6.6 Hz, 1H), 3.39 – 3.32 (m, 1H), 3.17 – 3.10 (m, 1H), 2.89 (dd, J = 8.1, 6.7 Hz, 2H), 2.80 – 2.64 (m, 3H), 2.11 (dtd, J = 11.0, 7.3, 3.3 Hz, 1H), 1.83 – 1.71 (m, 2H). MS (ESI+) m/z: 477.2 (M+1). HPLC purity: 95%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-N-(2,6-difluorobenzyl)-2-fluorobenzamide hydrochloride (CCG208947) (14ad).
Prepared from Intermediate 13ad as described for compound 14aa (78 % yield). 1H NMR (CDCl3, 400MHz) δ 7.98 (dd, J = 7.6, 2.4 Hz, 1H), 7.26 (s, 5H), 7.14 (s, 1H), 7.02 (dd, J = 11.8, 8.4 Hz, 1H), 6.91 (t, J = 7.8 Hz, 2H), 6.60 (d, J = 8.4 Hz, 1H), 6.10 (dd, J = 8.5, 2.5 Hz, 1H), 5.86 (s, 2H), 4.77 (d, J = 5.7 Hz, 2H), 3.53– 3.40 (m, 3H), 3.19– 3.16 (m, 1H), 2.70 – 2.63 (m, 2H), 2.12 (s, 1H), 1.77– 1.67 (m, 3H). MS (ESI+) m/z: 499.3 (M+1). HPLC purity: 97%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-N-(2,6-difluorophenethyl)-2-fluorobenzamide (CCG211990) (14ae).
Prepared from Intermediate 13ae as described for compound 14aa (50% yield). 1H NMR (CDCl3, 400MHz) δ 7.98 (dd, J = 7.6, 2.4 Hz, 1H), 7.26 (s, 5H), 7.14 (s, 1H), 7.02 (dd, J = 11.8, 8.4 Hz, 1H), 6.91 (t, J = 7.8 Hz, 2H), 6.60 (d, J = 8.4 Hz, 1H), 6.10 (dd, J = 8.5, 2.5 Hz, 1H), 5.86 (s, 2H), 4.77 (d, J = 5.7 Hz, 2H), 3.53– 3.40 (m, 3H), 3.19– 3.16 (m, 1H), 2.70 – 2.63 (m, 2H), 2.12 (s, 1H), 1.77– 1.67 (m, 3H). MS (ESI+) m/z: 513.4 (M+1). HPLC purity: 95%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluoro-N-(2-(trifluoromethyl)benzyl)benzamide (CCG211991) (14af).
Prepared from Intermediate 13af as described for compound 14aa (83% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.75 (s, 2H), 7.81 – 7.01 (m, 9H), 6.94 (d, J = 11.2 Hz, 2H), 6.37 (s, 2H), 5.91 (d, J = 35.8 Hz, 2H), 4.77 (s, 3H), 4.06 – 3.33 (m, 5H), 2.88 (s, 3H), 2.05 (s, 2H), 1.78 (s, 2H), 1.41 – 0.82 (m, 9H), 5.01 – 4.91 (m, 1H). ESI+MS m/z 530.1 (M+H +) HPLC purity: 96%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-N-(2,6-dimethylbenzyl)-2-fluorobenzamide hydrochloride (CCG232403) (14ag).
Prepared from Intermediate 13ag as described for compound 14aa (92% yield) 1H NMR (500 MHz, Methanol-d4) δ 7.54 (dd, J = 6.7, 2.2 Hz, 1H), 7.41 (ddd, J = 8.5, 4.7, 2.3 Hz, 1H), 7.13 (dd, J = 10.2, 8.5 Hz, 1H), 7.08 (dd, J = 8.6, 6.2 Hz, 1H), 7.02 (d, J = 8.2 Hz, 2H), 6.60 (d, J = 8.4 Hz, 1H), 6.38 (d, J = 2.4 Hz, 1H), 6.17 (dd, J = 8.5, 2.5 Hz, 1H), 5.85 – 5.83 (m, 2H), 4.60 (s, 2H), 3.69 – 3.65 (m, 2H), 3.54 (ddd, J = 12.6, 9.8, 4.2 Hz, 2H), 3.16 (td, J = 12.2, 5.2 Hz, 2H), 3.03 (td, J = 11.2, 5.4 Hz, 1H), 2.45 (dddd, J = 14.6, 11.2, 6.2, 3.2 Hz, 1H), 2.38 (s, 6H), 2.10 – 2.02 (m, 2H). MS (ESI+) m/z: 491.2 (M+1). HPLC purity: 98%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-N-(2,6-dichlorobenzyl)-2-fluorobenzamide (14ah).
Prepared from Intermediate 13ah as described for compound 14aa. 1H NMR (CD3OD, 400 MHz) δ 7.57 (dd, J = 6.9, 2.4 Hz, 1H), 7.45–7.37 (m, 3H), 7.30 (dd, J = 8.6, 7.6 Hz, 1H), 7.15 (dd, J = 10.4, 8.5 Hz, 1H), 6.61 (d, J = 8.5 Hz, 1H), 6.39 (d, J = 2.4 Hz, 1H), 6.17 (dd, J = 8.5, 2.5 Hz, 1H), 5.86 (d, J = 1.2 Hz, 2H), 4.87 (s, 2H), 3.70 – 3.63 (m, 2H), 3.61 – 3.44 (m, 2H), 3.16 (td, J = 12.5, 3.9 Hz, 2H), 3.03 (td, J = 11.5, 4.6 Hz, 1H), 2.41 (tq, J = 8.5, 3.8 Hz, 1H), 2.10–1.96 (m, 2H). MS (ESI+) m/z: 531.0 (M+1). HPLC purity: 98%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-N-(2,6-dimethoxybenzyl)-2-fluorobenzamide hydrochloride (CCG232402) (14ai).
Prepared from Intermediate 13ai as described for compound 14aa. (91% yield) 1H NMR (500 MHz, Methanol-d4) δ 7.67 (dd, J = 7.0, 2.4 Hz, 1H), 7.41 (ddd, J = 8.4, 4.8, 2.4 Hz, 1H), 7.25 (t, J = 8.4 Hz, 1H), 7.15 (dd, J = 11.0, 8.5 Hz, 1H), 6.65 (d, J = 8.3 Hz, 2H), 6.60 (d, J = 8.5 Hz, 1H), 6.37 (d, J = 2.4 Hz, 1H), 6.17 (dd, J = 8.5, 2.5 Hz, 1H), 5.84 (t, J = 1.6 Hz, 2H), 4.66 (d, J = 2.5 Hz, 2H), 3.84 (s, 6H), 3.67 (dd, J = 9.6, 3.3 Hz, 2H), 3.62 – 3.49 (m, 2H), 3.16 (td, J = 12.3, 3.3 Hz, 2H), 3.02 (td, J = 11.2, 5.5 Hz, 1H), 2.44 (ddd, J = 14.7, 9.9, 5.6 Hz, 1H), 2.09 – 1.96 (m, 2H). MS (ESI+) m/z: 523.2 (M+1). HPLC purity: 95%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluoro-N-(2-methoxybenzyl)benzamide hydrochloride (CCG 211993) (14aj).
Prepared from Intermediate 13aj as described for compound 14aa. (68%yield) 1H NMR (CDCl3, 400MHz) δ 7.97 (m, 1H), 7.44 (m, 1H), 7.38 – 7.22 (m, 4H), 7.04 (m, 2H), 6.98 – 6.79 (m, 2H), 6.20 (s, 1H), 5.87 (s, 2H), 4.67 (d, J = 5.9 Hz, 2H), 3.89 (s, 3H), 3.57 (m, 1H), 3.48– 3.39 (m, 3H), 3.19 (m, 1H), 2.72 – 2.57 (m, 1H), 2.17 (m, 1H), 1.78–1.76 (s, 1H), 1.45 – 1.15 (m, 3H). MS (ESI+) m/z: 493.3 (M+1). HPLC purity: 97%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluoro-N-(pyridin-2-ylmethyl)benzamide hydrochloride (CCG211998) (14ak).
Prepared from Intermediate 13ak as described for compound 14aa. (34% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.80 (s, 1H), 8.58 (t, J = 7.8 Hz, 1H), 8.06 (m, 1H), 7.98 (m, 1H), 7.91 – 7.67 (m, 1H), 7.51 (m, 1H), 7.26 (dd, J = 10.9, 8.5 Hz, 1H), 6.61 (d, J = 8.4 Hz, 1H), 6.39 (m, 1H), 6.18 (m, 1H), 5.85 (s, 2H), 4.92 (s, 2H), 3.68 – 3.66 (m, 2H), 3.58– 3.51 (m, 2H), 3.21–3.14 (m, 2H), 2.48 (m, 1H), 2.07–1.28 (m, 3H). MS (ESI+) m/z: 464.2 (M+1). HPLC purity: 95%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluoro-N-(pyridin-3-ylmethyl)benzamide (CCG258203). (14al).
