

Abstract
Listeria monocytogenes is a food-borne pathogen able to survive and grow in different environments including food processing plants where it can persist for month or years. In the present study the discriminatory power of Whole Genome Sequencing (WGS)-based analysis (cgMLST) was compared to that of molecular typing methods on 34 L. monocytogenes isolates collected over one year in the same rabbit meat processing plant and belonging to three genotypes (ST14, ST121, ST224). Each genotype included isolates indistinguishable by standard molecular typing methods. The virulence potential of all isolates was assessed by Multi Virulence-Locus Sequence Typing (MVLST) and the investigation of a representative database of virulence determinant genes. The whole genome of each isolate was sequenced on a MiSeq platform. The cgMLST, MVLST, and in silico identification of virulence genes were performed using publicly available tools. Draft genomes included a number of contigs ranging from 13 to 28 and N50 ranging from 456298 to 580604. The coverage ranged from 41 to 187X. The cgMLST showed a significantly superior discriminatory power only in comparison to ribotyping, nevertheless it allows the detection of two singletons belonging to ST14 that were not observed by other molecular methods. All ST14 isolates belonged to VT107, which 7-loci concatenated sequence differs for only 4 nucleotides to VT1 (Epidemic clone III). Analysis of virulence genes showed the presence of a fulllength inlA version in all ST14 isolates and of a mutated version including a premature stop codon (PMSC) associated to attenuated virulence in all ST121 isolates.
Key words: Whole genome sequencing, Listeria monocytogenes, Rabbit meat, cgMLST, Virulence genes
Introduction
Listeria monocytogenes is a food-borne pathogen adapted to survive, grow and ultimately persist in different environments (Gray et al., 2006). In food processing plants, L. monocytogenes has been repeatedly isolated from food samples as well as food processing environment for months or years (Stasiewicz et al., 2015; Véghová et al., 2017; Leong et al., 2014). Through the contamination of food, L. monocytogenes can be transmitted to humans where it can switch from saprophyte to intracellular pathogen associated to approximately 2000 confirmed cases of Listeriosis each year in Europe, with a fatality rate of 17 % (EFSA and ECDC, 2016).
Within L. monocytogenes biodiversity, isolates of lineage II and serotype 1/2a have been isolated from food and food processing plants more frequently than isolates belonging to lineage I (Orsi et al., 2011). Clonal complex (CC) 121 belonging to serotype 1/2a has been described as the most prevalent clone with a strong association to food origin. In particular, CC121 showed an overall prevalence of 17.6% over 6633 tested isolates of L. monocytogenes collected over nine years from food and clinical sources, with a statistical significant over-representation in food sources in comparison to clinical ones (92.9% vs 7.0%) (Maury et al., 2016). Comparable values of prevalence of human CC121 (11/116, 9.5%) were observed also within the Institute Pasteur L. monocytogenes database (http://bigsdb.pasteur.fr/listeria/). The low frequency of L. monocytogenes in humans might be linked to the attenuated virulence of this CC. Premature Stop Codons (PMSC) in key virulence genes, such as inlA and prfA, as well as production of truncated InlA and PrfA proteins, were associated to attenuated virulence (Nightingale et al., 2005; Van Stelten et al., 2011; Cruz et al., 2014; Kanki et al., 2015). Compared to CC121, CC14 has been isolated rarely in food. Maury and colleagues found 92 isolates belonging to CC14 over 6633 tested isolates (1.4%) (Maury et al., 2016). Lower detection values were described also in 19 meat processing plants located in Northern Italy (5.7% over 69 tested isolates) in comparison to CC121 (23%) (Morganti et al., 2015). Besides a low prevalence, CC14 was described to be associated to higher infection rates. The clinical frequency of CC14 reached a not negligible 29.5% among isolates of clinical sources (Maury et al., 2016). Within CC14, a ST14 strain isolated from a case of invasive listeriosis was molecular characterised as belonging to epidemic clone (EC) III (corresponding to Multi-Virulence Locus Type 1 (VT1)) previously associated to a sporadic case occurred in United States in 1988, as well as a multi-state outbreak occurred in United States in 2000 (Mammina et al., 2013; Kathariou et al., 2002) (https://sites.google.com/site/mvlstdatabase/protocol-for-mvlst).