Prepared from Intermediate 13al as described for compound 14aa (87% yield) 1H NMR (400 MHz, Methanol-d4) δ 8.57 (d, J = 2.2 Hz, 1H), 8.44 (dd, J = 4.9, 1.6 Hz, 1H), 7.85 (dt, J = 8.0, 1.9 Hz, 1H), 7.65 (dd, J = 6.9, 2.4 Hz, 1H), 7.44 (ddd, J = 12.8, 7.9, 4.8 Hz, 2H), 7.20 (dd, J = 10.5, 8.5 Hz, 1H), 6.61 (d, J = 8.5 Hz, 1H), 6.38 (d, J = 2.5 Hz, 1H), 6.17 (dd, J = 8.5, 2.5 Hz, 1H), 5.85 (s, 2H), 4.61 (s, 2H), 3.64 (td, J = 11.6, 10.6, 3.1 Hz, 2H), 3.55 (dd, J = 9.9, 6.0 Hz, 1H), 3.47 (d, J = 12.8 Hz, 1H), 3.19 – 3.04 (m, 2H), 3.07 – 2.96 (m, 1H), 2.45 – 2.34 (m, 1H), 2.04 (d, J = 3.4 Hz, 1H). MS (ESI+) m/z: 464.2 (M+1). HPLC purity: 96%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluoro-N-(pyridin-4-ylmethyl)benzamide (CCG258204) (14am).
Prepared from Intermediate 13am as described for compound 14aa (57% yield) 1H NMR (400 MHz, Methanol-d4) δ 8.49 – 8.44 (m, 2H), 7.65 (dd, J = 6.9, 2.4 Hz, 1H), 7.44 (ddd, J = 8.5, 4.8, 2.4 Hz, 1H), 7.41 – 7.37 (m, 2H), 7.18 (dd, J = 10.6, 8.5 Hz, 1H), 6.59 (d, J = 8.5 Hz, 1H), 6.34 (d, J = 2.5 Hz, 1H), 6.14 (dd, J = 8.5, 2.5 Hz, 1H), 5.83 (d, J = 1.4 Hz, 2H), 4.62 (s, 2H), 3.58 (dd, J = 9.7, 3.0 Hz, 1H), 3.49 (dd, J = 9.7, 6.4 Hz, 1H), 3.37 (dd, J = 12.7, 3.8 Hz, 1H), 3.22 – 3.14 (m, 1H), 2.85 – 2.69 (m, 3H), 2.21 – 2.11 (m, 1H), 1.86 – 1.75 (m, 2H). MS (ESI+) m/z: 464.2 (M+1). HPLC purity: 95%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluoro-N-(2-(pyridin-2-yl)ethyl)benzamide (CCG258205) (14an).
Prepared from Intermediate 13an as described for compound 14aa (24% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.46 (ddd, J = 5.0, 1.8, 1.0 Hz, 1H), 7.76 (td, J = 7.7, 1.8 Hz, 1H), 7.59 (dd, J = 6.9, 2.4 Hz, 1H), 7.42 (ddd, J = 8.5, 4.7, 2.4 Hz, 1H), 7.36 (dt, J = 7.8, 1.1 Hz, 1H), 7.27 (ddd, J = 7.5, 5.0, 1.2 Hz, 1H), 7.17 (dd, J = 10.5, 8.6 Hz, 1H), 6.63 (d, J = 8.5 Hz, 1H), 6.40 (d, J = 2.5 Hz, 1H), 6.19 (dd, J = 8.5, 2.5 Hz, 1H), 5.85 (s, 2H), 3.74 (t, J = 7.1 Hz, 2H), 3.71 – 3.63 (m, 2H), 3.60 – 3.49 (m, 2H), 3.21 – 3.11 (m, 2H), 3.08 (t, J = 7.1 Hz, 2H), 3.02 (dd, J = 11.1, 5.8 Hz, 1H), 2.48 – 2.37 (m, 1H), 2.09 – 2.00 (m, 2H). MS (ESI+) m/z: 478.2 (M+1). HPLC purity: 97%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluoro-N-(2-(pyridin-3-yl)ethyl)benzamide (CCG258207) (14ao).
Prepared from Intermediate 13ao as described for compound 14aa (82% yield) 1H NMR (400 MHz, Methanol-d4) δ 8.45 (d, J = 1.7 Hz, 1H), 8.39 (dd, J = 5.0, 1.6 Hz, 1H), 7.78 (dt, J = 7.9, 1.9 Hz, 1H), 7.52 (dd, J = 6.9, 2.4 Hz, 1H), 7.38 (dddd, J = 10.7, 7.8, 4.9, 1.7 Hz, 2H), 7.13 (dd, J = 10.5, 8.5 Hz, 1H), 6.61 (d, J = 8.5 Hz, 1H), 6.36 (d, J = 2.5 Hz, 1H), 6.16 (dd, J = 8.5, 2.5 Hz, 1H), 5.84 (d, J = 1.2 Hz, 2H), 3.64 (t, J = 7.0 Hz, 2H), 3.59 (dd, J = 9.7, 3.0 Hz, 1H), 3.53 – 3.42 (m, 2H), 3.27 (d, J = 12.7 Hz, 1H), 2.95 (t, J = 7.0 Hz, 2H), 2.86 (qd, J = 12.4, 11.8, 3.8 Hz, 3H), 2.21 (tq, J = 7.8, 4.4, 3.8 Hz, 1H), 1.91 – 1.79 (m, 2H). MS (ESI+) m/z: 478.2 (M+1). HPLC purity: 96%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluoro-N-(2-(pyridin-4-yl)ethyl)benzamide (CCG258206) (14ap).
Prepared from Intermediate 13ap as described for compound 14aa (90% yield). 1H NMR (500 MHz, Methanol-d4) δ 8.44 (d, J = 5.1 Hz, 2H), 7.55 (dd, J = 6.8, 2.4 Hz, 1H), 7.43 (ddd, J = 8.5, 4.8, 2.4 Hz, 1H), 7.39 – 7.35 (m, 2H), 7.17 (dd, J = 10.5, 8.5 Hz, 1H), 6.63 (d, J = 8.5 Hz, 1H), 6.41 (d, J = 2.5 Hz, 1H), 6.19 (dd, J = 8.5, 2.5 Hz, 1H), 5.85 (s, 2H), 3.72 – 3.64 (m, 4H), 3.59 – 3.50 (m, 2H), 3.22 – 3.14 (m, 2H), 3.04 (ddd, J = 11.4, 9.3, 6.9 Hz, 1H), 2.98 (t, J = 7.1 Hz, 2H), 2.45 (dddt, J = 11.5, 8.4, 5.9, 3.2 Hz, 1H), 2.09 – 2.02 (m, 2H). MS (ESI+) m/z: 478.2 (M+1). HPLC purity: 96%.
N-((1H-imidazol-2-yl)methyl)-5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluorobenzamide (CCG258209) (14aq).
Prepared from Intermediate 13aq as described for compound 14aa (78% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.72 (dd, J = 6.9, 2.4 Hz, 1H), 7.43 (ddd, J = 8.6, 4.8, 2.4 Hz, 1H), 7.17 (dd, J = 10.7, 8.5 Hz, 1H), 6.98 (s, 2H), 6.60 (d, J = 8.5 Hz, 1H), 6.37 (d, J = 2.5 Hz, 1H), 6.16 (dd, J = 8.5, 2.5 Hz, 1H), 5.84 (d, J = 1.4 Hz, 2H), 4.62 (s, 2H), 3.62 (td, J = 11.0, 10.4, 3.2 Hz, 2H), 3.54 (dd, J = 9.8, 6.3 Hz, 1H), 3.40 (d, J = 12.6 Hz, 1H), 3.08 – 2.90 (m, 3H), 2.40 – 2.29 (m, 1H), 2.01 – 1.90 (m, 2H). MS (ESI+) m/z: 453.2 (M+1). HPLC purity: 95%.
N-(2-(1H-imidazol-4-yl)ethyl)-5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluorobenzamide (CCG258210) (14ar)
Prepared from Intermediate 13ar as described for compound 14aa (72% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.57 (dd, J = 6.9, 2.2 Hz, 1H), 7.40 (ddd, J = 8.4, 4.8, 2.4 Hz, 1H), 7.14 (dd, J = 10.6, 8.5 Hz, 1H), 7.62 – 7.59 (m, 1H), 6.89 (s, 1H), 6.62 (d, J = 8.5 Hz, 1H), 6.37 (d, J = 2.5 Hz, 1H), 6.17 (dd, J = 8.5, 2.5 Hz, 1H), 5.84 (s, 2H), 3.66 – 3.59 (m, 3H), 3.57 – 3.47 (m, 2H), 3.33 (d, J = 12.3 Hz, 2H), 3.01 – 2.84 (m, 5H), 2.31 – 2.22 (m, 1H), 1.96 – 1.89 (m, 2H). MS (ESI+) m/z: 467.2 (M+1). HPLC purity: 95%.
N-((1H-pyrazol-5-yl)methyl)-5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluorobenzamide (CCG258208) (14as).
Prepared from Intermediate 13as as described for compound 14aa (81% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.64 (dd, J = 6.9, 2.4 Hz, 1H), 7.57 (s, 1H), 7.42 (ddd, J = 8.5, 4.8, 2.4 Hz, 1H), 7.15 (dd, J = 10.6, 8.5 Hz, 1H), 6.60 (d, J = 8.5 Hz, 1H), 6.36 (d, J = 2.5 Hz, 1H), 6.29 (d, J = 2.2 Hz, 1H), 6.15 (dd, J = 8.5, 2.5 Hz, 1H), 5.84 (q, J = 1.1 Hz, 2H), 4.60 (s, 2H), 3.60 (dd, J = 9.7, 3.0 Hz, 1H), 3.51 (dd, J = 9.8, 6.4 Hz, 1H), 3.45 (dd, J = 12.7, 3.8 Hz, 1H), 3.26 (d, J = 12.9 Hz, 1H), 2.92 – 2.80 (m, 3H), 2.27 – 2.16 (m, 1H), 1.92 – 1.79 (m, 2H). MS (ESI+) m/z: 453.2 (M+1). HPLC purity: 95%.
N-(2-(1H-pyrazol-3-yl)ethyl)-5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluorobenzamide (14at).
Prepared from Intermediate 13at as described for compound 14aa (90% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.60 (dd, J = 6.9, 2.4 Hz, 1H), 7.52 (d, J = 2.1 Hz, 1H), 7.41 (ddd, J = 8.4, 4.8, 2.4 Hz, 1H), 7.17 (dd, J = 10.6, 8.5 Hz, 1H), 6.62 (d, J = 8.5 Hz, 1H), 6.39 (d, J = 2.5 Hz, 1H), 6.21 (d, J = 2.1 Hz, 1H), 6.18 (dd, J = 8.5, 2.5 Hz, 1H), 5.85 (s, 2H), 3.71 – 3.61 (m, 4H), 3.58 – 3.48 (m, 2H), 3.16 (td, J = 12.5, 3.8 Hz, 2H), 3.08 – 2.99 (m, 1H), 2.96 (t, J = 7.2 Hz, 2H), 2.41 (bs, 1H), 2.10 – 1.98 (m, 2H). MS (ESI+) m/z: 467.2 (M+1). HPLC purity: 97%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluoro-N-((5-methyl-1H-pyrazol-3-yl)methyl)benzamide (14au).
Prepared from Intermediate 13au as described for compound 14aa (85% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.66 (dd, J = 6.9, 2.3 Hz, 1H), 7.42 (ddd, J = 7.5, 4.7, 2.4 Hz, 1H), 7.18 (dd, J = 10.5, 8.5 Hz, 1H), 6.61 (d, J = 8.5 Hz, 1H), 6.39 (d, J = 2.5 Hz, 1H), 6.18 (dd, J = 8.5, 2.5 Hz, 1H), 6.04 (s, 1H), 5.85 (s, 2H), 4.52 (s, 2H), 3.72 – 3.62 (m, 2H), 3.61 – 3.48 (m, 2H), 3.16 (t, J = 12.4 Hz, 2H), 3.03 (td, J = 11.5, 4.5 Hz, 1H), 2.41 (bs, 1H), 2.25 (s, 3H), 2.04 (d, J = 13.0 Hz, 3H). MS (ESI+) m/z: 467.2 (M+1). HPLC purity: 97%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluoro-N-((1-methyl-1H-pyrazol-3-yl)methyl)benzamide (14av).
Prepared from Intermediate 13av as described for compound 14aa (69% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.65 (dd, J = 6.9, 2.4 Hz, 1H), 7.51 (d, J = 2.3 Hz, 1H), 7.44 – 7.39 (m, 1H), 7.18 (dd, J = 10.6, 8.5 Hz, 1H), 6.61 (d, J = 8.5 Hz, 1H), 6.39 (d, J = 2.5 Hz, 1H), 6.23 (d, J = 2.3 Hz, 1H), 6.17 (dd, J = 8.5, 2.5 Hz, 1H), 5.85 (d, J = 0.8 Hz, 2H), 4.54 (s, 2H), 3.85 (s, 3H), 3.65 (td, J = 9.5, 9.0, 3.8 Hz, 2H), 3.56 (dd, J = 9.9, 6.1 Hz, 1H), 3.49 (d, J = 12.5 Hz, 1H), 3.18 – 3.08 (m, 2H), 3.01 (td, J = 11.7, 4.3 Hz, 1H), 2.37 (bs, 1H), 2.09 – 1.92 (m, 2H). MS (ESI+) m/z: 467.2 (M+1). HPLC purity: 95%.
5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluoro-N-((1-methyl-1H-pyrazol-5-yl)methyl)benzamide (14aw).
Prepared from Intermediate 13aw as described for compound 14aa (70% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.65 (dd, J = 6.8, 2.4 Hz, 1H), 7.51 (d, J = 2.4 Hz, 1H), 7.42 (ddd, J = 7.6, 4.7, 2.4 Hz, 1H), 7.18 (dd, J = 10.6, 8.5 Hz, 1H), 6.61 (d, J = 8.5 Hz, 1H), 6.39 (d, J = 2.6 Hz, 1H), 6.23 (d, J = 2.3 Hz, 1H), 6.17 (dd, J = 8.5, 2.6 Hz, 1H), 5.85 (d, J = 1.5 Hz, 2H), 4.54 (s, 2H), 3.85 (s, 3H), 3.65 (td, J = 9.6, 3.7 Hz, 2H), 3.56 (dd, J = 9.9, 6.1 Hz, 1H), 3.49 (d, J = 12.4 Hz, 1H), 3.12 (td, J = 12.5, 3.3 Hz, 2H), 3.07 – 2.94 (m, 1H), 2.37 (bs, 1H), 2.10 – 1.90 (m, 2H). MS (ESI+) m/z: 467.2 (M+1). HPLC purity: 97%.
N-((1H-pyrazol-4-yl)methyl)-5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluorobenzamide (14ax).
Prepared from Intermediate 13ax as described for compound 14aa (85% yield). 1H NMR (400 MHz, Methanol-d4) δ 7.65 – 7.58 (m, 3H), 7.41 (ddd, J = 8.5, 4.9, 2.4 Hz, 1H), 7.17 (dd, J = 10.5, 8.5 Hz, 1H), 6.61 (d, J = 8.5 Hz, 1H), 6.39 (d, J = 2.5 Hz, 1H), 6.18 (dd, J = 8.5, 2.5 Hz, 1H), 5.85 (s, 2H), 4.47 (s, 2H), 3.67 (dd, J = 13.3, 3.4 Hz, 2H), 3.58 – 3.48 (m, 2H), 3.16 (t, J = 12.4 Hz, 2H), 3.03 (td, J = 11.6, 4.2 Hz, 1H), 2.46 – 2.32 (m, 1H), 2.11 – 1.94 (m, 2H). MS (ESI+) m/z: 453.2 (M+1). HPLC purity: 95%.
N-(2-(1H-pyrazol-4-yl)ethyl)-5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluorobenzamide hydrochloride (CCG 224060) (14ay).
Prepared from Intermediate 13ay as described for compound 14aa (46% yield). 1H NMR (500 MHz, Methanol-d4) δ 7.54 (dd, J = 6.9, 2.4 Hz, 1H), 7.50 (s, 2H), 7.39 (ddd, J = 8.5, 4.9, 2.4 Hz, 1H), 7.12 (dd, J = 10.6, 8.5 Hz, 1H), 6.60 (d, J = 8.5 Hz, 1H), 6.34 (d, J = 2.5 Hz, 1H), 6.14 (dd, J = 8.5, 2.5 Hz, 1H), 5.83 (s, 2H), 3.60 – 3.52 (m, 3H), 3.49 (dd, J = 9.7, 6.6 Hz, 1H), 3.36 (dd, J = 12.6, 3.9 Hz, 1H), 3.17 (dt, J = 12.6, 3.0 Hz, 1H), 2.83 – 2.69 (m, 5H), 2.13 (dddt, J = 14.6, 10.9, 6.8, 3.5 Hz, 1H), 1.87 – 1.71 (m, 2H). MS (ESI+) m/z: 467.2 (M+1). HPLC purity: 95%.
(3S,4R)-tert-butyl-3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluoro-3-formylphenyl)piperidine-1-carboxylate (8ba).
N1,N1,N2,N2-tetramethylethylene-1,2-diamine (0.548 g, 4.72 mmol) was added to THF (10 mL) in a 50 mL round bottom flask cooled to −78 °C under nitrogen. Sec-buytllithium (3.63 mL, 4.72 mmol) was added and the reaction mixture was stirred for ten minutes then (3S,4R)-tert-butyl 3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluorophenyl)piperidine-1-carboxylate (0.998 g, 2.36 mmol) in THF (10 mL) was added and the solution was stirred at −78 °C for 1 hour. A solution of anhydrous DMF (2 ml) in THF (5 mL) was added and the reaction was allowed to warm to room temperature (4 hours). The reaction was quenched with NH4Cl (10 mL), extracted with ether 2x, washed with NaCl 3x, dried over MgSO4, concentrated in vacuo, and purified using flash chromatography with a 1%−10% EtOAc/Hexane gradient to give (3S,4R)-tert-butyl-3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluoro-3-formyl-phenyl)piperidine-1-carboxylate (1.075g crude, took all crude to next step, Rf:0.46 (10% EtOAc/Hexanes)). 1H NMR (DMSO – d6, 400 MHz) δ: 10.20 (s, 1H), 7.71–7.26 (m, 2H), 7.35 (t, J=8.0 Hz, 1H), 4.27 (d, J=12.0 Hz, 1H), 4.05 (br s, 1H), 3.55–3.10 (m, 4H), 2.63–2.55 (m, 1H), 1.83–1.52 (m, 3H), 1.57 (s, 9H), 0.82 (s, 9H), −0.12 (s, 6H); HPLC purity: 95%; MS (ESI+) m/z: 452.4 (M+1).