Whole Genome sequencing (WGS) based analyses have recently revealed a valuable potential in pathogen fingerprinting and identification of novel gene/features related to specific phenotypes. Based on the nucleotide sequence of the whole genome, an unprecedented discrimination power has been achieved by core genome Multi Locus Sequence Typing (cgMLST). This is particularly relevant in studies of repeatedly reisolated strains showing high genetic similarity (same Pulsed Field Gel Electrophoresis (PFGE) profile or 7-locus Multi Locus Sequence Typing (7-locus MLST)) in which a superior discrimination power is required, especially to differentiate true persistent strains to sporadic strains as well as strains repeatedly reintroduced into the food processing plant through contaminated raw materials (Ferreira et al., 2014). Moreover, starting from the same sequence data, WGS offers the great potential for multiple investigations of novel genetic determinants as well as genetic determinants already known as linked to specific phenotypes (i.e. virulence). In a previous study on prevalence of L. monocytogenes in four Italian rabbit meat processing plants (A, B, C, D), isolates sharing the same 7-locus MLST (ST), Multi Locus Tandem Repeat Analysis (MLVA) and ApaI-PFGE profiles were repeatedly collected over time from carcasses, meat cuts, meat products and the food processing environment (De Cesare et al., 2017). This study provided an interesting dataset to investigate further specific ST-types over time. For this purpose, a specific focus was put on ST14 (belonging to CC14) and ST121 (belonging to CC121) isolates, repeatedly collected over one year and six month, respectively, from different rabbit meat and environmental sources of the same processing plant (A). The aims of the present study were: 1) to evaluate the discriminatory power of cgMLST in comparison to molecular typing methods; 2) to characterize the virulence potential of ST14 and ST121 sequenced isolates by comparative genomic analyses.
Materials and Methods
L. monocytogenes isolates included in the present study are a subset of isolates collected over one year within a previous study on prevalence of L. monocytogenes in rabbit meat processing plants (De Cesare et al., 2017). In particular, isolates sharing the same genotype and collected more than six times over a period of more than six months from different sources (rabbit carcasses, rabbit meat cuts, rabbit meat products and food processing environment) in the same plant were considered as potentially persistent. With this definition, 33 isolates of L. monocytogenes isolates of lineage II, serotype 1/2a and belonging to two 7-locus Multi Locus Sequence Types (MLST) ST14 and ST121 were selected as potentially persistent. An additional sporadic isolate, belonging to lineage I, serotype 1/2b, ST224 was also included for backward comparability. Isolates sharing the same 7– locus MLST were indistinguishable also by ApaI-Pulsed Field Eletrophoresis (PFGE), automated ribotyping and Multi Locus Variable number tandem repeat Analysis (MLVA) (De Cesare et al., 2017).
Whole-genomic DNA was extracted using the MagAttract HMW DNA Kit (Qiagen, Hilden, Germany). The purified DNA concentration and the quality parameter ratio 260/280 were measured by BioSpectrometer fluorescence (Eppendorf). The whole genome of selected 34 L. monocytogenes isolates was sequenced using Illumina MiSeq platform (TrueSeq library, paired-end reads). Reads were quality checked and de novo assembled using the INNUca pipeline (https://github.com /INNUENDOCON/INNUca).
Briefly, INNUca calculates if the sample raw data fulfill the expected coverage (minimum default 15X). Then, after a read quality analysis using FASTQC and trimming using TRIMMOMATIC, de novo draft genome assembly is performed with SPAdes, which is subsequently improved using PILON to correct bases and fix misassemblies.
Core genome Multi Locus Sequence Typing (cgMLST) was inferred based on in silico sequence alignment of 1748 loci of corresponding core genes. Briefly, after alignment, similarly to 7-locus MLST, an allele number is assigned to each locus. A cluster type, representative of all allele numbers, is then assigned (Moura et al., 2016).