(3S,4R)-tert-butyl-3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluoro-3-(hydroxymethyl)phenyl)piperidine-1-carboxylate (8bb).
(3S,4R)-tert-butyl 3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluoro-3-formylphenyl)-peridine-1-carboxylate (crude, 1.075 g, 2.38 mmol) was added to THF (15 mL) in a 50 mL round bottom flask cooled to 0 °C. Sodium borohydride (0.0262 g, 1.19 mmol) was added and the reaction mixture was allowed to come to room temperature and stirred 3 hours. Methanol (10 mL) and citric acid (5%, 10 mL) were added to the reaction mixture and allowed to stir 30 minutes. The reaction was extracted with ether/ethyl acetate 2x, washed with NaCl 2x, dried over MgSO4, concentrated in vacuo, and purified using flash chromatography (80 mL silica gel) of 20% EtOAc/Hexane to give (3S,4R)-tert-butyl 3-(((tert-butyldi- methylsilyl)oxy)methyl)-4-(4-fluoro-3-(hydroxymethyl)phenyl)piperidine-1-carboxylate (0.627 g, Rf:0.22 (20% EtOAc/Hexanes), 59% yield from 2). 1H NMR (DMSO – d6, 400 MHz) δ: 7.30 (d, J=8.0 Hz, 1H) 7.15–7.08 (m, 1H) 7.05 (t, J=8.0 Hz, 1H), 5.24 (s, 1H), 4.52 (s, 2H), 4.30 (d, J=12.0 Hz, 1H), 4.05 (br s, 1H), 3.35 (s, 1H), 3.24 (s, 1H), 3.14 (t, J=8.0 Hz, 1H), 2.72 (s, 2H), 1.82–1.46 (m, 3H), 1.42 (s, 9H), 0.83 (s, 9H), 0.09 (s, 6H); HPLC purity: 95%; MS (ESI+) m/z: 454.4 (M+1), 476.4 (M+Na+).
(3S,4R)-tert-butyl-3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(3-(cyanomethyl)-4-fluorophenyl)piperidine-1-carboxylate (9b).
(3S,4R)-tert-butyl 3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(4-fluoro-3-(hydroxymethyl)phenyl)piperidine-1-carboxylate (0.344 g, 0.759 mmol) was added to anhydrous dichloromethane (8 mL) in a 50 mL round bottom flask cooled to 0 °C under nitrogen. Diisopropylethylamine (0.265 mL, 1.517 mmol) was added to the reaction mixture followed by methanesulfonic anhydride and the reaction mixture was stirred for 0.5 hours. The reaction mixture was washed 1x with distilled water, dried over MgSO4, and concentrated in vacuo. Anhydrous dimethyl sulfoxide (8 mL) was added to the 50 mL flask followed by sodium cyanide (0.112 g, 2.276 mmol) and stirred overnight at room temperature under nitrogen. The reaction was diluted with ether and washed with NaCl (6x). The aqueous layers were then washed 2x with ether. The organic layers were combined, dried over MgSO4, concentrated in vacuo, and purified using flash chromatography (30 mL silica gel) of 10–20% gradient of EtOAc/Hexane to give (3S,4R)-tert-butyl-3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(3-(cyanomethyl)-4-fluorophenyl)piperidine-1-carboxylate (0.210g, Rf:0.51 (20% EtOAc/Hexanes), 60% yield). 1H NMR (CDCl3, 400 MHz) δ: 7.25–7.18 (m, 1H) 7.13 (s, 1H) 7.03 (t, J=9.0 Hz, 1H), 4.37–4.16 (m, 2H), 3.74 (s, 2H), 3.37–3.23 (m, 1H), 3.19–3.08 (m, 1H), 2.68 (br s, 2H), 1.82–1.56 (m, 4H), 1.48 (s, 9H), 1.24 (s, 9H), −0.07 (s, 6H); HPLC purity: 99%; MS (ESI+) m/z: 463.1 (M+1), 485.1 (M+Na+).
(3S,4R)-tert-butyl-4-(3-(cyanomethyl)-4-fluorophenyl)-3-(hydroxymethyl)piperidine-1-carboxylate (10b).
(3S,4R)-tert-butyl 3-(((tert-butyldimethylsilyl)oxy)methyl)-4-(3-(cyanomethyl)-4-fluoropheny l)piperidine-1-carboxylate (0.501 g, 1.081 mmol) and tetra-n-butyl ammonium fluoride (1.30mL, 1.30 mmol) were added to THF (10 mL) in a 50 mL round bottom flask at room temperature and allowed to stir overnight. The next day another equivalent of tetra-n-butylammonium fluoride (1.30 mL, 1.30 mmol) was added to the reaction mixture and stirred for 3.0 hours. The reaction mixture was quenched with NH4Cl, extracted with ether/ethyl acetate 1x and washed with NaCl (2x). The organic layers were combined, dried over MgSO4, concentrated in vacuo, and purified using flash chromatography (25mL silica gel) of 30–50% gradient of EtOAc/Hexane to give (3S,4R)-tert-butyl-4-(3-(cyanomethyl)-4-fluorophenyl)-3-(hydroxymethyl) piperidine −1-carboxylate (0.240g, Rf:0.33 (30% EtOAc/Hexanes), 64% yield). 1H NMR (CDCl3, 400 MHz) δ: 7.24 (dd, J1=2.4 Hz, J2=7.2 Hz, 1H) 7.18–7.12 (m, 1H) 7.03 (t, J=9.0 Hz, 1H), 4.41–4.32 (m, 2H), 4.19 (br s, 1H), 3.74 (s, 1H), 3.42 (dd, J1=3.0 Hz, J2=11.0 Hz, 1H), 3.24 (q, J=6.4 Hz, 1H), 2.87–2.52 (m, 3H), 1.87–1.57 (m, 4H), 1.48 (s, 9H); HPLC purity: 89%; MS (ESI+) m/z: 349.3 (M+1), 371.3 (M+Na+).
(3S,4R)-tert-butyl 3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(3-(cyanomethyl)-4-fluorophenyl)piperidine-1-carboxylate (11b).
(3S,4R)-tert-butyl-4-(3-(cyanomethyl)-4-fluorophenyl)-3-(hydroxymethyl)piperidine-1-carboxylate was added to a dry 25 mL round bottom flask, with 5 mL anhydrous dichloromethane cooled to 0 °C followed by DIEA (0.346 mL, 1.979 mmol) and lastly, methanesulfonic anhydride (0.230 g, 1.319 mmol) and stirred for 2 hours. The reaction mixture was washed 1x with NaCl, dried of MgSO4, and concentrated in vacuo. To a separate 25 mL round bottom flask sodium hydride (0.058 g, 1.451 mmol) was added with DMF under nitrogen at 0°C followed by sesamol (0.1913 g, 1.385 mmol) in 2 mL DMF and stirred for 20 minutes. The dried, mesylated (3S,4R)-tert-butyl-4-(3-(cyanomethyl)-4-fluorophenyl)-3-(hydroxymethyl)piperidine-1-carboxylate was then added with 2 mL DMF to the reaction vessel with the activated sesamol, heated to 70°C, and stirred for 10 minutes. The reaction was quenched with NH4Cl, extracted with EtOAc/ether 2x, washed with brine 3x, dried over MgSO4 and purified using flash chromatography with a 0% to 80% gradient of EtOAc/Hexanes to give (3S,4R)-tert-butyl 3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(3-(cyanomethyl)-4-fluorophenyl) piperidine-1-carboxylate (0.138 g, Rf: 0.40 (40% EtOAc/Hexanes), 46% yield). 1H NMR (CDCl3, 400 MHz) δ: 7.25–7.21 (m, 1H), 7.16–7.12 (m, 1H), 7.02 (t, J=8.8 Hz, 1H), 6.63 (d, J=8.8, 1H), 6.35 (d, J=2.4, 1H), 6.14 (dd, J1=2.4 Hz, J2=8.4 Hz, 1H), 5.88 (s, 2H), 4.43 (broad s, 1H), 4.25 (broad s, 1H), 3.72 (s, 2H), 3.62–3.59 (m, 1H), 3.46–3.42 (m, 1H), 2.90–2.67 (m, 3H), 2.10–1.60 (m, 3H), 1.50 (s, 9H) ; HPLC purity: 98%; MS (ESI+) m/z: 469.1 (M+1), 491.1 (M+Na+).
2-(5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-1-(tert-butoxycarbonyl) piperidin-4-yl)-2-fluorophenyl)acetic acid (12b).
(3S,4R)-tert-butyl 3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(3-(cyanomethyl)-4-fluorophenyl) piperidine-1-carboxylate (0.459 g) in EtOH (3.0 mL) was added to a 25 mL pressure vessel followed by 50% NaOH (0.26 mL) and heated to 98 °C overnight. The following day further excess 50% NaOH (0.10 mL) was added and the reaction was again stirred overnight. The reaction mixture was cooled to room temperature and diluted with H2O then 2N HCl was added to reach pH 2.0. The resulting suspension was extracted with dichloromethane 3x, washed with NaCl 2x, dried with MgSO4, and concentrated in vacuo to give 2-(5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-1-(tert-butoxycarbonyl)piperidin-4-yl)-2-fluorophenyl)acetic acid without further purification (0.372 g, 78% yield). 1H NMR (CDCl3, 400 MHz) δ: 7.11–7.04 (m, 2H), 7.00 (t, J=9.0 Hz, 1H), 6.20 (d, J=8.8, 1H), 6.34 (2, 1H), 6.14 (d, J=8.4 Hz, 1H), 5.88 (s, 2H), 4.42 (broad s, 1H), 4.23 (broad s, 1H), 3.66 (s, 2H), 3.64–3.58 (m, 1H), 3.48–3.42 (m, 1H), 2.88–2.72 (m, 2H), 2.71–2.60 (m, 1H), 2.04–1.92 (m, 1H), 1.85–1.63 (m, 3H), 1.49 (s, 9H) ; HPLC purity: 87%; MS (ESI+) m/z: 488.2 (M+1), 410.2 (M+Na+); (ESI-) m/z: 486.2 (M-1).
(3S,4R)-tert-butyl-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-(2-((2-methoxybenzyl)amino)-2-oxoethyl)phenyl)piperidine-1-carboxylate (1–93) (13ba).
2-(5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-1-(tert-butoxycarbonyl)piperidin-4-yl)-2-fluorophenyl)acetic acid 12b (0.060 g, 0.123 mmol), 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (0.027 g, 0.172 mmol), N,N-diisopropylethylamine (0.021mL, 0.123 mmol), 1-hydroxybenzotriazole (0.008 g, 0.062 mmol) and 2-methoxybenzylamine (0.038 mL, 0.369 mmol) were added to THF (5 mL) in a 15 mL round bottom flask and stirred overnight at room temperature. Diluted with ethyl acetate and saturated sodium bicarbonate and then separated the layers. The organic layer was then washed with 10% citric acid (1x), brine (2x), dried with MgSO4, and concentrated in vacuo. Purified using flash chromatography in a gradient of 20% - 50% EtOAc/Hexanes to give as an amorpous solid (3S,4R)-tert-butyl 3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-(2-((2-methoxybenzyl)amino)-2-oxoethyl)phenyl)piperidine-1-carboxylate (0.030 g, 41% yield). 1H NMR (CDCl3, 400 MHz) δ: 7.24 (dd, J = 8.0, 4.0 Hz, 1H), 7.20 (dd, J = 8.0, 4.0 Hz, 1H), 7.11–7.04 (m. 2H), 7.0 – 6.95 (m, 1H), 6.88 (t, J = 6.0 Hz, 1H), 6.83 (d, J = 8.0 Hz, 1H), 6.61 (d, J = 8.0 Hz, 1H), 6.33 (d, J = 2.0 Hz, 1H), 6.12 (dd, J = 8.0, 4.0 Hz, 1H), 6.04 (bs, 1H), 5.87 (s, 2H), 4.42–4.39 (m, 2H), 4.23 (bs, 1H), 3.75 (s, 3H), 3.65–3.44 (m, 3H), 3.41 (dd, J = 10.0, 8.0 Hz, 1H), 2.78 (bs, 2H), 2.63 (t, J = 12.0 Hz, 1H), 1.98 (bs, 1H), 1.88–1.63 (m, 3H), 1.50 (s, 9H).
(3S,4R)-tert-butyl 3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-(2-oxo-2-((pyridin-2-ylmethyl)amino)ethyl)phenyl)piperidine-1-carboxylate (13bb).
Compound 13bb was prepared as described for compound 13ba replacing 2-methoxy benzyl amine with pyridin-2-ylmethanamine (36% yield). 1H NMR (400 MHz, Chloroform-d) δ 8.44 (d, J = 4.9 Hz, 1H), 7.65 (t, J = 7.7 Hz, 1H), 7.24 (d, J = 8.1 Hz, 1H), 7.20 – 7.11 (m, 2H), 7.07 (s, 1H), 6.99 (t, J = 8.9 Hz, 1H), 6.89 (d, J = 5.2 Hz, 1H), 6.59 (d, J = 8.1 Hz, 1H), 6.33 (d, J = 2.6 Hz, 1H), 6.17 – 6.08 (m, 1H), 5.87 (s, 2H), 4.52 (d, J = 4.9 Hz, 2H), 4.43 (s, 1H), 4.24 (s, 1H), 3.65 – 3.57 (m, 2H), 3.45 (m, 1H), 2.80 (m, 2H), 2.65 (dd, J = 14.0, 10.2 Hz, 1H), 2.13 (bs, 1H), 2.0 (m, 1H), 1.78 (m, 1H), 1.69 (dd, J = 12.6, 4.2 Hz, 1H), 1.49 (s, 9H).
(3S,4R)-tert-butyl 3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-(2-oxo-2-((2-(pyridin-2-yl)ethyl)amino)ethyl)phenyl)piperidine-1-carboxylate (13bc).
Compound 13bc was prepared as described for compound 13ba replacing 2-methoxy benzyl amine with 2-(pyridin-2-yl)ethan-1-amine (17% yield). 1H NMR (400 MHz, Chloroform-d) δ 8.40 (s, 1H), 7.79 – 7.68 (m, 1H), 7.08 – 6.99 (m, 2H), 6.91 (t, J = 9.0 Hz, 1H), 6.58 (d, J = 8.3 Hz, 1H), 6.30 (s, 1H), 6.09 (d, J = 8.5 Hz, 1H), 5.84 (s, 2H), 4.40 (s, 1H), 4.21 (s, 1H), 3.65 – 3.33 (m, 4H), 3.05 – 2.55 (m, 7H), 1.99 (s, 1H), 1.78 – 1.56 (m, 2H), 1.48 (s, 9H).
(3S,4R)-tert-butyl 4-(3-(2-((2-(1H-pyrazol-4-yl)ethyl)amino)-2-oxoethyl)-4-fluorophenyl)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidine-1-carboxylate (13bd).
Compound 13bd was prepared as described for compound 13ba replacing 2-methoxy benzyl amine with 2-(1H-pyrazol-4-yl)ethan-1-amine (65% yield). 1H NMR (400 MHz, Chloroform-d) δ 7.32 (s, 2H), 7.05 (s, 2H), 6.98 (t, J = 8.6 Hz, 1H), 6.62 (d, J = 8.3 Hz, 1H), 6.33 (s, 1H), 6.13 (d, J = 8.1 Hz, 1H), 5.87 (s, 2H), 5.55 (s, 1H), 4.42 (s, 1H), 4.24 (s, 1H), 3.57 (d, J = 8.6 Hz, 2H), 3.50 (d, J = 6.9 Hz, 3H), 3.41 (s, 3H), 2.79 (s, 2H), 2.65 (s, 2H), 1.93 (s, 1H), 1.77 (d, J = 12.5 Hz, 1H), 1.51 (s, 9H).
2-(5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluorophenyl)-N-(2-methoxybenzyl)acetamide hydrochloride (CCG 215140) (14ba).
(3S,4R)-tert-butyl 3-((benzo[d][1,3]dioxol-5-yloxy)methyl)-4-(4-fluoro-3-(2-((2-methoxybenzyl)amino)-2-oxoethyl)phenyl)piperidine-1-carboxylate 13ba (0.029 g, 0.048 mmol) was dissolved in 1,4-dioxanes (0.5 mL) followed by 4M HCl/1,4-dioxanes (0.186 mL) and stirred at room temperature for one hour. The reaction was concentrated in vacuo, then diethyl ether was added to precipitate a white solid which was then concentrated to give 2-(5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluorophenyl)-N-(2-methoxybenzyl)acetamide hydrochloride as a white solid (0.018 g, 0.037 mmol, 77% yield) (95% purity). 1H NMR (400 MHz, Chloroform-d) δ 9.82 (s, 1H), 7.18 – 7.02 (m, 3H), 6.95 (d, J = 8.4 Hz, 1H), 6.86 – 6.72 (m, 2H), 6.54 (d, J = 7.7 Hz, 1H), 6.26 (s, 1H), 6.09 – 5.95 (m, 2H), 5.81 (s, 2H), 4.32 (bs, 2H), 3.71 (s, 3H), 3.66 – 3.30 (m, 4H), 3.16 – 2.75 (m, 3H), 2.57 (s, 1H), 2.30 (s, 1H), 1.96 (s, 1H). MS (ESI+) m/z: 506.9 (M+1), 528.9 (M+Na). HPLC purity: 95%
2-(5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluorophenyl)-N-(pyridin-2-ylmethyl)acetamide hydrochloride (CCG215143) (14bb).