The Multi Virulence Locus Sequence Type (MVLST) was inferred based on in silico sequence alignment of seven virulence determinant gene loci: clpP, dal, inlB, inlC, lisR, prfA (https://sites.google.com /site/mvlstdatabase ) (Zhang et al., 2004).
Analyses of virulence was performed using VirulenceFinder 1.5 (https://cge.cbs.dtu.dk/services/VirulenceFinder) (Joensen et al., 2014). With this tool a BLAST search of a database of 82 L. monocytogenes virulence determinant genes was applied to all ST14 and ST121 draft genomes included in the present study, as well as to 23 publicly available genomes of L. monocytogenes belonging to ST14 and ST121 and isolated from humans or food processing plants. Publicly available genome of EGD-e (Genbank accession number NC_003210.1) was included as reference. The default parameters used were 90% ID threshold and 60% of minimum length. Upon detection of no gene the analysis was repeated with 85% ID threshold and 20% minimum length. Alignment of inlA and prfA genes was performed by Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo).
The discriminatory power of each typing method was assessed by the Simpson’s Index of Diversity (ID). ID values with P<0.05 were considered statistical significant different (Hunter and Gaston, 1988).
Results
Thirty four isolates of L. monocytogenes of ST14, ST121 and ST224 collected over one year of sampling in a rabbit meat processing plant from environment, meat cuts and meat products were sequenced and de novo assembled. Extracted DNA had a yield ranging from 14.2 to 81.3 μg/mL and a 260/280 ratio ranging from 1.81 to 1.88. The generated draft whole genomes sizes ranged from 2,925,197 to 3,068,560 bp. Draft genomes included a number of contigs ranging from 13 to 28 and N50 ranging from 456298 to 580604. Finally, coverage ranged from 42 to 187X (Table 1).
Table 1.
Sequence type and assembly statistics of the 34 sequenced Listeria monocytogenes isolates.
| Isolate ID | ST | No. of contigs | N50 | Genome size (bp) | Fold coverage |
|---|---|---|---|---|---|
| LM1 | 14 | 13 | 579785 | 3022050 | 84,03 |
| LM2 | 14 | 14 | 456298 | 3022428 | 41,78 |
| LM3 | 14 | 13 | 580466 | 3022420 | 114,86 |
| LM5 | 14 | 15 | 579913 | 3022168 | 57,83 |
| LM8 | 14 | 14 | 580478 | 3023685 | 176,68 |
| LM9 | 14 | 13 | 580478 | 3023308 | 121,54 |
| LM10 | 14 | 14 | 580478 | 3023687 | 167,18 |
| LM11 | 224 | 13 | 511384 | 2925197 | 159 |
| LM15 | 14 | 13 | 580472 | 3022932 | 146,98 |
| LM16 | 14 | 14 | 580454 | 2986892 | 131,2 |
| LM17 | 14 | 14 | 580454 | 2986900 | 149,23 |
| LM18 | 14 | 14 | 580454 | 2986900 | 187,12 |
| LM19 | 14 | 15 | 580471 | 3023022 | 172,15 |
| LM22 | 14 | 13 | 580454 | 3022659 | 79,03 |
| LM27 | 121 | 26 | 529438 | 3060499 | 120,24 |
| LM29 | 121 | 28 | 530140 | 3061007 | 131,41 |
| LM31 | 121 | 25 | 530140 | 3060579 | 127,78 |
| LM35 | 14 | 13 | 580472 | 3022414 | 106,18 |
| LM39 | 14 | 15 | 580604 | 3022475 | 120,54 |
| LM41 | 14 | 13 | 580472 | 3022475 | 126,39 |
| LM44 | 14 | 13 | 580478 | 3022308 | 144,31 |
| LM46 | 14 | 13 | 580478 | 3022405 | 85,42 |
| LM47 | 14 | 13 | 580478 | 3022625 | 106,27 |
| LM50 | 121 | 24 | 530364 | 3068560 | 111,58 |
| LM51 | 121 | 25 | 530139 | 3060573 | 137,29 |
| LM53 | 121 | 21 | 530354 | 3062194 | 139,38 |
| LM54 | 14 | 13 | 580472 | 3022424 | 150,5 |
| LM55 | 14 | 13 | 580478 | 3022427 | 142,63 |
| LM56 | 14 | 13 | 580478 | 3022415 | 121,34 |
| LM57 | 14 | 13 | 580478 | 3022426 | 134,73 |
| LM58 | 14 | 13 | 580478 | 3022498 | 123,61 |
| LM59 | 14 | 13 | 580478 | 3022821 | 135,9 |
| LM60 | 14 | 13 | 580472 | 3022629 | 132,49 |
| LM61 | 14 | 13 | 580478 | 3022408 | 108,5 |
ST, sequence type.