Prepared from Intermediate 13bb as described for compound 14aa (95% yield) 1H NMR (400 MHz, Methanol-d4) δ 8.75 (d, J = 5.7 Hz, 1H), 8.54 (t, J = 7.9 Hz, 1H), 7.94 (t, J = 9.7 Hz, 2H), 7.30 (d, J = 7.0 Hz, 1H), 7.20 (d, J = 6.2 Hz, 1H), 7.06 (t, J = 9.1 Hz, 1H), 6.61 (d, J = 8.3 Hz, 1H), 6.39 (s, 1H), 6.18 (d, J = 8.4 Hz, 1H), 5.85 (s, 2H), 4.72 (s, 2H), 3.71 – 3.61 (m, 3H), 3.60 – 3.47 (m, 3H), 3.15 (t, J = 12.4 Hz, 2H), 2.95 (s, 1H), 2.45 (s, 1H), 2.13 – 1.99 (m, 2H). MS (ESI+) m/z: 477.9 (M+1), 499.9 (M+Na). HPLC purity: 97%
2-(5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluorophenyl)-N-(2-(pyridin-2-yl)ethyl)acetamide hydrochloride (CCG 215142) (14bc).
Prepared from Intermediate 13bc as described for compound 14aa (93% yield). 1H NMR (400 MHz, Methanol-d4) δ 8.70 (s, 1H), 8.47 – 8.38 (m, 1H), 7.88 (d, J = 7.5 Hz, 2H), 7.23 – 7.16 (m, 2H), 7.04 (t, J = 8.7 Hz, 1H), 6.63 (d, J = 8.3 Hz, 1H), 6.40 (s, 1H), 6.20 (d, J = 8.4 Hz, 1H), 5.85 (s, 2H), 3.71 – 3.45 (m, 8H), 3.23 – 3.14 (m, 4H), 2.43 (s, 1H), 2.14 (s, 1H), 2.04 (s, 1H), 1.29 (s, 1H). MS (ESI+) m/z: 491.9 (M+1), 513.9 (M+Na). HPLC purity: 98%.
N-(2-(1H-pyrazol-4-yl)ethyl)-2-(5-((3S,4R)-3-((benzo[d][1,3]dioxol-5-yloxy)methyl)piperidin-4-yl)-2-fluorophenyl)acetamide (CCG 222886) (14bd).
Prepared from Intermediate 13bd as described for compound 14aa (75% yield). 1H NMR (500 MHz, Methanol-d4) δ 7.41 (s, 2H), 7.20 – 7.13 (m, 2H), 7.02 (t, J = 9.3 Hz, 1H), 6.60 (d, J = 8.5 Hz, 1H), 6.34 (d, J = 2.5 Hz, 1H), 6.15 (dd, J = 8.5, 2.5 Hz, 1H), 5.82 (s, 2H), 3.59 (dd, J = 9.7, 2.9 Hz, 1H), 3.53 – 3.47 (m, 3H), 3.43 (dd, J = 12.6, 3.9 Hz, 1H), 3.38 – 3.30 (m, 4H), 3.27 – 3.19 (m, 1H), 2.87 – 2.77 (m, 2H), 2.73 (ddd, J = 16.5, 10.7, 5.2 Hz, 2H), 2.67 (t, J = 7.2 Hz, 2H), 2.17 (dtt, J = 11.2, 7.5, 3.3 Hz, 1H), 1.89 – 1.76 (m, 2H). MS (ESI+) m/z: 481.9 (M+1). HPLC purity: 95%.
Protein Purification:
A C-terminally His6-tagged S670A mutant of human GRK2 and a soluble variant of human Gβ1γ2 (Gγ2-C68S) were used for crystallographic studies with compounds 14ak and 14bd, as previously described.24 The 14as co-crystal structure used bovine GRK2, purified similarly, and wild-type human Gβ1γ2 containing an N-terminally hexahistidine tagged β subunit, which was expressed using a dual promoter insect cell expression vector in High Five cells and harvested 48 h after post infection. Gβ1γ2 was then purified from clarified lysate using nickel-nitrilotriacetic acid (NTA) affinity and anion exchange chromatography as described previously.35 Fractions containing Gβ1γ2 were subsequently pooled and gel filtered into 20 mM HEPES pH 8.0, 100 mM NaCl, 0.5 mM EDTA, 2 mM MgCl2, 10 mM CHAPS and 1 mM DTT using a Superdex 200 column (GE Healthcare Life Sciences). Pooled fractions from the S200 column were concentrated to 5 mg/ml, as determined by Bradford analysis, using a 30kD cut-off Amicon Ultra-15 Centrifugal Filter Unit, flash frozen in liquid nitrogen, and stored at −80 ˚C until future use. Enzymatic assays used bovine GRK1 (residues 1–535), bovine GRK2 (S670A), bovine GRK5, and mouse PKA catalytic subunits purified via the sequential chromatographic steps of NTA acid affinity, Source15S, and tandem S200 size exclusion as described previously (Yang, P., Glukhova, A., Tesmer,J.J., and Chen, Z.(2013) Membrane orientation and binding determinants of G protein-coupled receptor kinase 5 as assessed by combined vibrational spectroscopic studies. PLoS One 8, e82072; Huang, C. C., Yoshino-Koh, K., and Tesmer, J. J. (2009) A surface of the kinase domain critical for the allosteric activation of G protein-coupled receptor kinases. J. Biol. Chem. 284, 17206 –17215 )
Crystal Structure Determinations:
For 14ak and 14bd co-crystal structures, human GRK2 and Gβ1γ2C68S were mixed in a 1.2:1 molar ratio of GRK2:Gβγ with a final total protein concentration of 9–11 mg/mL. A final concentration of 500 μM inhibitor was added from a 50 mM stock in 100% DMSO. The complex was stored on ice for at least 30 min prior addition into the crystallization experiment. For the 14as co-crystal structure, bovine GRK2 and human Gβ1γ2 were added, in a 1:1 molar ratio, to a buffer solution composed of 20 mM HEPES pH 8, 100 mM NaCl, 10 mM CHAPS, 5 mM MgCl2 and 2 mM DTT. This solution was concentrated to approximately 12 mg/mL. A final concentration of 500 μM inhibitor was added to this concentrated solution from a 25 mM stock in 50% DMSO. This solution was then stored on ice for 1 hr and filtered through a 0.2 µm Nanosep Centrifugal Device prior to the assembly of crystal trays. All inhibitor complexes were crystallized at 4 ˚C by vapor diffusion using hanging drops consisting of 0.8 μL GRK2-Gβγ-inhibitor complex and 0.8 μL reservoir solution which contained 100 mM MES pH 6.4–6.7, 200 mM NaCl, and 8–10% PEG 3350. Crystals appeared after three days and continued to grow for 1–2 wk. Crystals were harvested in a cryoprotectant solution containing the contents of the reservoir solution supplemented with 25% ethylene glycol and 500 μM inhibitor and were flash frozen in liquid nitrogen. Diffraction data were collected at the Advanced Photon Source on the LS-CAT beamline 21-ID-G (14ak and 14bd) or beamline 21-ID-D (14as). Data were integrated and scaled using either HKL2000 (14ak and 14bd) or XDS (14as).36 Initial phases for each of the structures were calculated using Phaser molecular replacement with ligand-free 3V5W as the search model. Refinement was conducted either with REFMAC5 or phenix.refine, a part of the PHENIX suite, alternating with manual building in Coot. Early in refinement, the dihedral angles from 3V5W were used as restraints for the lower resolution 14as structure. The final models and structure factors for the 14ak, 14bd, and 14as co-crystal structures were validated with MolProbity37 prior to deposition into the Protein Data Bank under accession codes 5HE0, 5HE1, 5HE2, and 5HE3.
Enzymatic Assays:
PKA and ROCK1 inhibition assays were performed using the ADP-Glo Kinase Assay system (Promega) in accordance with the manufacturer’s recommendations and as described previously.27 Luminescence was measured using a BMG Labtech PHERAstar imaging system, and inhibition curves were analyzed using GraphPad Prism. Kinetic activity of the inhibitors with respect to the bovine GRKs were run in a buffer comprised of 20 mM HEPES (pH 7.0), 2 mM MgCl2, and 0.025% DDM with 50 nM GRK and 500 nM tubulin. ATP (500 μCi, 5μM) initiated the kinetic reactions which were then run for 5 min, at which time SDS loading buffer was added to quench the reactions and they were then separated via SDS-PAGE. The resulting gels were dried using a vacuum gel drying system, and exposed with a phosphorimaging screen. The images were then scanned and quantification via Typhoon imager and Image Quant respectively, as previously reported.27, 33 Data were analyzed and inhibition curves were fit via GraphPad Prism with a three variable dose-inhibitor response curve and a fixed Hill slope of 1.
Metabolic Stability in Mouse Liver Microsomes:
The metabolic stability was assessed using CD-1 mouse liver microsomes. 1 μM of each compound was incubated with 0.5 mg/mL microsomes and 1.7 mM cofactor β-NADPH in 0.1 M phosphate buffer (pH = 7.4) containing 3.3 mM MgCl2 at 37 °C. The DMSO concentration was less than 0.1% in the final incubation system. At 0, 5, 10, 15, 30, 45, and 60 min of incubation, 40 µL of reaction mixture were taken out, and the reaction is quenched by adding 3-fold excess of cold acetonitrile containing 100 ng/mL of internal standard for quantification. The collected fractions were centrifuged at 15000rpm for 10 min to collect the supernatant for LC−MS/MS analysis, from which the amount of compound remaining was determined. The natural log of the amount of compound remaining was plotted against time to determine the disappearance rate and the half-life of tested compounds.
Myocyte Shortening Measurements:
Cardiac myocytes were isolated from LV free wall and septum of C57/Bl6 mice as previously described.38 All cells were used within 2−8 h of isolation. Myocytes were plated on laminin-coated coverslips and were bathed in HEPES-buffered (20 mM, pH 7.4) medium 199 containing 1.8 mM extracellular Ca2+. When recording, coverslips containing myocytes were mounted in the Dvorak-Stotler chamber and bathed in 0.7 mL of fresh medium. Cells were paced at 1 Hz and imaged with a variable field-rate camera (Zeiss IM35, Ionoptix) by both edge detection and sarcomere length. Peak contraction was measured as the percentage of cell shortening. Cells were treated with isoproterenol (Iso, 0.5 μM) for 2 min for the recording of contraction, with pretreatment of either PBS as vehicle or 14ak (0.5 μM, 1 μM), 14as (0.1 μM, 0.5 μM, 1 μM), or 2 (0.1 μM, 0.5 μM, 1 μM) for 10 min.25
Pharmacokinetic Studies in Mice:
All animal experiments in this study were approved by the University of Michigan Committee on Use and Care of Animals and Unit for Laboratory Animal Medicine (ULAM). The abbreviated pharmacokinetics of paroxetine and Takeda101 (2) were determined in female CD-1 mice following intraperitoneal (ip) injection at 10 mg/kg. Compounds were dissolved in the vehicle containing 15% (v/v) DMSO, 15% (v/v) PEG-400, and 70% (v/v) PBS. Four blood samples (50 μL) were collected over 7 h (at 0.5h, 2h, 4h, and 7h), centrifuged at 35 00 rpm for 10 min, and plasma was frozen at −80˚C for later analysis. Plasma concentrations of the compounds were determined by the LC−MS/MS method developed and validated for this study. The LC−MS/MS method consisted of a Shimadzu HPLC system, and chromatographic separation of tested compound which was achieved using a Waters Xbridge-C18 column (5 cm × 2.1 mm, 3.5 μm). An AB Sciex QTrap 4500 mass spectrometer equipped with an electrospray ionization source (ABI-Sciex, Toronto, Canada) in the positive-ion multiple reaction monitoring (MRM) mode was used for detection. All pharmacokinetic parameters were calculated by noncompartmental methods using WinNonlin, version 3.2 (Pharsight Corporation, Mountain View, CA, USA).
Supplementary Material
Figure 4.
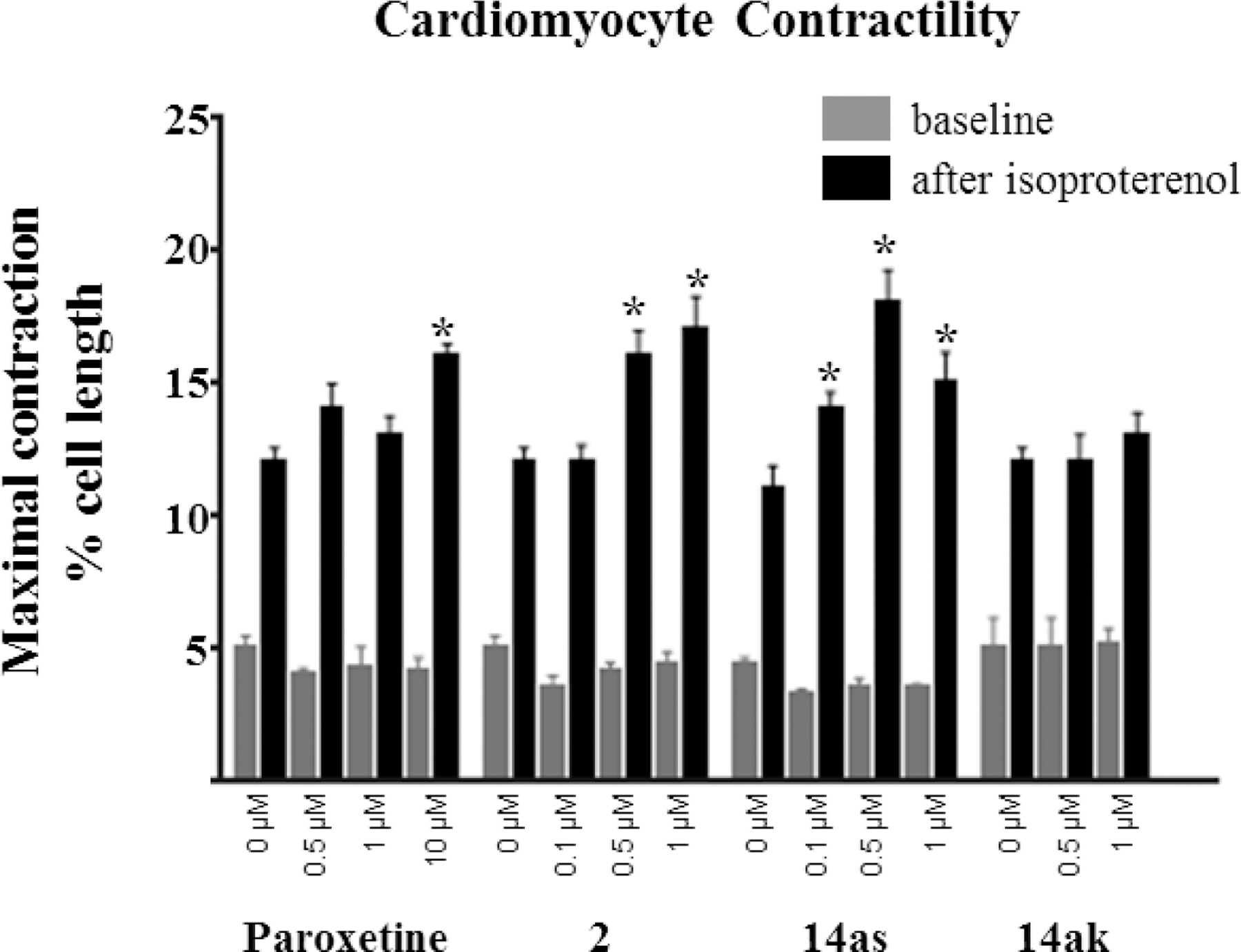
Contractility in murine cardiomyocytes of paroxetine, 2, 14as, and 14ak at varying concentrations. After addition of the βar agonist, isoproterenol, an increase in maximal contraction is observed (black bars) with respect to baseline, no isoproterenol stimulation (grey). Values represent the mean ± SEM for 6–8 cardiomyocytes. * denotes p-value > 0.02 vs control.
Acknowledgements
We acknowledge Kelly M. Larimore for technical assistance. The mouse liver microsomes stability and short pharmacokinetics studies were executed by The University of Michigan Pharmacokinetics Core. This work was supported by NIH grants HL071818, HL086865, and HL122416 (to J.T.), and American Heart Association grants N014938 (to K.T.H.) and 15PRE22730028 (to H.V.W.). Work at Temple (to W.J.K) was supported by NIH grants R37 HL061690, P01 HL075443 (Project 2), P01 HL108806 (Project 3) and P01 HL091799. J.T. and S.D.L. were supported by grants from the Center for Discovery of New Medicine, University of Michigan. M.C.C. and O.C. acknowledge training grant support from the University of Michigan Chemistry-Biology Interface (CBI) training program (NIH grant 5T32GM008597). Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357, and use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and Michigan Technology Tri-Corridor Grant [085P1000817].
Abbreviations used
- GRK
G-protein coupled receptor kinase
- GPCR
G protein-couple receptor
- P-loop
polyphosphate binding loop
- βARs
β-adrenergic receptors
- FDA
Federal Drug Administration
- Boc
tert – butyloxycarbonyl
- ROCK
rho associated coiled coil kinase
- PKA
protein kinase A
- AUC
area under the curve
Footnotes
Ancillary Information.
Supporting Information
The supporting information is available free of charge on the ACS publication website.
Crystal refinement statistics and complete data for the cardiomyocyte contractility experiments at varying concentrations.
PDB ID Codes
Authors will release the atomic coordinates and experimental data upon article publication.