The cgMLST showed a superior discriminatory power in comparison to ribotyping, 7-loci MLST and MLVA (0.439 vs 0.214, 0.348 and 0.348 respectively) although statistically significant only in comparison to ribotyping (P=0.01) (Table 2, Figure 1). In particular, by WGS based method, ST14 isolates were differentiated in three clusters, whereas the same isolates were indistinguishable by ApaI-PFGE, automated ribotyping and MLVA (De Cesare et al., 2017). According to cgMLST results, one cluster gathered the majority of putative persistent ST14 sequenced L. monocytogenes isolates (CT1701). The other two clusters (CT1702 and CT1703) included only one isolate each (LM15, LM10). Out of the 1748 loci of the cgMLST scheme, these two singletons carried 12 and 11 different loci respectively in comparison to CT1701. This finding suggests that three ST14 strains circulated in the rabbit-meat processing plant during the time of sampling: one persistent strain and two sporadic ones. All ST121 isolates were indistinguishable by cgMLST, confirming the high similarity of these isolates already suggested by molecular typing methods.
Table 2.
Simpson’s ID of Whole Genome Sequencing-based analysis in comparison with molecular typing methods on the 34 sequenced Listeria monocytogenes isolates.
| Typing method | N profiles | Simpson’s ID | CI (95%) |
|---|---|---|---|
| cgMLST | 5 | 0.439 | (0.253-0.624) |
| Ribotyping | 2 | 0.214 | (0.045-0.383) |
| ApaI-PFGE | 3 | 0.348 | (0.170-0.525) |
| MLST | 3 | 0.348 | (0.170-0.525) |
| MLVA | 3 | 0.348 | (0.170-0.525) |
CI, confidence interval; MLST, multilocus sequence typing; MLVA, multiple-locus variable number tandem repeat analysis.
Figure 1.
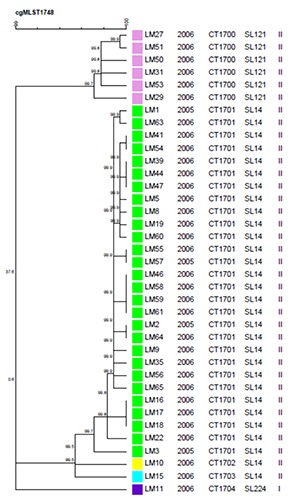
Whole Genome Sequencing-based analysis of the 34 sequenced Listeria monocytogenes isolates.
In order to investigate the virulence potential of 33 ST14 and ST121 isolates, repeatedly isolated over one year and six month, respectively, in a rabbit meat processing plant, MVLST was assessed along with the in silico detection of 82 Listeria virulence genes. In order to investigate potential associations of presence/absence of genes with STs or origin, virulence genes were additionally investigated in the reference genome EGD-e as well as in 23 publicly available genomes of L. monocytogenes belonging to ST14 and ST121 and isolated from humans or food processing environment. Regarding MVLST results, all sequenced ST14 isolates belonged to virulence type VT107, whereas all ST121 to VT94. Regarding in silico detection of virulence genes, the reference genome EGD-e and the analyzed ST14 and ST121 genomes carried 82, 80 and 80 virulence genes respectively indicating that those genes are core virulence genes of L. monocytogenes. The genes inlF and lmo2026 were not detected in any of the 56 ST14 and ST121 genomes analyzed. The gene inlJ was not found or found truncated in all 56 genomes analyzed. All ST14 carried a full-length version of the gene actA (1920 nucleotides), which was detected truncated in all the ST121 genomes. The gene ami was detected in a truncated version in some ST14 and ST121 genomes.