References
- 1.Rosenbaum DM; Rasmussen SGF; Kobilka BK, The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stevens RC; Cherezov V; Katritch V; Abagyan R; Kuhn P; Rosen H; Wuthrich K, The GPCR Network: a large-scale collaboration to determine human GPCR structure and function. Nat. Rev. Drug Discov 2013, 12, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gurevich EV; Tesmer JJG; Mushegian A; Gurevich VV, G protein-coupled receptor kinases: More than just kinases and not only for GPCRs. Pharmacol. Ther 2012, 133, 40–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lefkowitz RJ; Stadel JM; Caron MG, Adenylate cyclase-coupled beta-adrenergic receptors - structure and mechanisms of activation and desensitization. Annu. Rev. Biochem 1983, 52, 159–186. [DOI] [PubMed] [Google Scholar]
- 5.Kohout TA; Lefkowitz RJ, Regulation of G Protein-Coupled Receptor Kinases and Arrestins During Receptor Desensitization. Mol. Pharmcol 2003, 63, 9–18. [DOI] [PubMed] [Google Scholar]
- 6.Pitcher JA; Freedman NJ; Lefkowitz RJ, G Protein–Coupled Receptor Kinases. Annu. Rev. Biochem 1998, 67, 653–692. [DOI] [PubMed] [Google Scholar]
- 7.Ren XR; Reiter E; Ahn S; Kim J; Chen W; Lefkowitz RJ, Different G protein-coupled receptor kinases govern G protein and beta-arrestin-mediated signaling of V2 vasopressin receptor. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 1448–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wada Y; Sugiyama J; Okano T; Fukada Y, GRK1 and GRK7: Unique cellular distribution and widely different activities of opsin phosphorylation in the zebrafish rods and cones. J. Neurochem 2006, 98, 824–837. [DOI] [PubMed] [Google Scholar]
- 9.Claing A; Laporte SA; Caron MG; Lefkowitz RJ, Endocytosis of G protein-coupled receptors: roles of G protein-coupled receptor kinases and beta-arrestin proteins. Prog. Neurobiol 2002, 66, 61–79. [DOI] [PubMed] [Google Scholar]
- 10.Ribas C; Penela P; Murga C; Salcedo A; García-Hoz C; Jurado-Pueyo M; Aymerich I; Mayor F Jr, The G protein-coupled receptor kinase (GRK) interactome: Role of GRKs in GPCR regulation and signaling. BBA- Biomemb 2007, 1768, 913–922. [DOI] [PubMed] [Google Scholar]
- 11.Lymperopoulos A; Rengo G; Funakoshi H; Eckhart AD; Koch WJ, Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nature Med 2007, 13, 315–323. [DOI] [PubMed] [Google Scholar]
- 12.Cohn JN; Levine TB; Olivari MT; Garberg V; Lura D; Francis GS; Simon AB; Rector T, Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart-failure. N. Engl. Jour. Med 1984, 311, 819–823. [DOI] [PubMed] [Google Scholar]
- 13.Port JD and Bristow MR, Altered Beta-adrenergic receptor gene regulation and signaling in chronic heart failure. J. Mol. Cell. Cardiol 2001, 33, 887–905. [DOI] [PubMed] [Google Scholar]
- 14.Sutherland EW; Robison GA; Butcher RW, Some Aspects of the Biological Role of Adenosine 3’,5’-monophosphate (Cyclic AMP). Circulation 1968, 37, 279–306. [Google Scholar]
- 15.Rockman HA; Koch WJ; Lefkowitz RJ, Seven-transmembrane-spanning receptors and heart function. Nature 2002, 415, 206–212. [DOI] [PubMed] [Google Scholar]
- 16.Huang ZM; Gold JI; Koch WJ, G protein-coupled receptor kinases in normal and failing myocardium. Fron. Biosci. (Landmark Ed.) 2011, 16, 3047–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ungerer M; Böhm M; Elce JS; Erdmann E; Lohse MJ, Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation 1993, 87, 454–463. [DOI] [PubMed] [Google Scholar]
- 18.Eschenhagen T, beta-adrenergic signaling in heart failure - adapt or die. Nature Med 2008, 14, 485–487. [DOI] [PubMed] [Google Scholar]
- 19.Hullmann J; Traynham CJ; Coleman RC; Koch WJ, The expanding GRK interactome: Implications in cardiovascular disease and potential for therapeutic development. Pharmacol. Res 2016, 110, 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raake PW; Vinge LE; Gao EH; Boucher M; Rengo G; Chen XW; DeGeorge BR; Matkovich S; Houser SR; Most P; Eckhart AD; Dorn GW; Koch WJ, G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ. Res 2008, 103, 413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raake PWJ; Schlegel P; Ksienzyk J; Reinkober J; Barthelmes J; Schinkel S; Pleger S; Mier W; Haberkorn U; Koch WJ; Katus HA; Most P; Muller OJ, AAV6.beta ARKct cardiac gene therapy ameliorates cardiac function and normalizes the catecholaminergic axis in a clinically relevant large animal heart failure model. Eur. Heart J 2013, 34, 1437–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ikeda S, K. M., and Fujiwara S, et al. Cardiotonic agent comprising GRK inhibitor. WO2007034846, March 29 2007, 2007. [Google Scholar]
- 23.Thal DM; Yeow RY; Schoenau C; Huber J; Tesmer JJG, Molecular Mechanism of Selectivity among G Protein-Coupled Receptor Kinase 2 Inhibitors. Mol. Pharmcol 2011, 80, 294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waldschmidt HV; Homan KT; Cruz-Rodríguez O; Cato MC; Waninger-Saroni J; Larimore KM; Cannavo A; Song J; Cheung JY; Kirchhoff PD; Koch WJ; Tesmer JJG; Larsen SD, Structure-Based Design, Synthesis, and Biological Evaluation of Highly Selective and Potent G Protein-Coupled Receptor Kinase 2 Inhibitors. J. Med. Chem 2016, 59, 3793–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thal DM; Homan KT; Chen J; Wu EK; Hinkle PM; Huang ZM; Chuprun JK; Song J; Gao E; Cheung JY; Sklar LA; Koch WJ; Tesmer JJG, Paroxetine Is a Direct Inhibitor of G Protein-Coupled Receptor Kinase 2 and Increases Myocardial Contractility. ACS Chem. Biol 2012, 7, 1830–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schumacher SM; Gao E; Zhu W; Chen X; Chuprun JK; Feldman AM; Tesmer G, J. J.; Koch WJ, Paroxetine-mediated GRK2 inhibition reverses cardiac dysfunction and remodeling after myocardial infarction. Sci. Transl. Med 2015, 7, 277ra31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Homan KT; Larimore KM; Elkins JM; Szklarz M; Knapp S; Tesmer JJG, Identification and Structure–Function Analysis of Subfamily Selective G Protein-Coupled Receptor Kinase Inhibitors. ACS Chem. Biol 2014, 10, 310–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sehon CA; Wang GZ; Viet AQ; Goodman KB; Dowdell SE; Elkins PA; Semus SF; Evans C; Jolivette LJ; Kirkpatrick RB; Dul E; Khandekar SS; Yi T; Wright LL; Srnith GK; Behm DJ; Bentley R; Doe CP; Hu E; Lee D, Potent, Selective and Orally Bioavailable Dihydropyrimidine Inhibitors of Rho Kinase (ROCK1) as Potential Therapeutic Agents for Cardiovascular Diseases. J. Med. Chem 2008, 51, 6631–6634. [DOI] [PubMed] [Google Scholar]
- 29.Lipinski CA; Lombardo F; Dominy BW; Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- 30.Gangula S; Kolla NK; Elati C; Dongamanti A; Bandichhor R, Improved Process for Paroxetine Hydrochloride Substantially Free from Potential Impurities. Syn. Comm 2012, 42, 3344–3360. [Google Scholar]
- 31.Bridges J, A.; Lee A; Maduakor EC; Schwartz CE, Fluorine as an ortho-directing group in aromatic metalation: Generality of the reaction and the high position of fluorine in the Dir-Met potency scale. Tet. Lett 1992, 33, 7495–7498. [Google Scholar]
- 32.Homan KT; Wu E; Wilson MW; Singh P; Larsen SD; Tesmer JJG, Structural and Functional Analysis of G Protein–Coupled Receptor Kinase Inhibition by Paroxetine and a Rationally Designed Analog. Mol. Pharmacol 2014, 85, 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Homan KT; Waldschmidt HV; Glukhova A; Cannavo A; Song J; Cheung JY; Koch WJ; Larsen SD; Tesmer JJG, Crystal Structure of G Protein-Coupled Receptor Kinase 5 in Complex with a Rationally Designed Inhibitor. J. Biol. Chem 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Komolov KE; Bhardwaj A; Benovic JL, Atomic Structure of GRK5 Reveals Distinct Structural Features Novel for G Protein-coupled Receptor Kinases. J. Biol. Chem 2015, 290, 20629–20647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kozasa T, Purification of G Protein Subunits from Sf9 Insect Cells Using Hexahistidine-Tagged α and βγ Subunits In G Protein Signaling, Smrcka A, Ed. Humana Press; 2004; Vol. 237, pp 21–38. [DOI] [PubMed] [Google Scholar]
- 36.Kabsch W, XDS. Acta Crystallogr. Sect. D 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen VB; Arendall WB III; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song J; Zhang X-Q; Wang J; Cheskis E; Chan TO; Feldman AM; Tucker AL; Cheung JY, Regulation of cardiac myocyte contractility by phospholemman: Na+/Ca2+ exchange versus Na+-K+-ATPase. Am. J. Physiol. Heart Circ. Physiol 2008, 295, H1615–H1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.