No mutations were found in prfA genes of all sequenced genomes as well as of inlA of ST14 genomes. In contrast, the inlA gene of all ST121 genomes showed a mutation leading to a premature stop codon (PMSC) predicting the translation of a truncated InlA protein of 492 aa instead of the fulllength InlA of 800 aa (Figure 2). This mutation has been already described as PMSC of type 6 which was specifically associated to attenuated virulence of L. monocytogenes (Van Stelten et al., 2010).
Figure 2.

Predicted amino acid sequence alignment of InlA in ST121 Listeria monocytogenes isolates and EGD-e (GenBank accession number NC_003210.1). The mutation leading to the truncated InlA protein is highlighted in bold.
No relevant differences in carriage of virulence genes were found in relation to food or human origin.
Discussion
Whole-genome based analyses have recently revealed an unprecedented potential as a powerful tool for multiple analyses, including typing and gene characterization of food-borne pathogens. In the present study, WGS revealed its great potential as a one-serve-all approach. Based on the same genome sequence data, cgMLST typing, in silico MVLST and in silico detection of 82 virulence genes were performed in parallel. However, it must be noted that, in comparison to standard molecular techniques, this approach requires great bioinformatics skills, standardized protocols, agreed cut off and parameter values in order to obtain robust and comparable results.
The cgMLST revealed a superior discriminatory power in comparison to molecular tools, although significant only in comparison to ribotyping. The cgMLST confirmed only in part results of molecular typing, suggesting that most, but not all, of the ST14 isolates, collected over one year in the same rabbit-meat processing plant, belonged to the same persistent event. Two cgMLST clusters included only one isolate each, suggesting the detection of these two isolates as a sporadic event in the rabbitmeat processing plant.
Persistent L. monocytogenes isolates collected in food processing plants represent a public health concern, due to their potential transfer to humans via the food chain. In order to investigate the virulence potential of sequenced ST14 and ST121 L. monocytogenes isolates, MVLST as well as an in silico detection of 82 virulence determinant genes was carried out. All ST14 belonged to VT107. This VT is particularly interesting since the concatenated sequence of the 7 virulence loci differs to VT1 (Epidemic Clone III) for only 4 nucleotides, suggesting the high similarity of sequenced ST14 isolates to this epidemic clone (Murugesan et al., 2015). Interestingly, three years after sampling of the rabbitmeat processing plant, a case of invasive Listeriosis in the North of Italy associated to ST14 strain belonging to VT1 has been described (Mammina et al., 2013).
Eighty out of 82 virulence determinant genes were found in the 57 L. monocytogenes genomes analyzed. This finding suggests that the presence/absence of these 82 genes can hardly explain the high diversity between ST14 and ST121 which have been shown to occur with a high and low frequency respectively in clinical samples (Maury et al., 2016, Morganti et al., 2015). The genes inlF and lmo2026 were not detected. The gene inlF belong to the internalin family, which includes genes exclusive of the genus Listeria and associated to the adehesion and invasion of host cells (Vázquez-Boland et al., 2001). The gene lmo2026, was recently identified as an internalin gene, inlL, associated to biofilm formation and adhesion to mucin (Popowska et al., 2017). Besides presence/absence of virulence genes the regulation of expression as well as the presence of truncated genes or the presence of mutations leading to truncated versions of their translated proteins might have a crucial role in the virulence potential of ST14 and ST121. Regarding truncated virulence genes, the genes inlJ, actA and ami were found truncated. The gene inlJ belong to the internalin family (Vázquez-Boland et al., 2001; Sabet et al., 2008). The actA gene found truncated in all ST121 genomes, encodes the surface protein ActA, the factor responsible for actin-based motility and cell-to-cell spread (Smith and Portnoy, 1997). The gene ami codes for an autolysin which contributes to the adhesion of L. monocytogenes to eukaryotic cells by anchoring its cell wall (Milohanic et al., 2001). Regarding point mutations, a premature stop codon (PMSC), already described as associated to attenuated virulence of ST121, was detected in all the sequenced ST121 genomes (Van Stelten et al., 2010). The attenuated virulence potential of sequenced ST121 isolates is also supported by the detection of a truncated version of the gene actA, which was described as indispensable for L. monocytogenes pathogenicity (Smith and Portnoy, 1997). An attenuated virulence of ST121 might explain the low frequency of its corresponding clonal complex CC in clinical samples (Maury et al., 2016).
Conclusions
Although not yet fully standardized, Whole genome Sequencing (WGS) can be applied for multiple investigations, such as typing and prediction of virulence. In the present study, WGS-based analyses revealed higher discriminatory power in comparison to molecular typing methods, although significant only in comparison to ribotyping, on 34 L. monocytogenes isolates collected over one year in a rabbit meat processing plant. In particular 25 ST14 and 6 ST121 isolates were confirmed to belong to two persistent strains. Moreover, based on the same genome sequence used for typing purposes, in silico analyses were useful to predict the potential virulence of L. monocytogenes isolates. Multi Virulence Locus Sequence typing (MVLST) results, as well as analysis of virulence genes, suggest a higher virulence potential of ST14 sequenced isolates in comparison to ST121. Additional in vitro investigations should be performed to confirm the virulence of ST14 and ST121 isolates.
Acknowledgments
We acknowledge the team of curators of the Institut Pasteur cgMLST system (Paris, France) (http://bigsdb.pasteur.fr/listeria/) for assistance in CT assignments.
Funding Statement
The work was supported by the EU founded project COMPARE (Grant Agreement N° 643476).
References
- Cruz CD, Pitman AR, Harrow SA, Fletcher GC, 2014. Listeria monocytogenes associated with New Zealand seafood production and clinical cases: unique sequence types, truncated InlA, and attenuated invasiveness. Appl Environ Microbiol 80:1489-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cesare A, Parisi A, Mioni R, Comin D, Lucchi A, Manfreda G, 2017. Listeria monocytogenes circulating in rabbit meat products and slaughterhouses in Italy: prevalence data and comparison among typing results. Foodborne Pathog Dis 14:167-76. [DOI] [PubMed] [Google Scholar]
- EFSA-ECDC, 2016. The European Union summary report on trends and sources of zoonoses, zoonotic agents and foodborne outbreaks in 2015. EFSA J 14:4634. [Google Scholar]
- Ferreira V, Wiedmann M, Teixeira P, Stasiewicz MJ, 2014. Listeria monocytogenes persistence in food-associated environments: epidemiology, strain characteristics, and implications for public health. J Food Protect 77:150-70. [DOI] [PubMed] [Google Scholar]
- Gray MJ, Freitag NE, Boor KJ, 2006. How the bacterial pathogen Listeria monocytogenes mediates the switch from environmental Dr. Jekyll to pathogenic Mr. Hyde. Infect Immun 74:2505-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter PR, Gaston MA, 1988. Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. J Clin Microbiol 26:2465-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joensen KG, Scheutz F, Lund O, Hasman H, Kaas RS, Nielsen EM, Aarestrup FM, 2014. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J Clin Microbiol 52:1501-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki M, Naruse H, Taguchi M, Kumeda Y, 2015. Characterization of specific alleles in InlA and PrfA of Listeria monocytogenes isolated from foods in Osaka, Japan and their ability to invade Caco-2 cells. Int J Food Microbiol 211:18-22. [DOI] [PubMed] [Google Scholar]
- Kathariou S, 2002. Listeria monocytogenes virulence and pathogenicity, a food safety perspective. J Food Prot 65:1811-29. [DOI] [PubMed] [Google Scholar]
- Leong D, Alvarez-Ordóñez A, Jordan K, 2014. Monitoring occurrence and persistence of Listeria monocytogenes in foods and food processing environments in the Republic of Ireland. Front Microbiol 5:436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammina C, Parisi A, Guaita A, Aleo A, Bonura C, Nastasi A, Pontello M, 2013. Enhanced surveillance of invasive listeriosis in the Lombardy region, Italy, in the years 2006-2010 reveals major clones and an increase in serotype 1/2a. BMC Infect Dis 13:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammina C, Parisi A, Guaita A, Aleo A, Bonura C, Nastasi A, Pontello M, 2013. Enhanced surveillance of invasive listeriosis in the Lombardy region, Italy, in the years 2006-2010 reveals major clones and an increase in serotype 1/2a. BMC Infect Dis 13:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milohanic E, Jonquières R, Cossart P, Berche P, Gaillard JL, 2001. The autolysin Ami contributes to the adhesion of Listeria monocytogenes to eukaryotic cells via its cell wall anchor. Mol Microbiol 39:1212-24. [DOI] [PubMed] [Google Scholar]
- Morganti M, Scaltriti E, Cozzolino P, Bolzoni L, Casadei G, Pierantoni M, Foni E, Pongolini S., 2015. Processing- Dependent and Clonal Contamination Patterns of Listeria monocytogenes in the Cured Ham Food Chain Revealed by Genetic Analysis. Appl Environ Microbiol 82:822-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura A Criscuolo A Pouseele H Maury MM Leclercq A Tarr C Björkman JT Dallman T Reimer A Enouf V Larsonneur E Carleton H Bracq-Dieye H Katz LS Jones L Touchon M Tourdjman M Walker M Stroika S Cantinelli T, Chenal-Francisque V Kucerova Z Rocha EPC Nadon C Grant K Nielsen EM Pot B Gerner-Smidt P Lecuit M Brisse S. 2016. Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nature Microbiol 2:16185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugesan L, Kucerova Z, Knabel SJ, LaBorde LF, 2015. Predominance and distribution of a persistent Listeria monocytogenes clone in a commercial fresh mushroom processing environment. J Food Protect 78:1988-98. [DOI] [PubMed] [Google Scholar]
- Nightingale KK, Windham K, Martin KE, Yeung M, Wiedmann M, 2005. Select Listeria monocytogenes subtypes commonly found in foods carry distinct nonsense mutations in inlA, leading to expression of truncated and secreted internalin A, and are associated with a reduced invasion phenotype for human intestinal epithelial cells. Appl Environ Microbiol 71:8764-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi RH, den Bakker HC, Wiedmann M, 2011. Listeria monocytogenes lineages: genomics, evolution, ecology, and phenotypic characteristics. Int J Med Microbiol 301:79-96. [DOI] [PubMed] [Google Scholar]
- Popowska M, Krawczyk-Balska A, Ostrowski R, Desvaux M, 2017. InlL from Listeria monocytogenes is involved in Biofilm formation and adhesion to Mucin. Front Microbiol 8:660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabet C, Toledo-Arana A, Personnic N, Lecuit M, Dubrac S, Poupel O, Gouin E, Nahori MA, Cossart P, Bierne H, 2008. The Listeria monocytogenes virulence factor InlJ is specifically expressed in vivo and behaves as an adhesin. Infect Immun 76:1368-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GA, Portnoy DA, 1997. How the Listeria monocytogenes ActA protein converts actin polymerization into a motile force.Trends Microbiol 272-76. [DOI] [PubMed] [Google Scholar]
- Stasiewicz MJ, Oliver HF, Wiedmann M, den Bakker HC, 2015. Whole-Genome Sequencing allows for improved identification of persistent Listeria monocytogenes in food-associated Environments. Appl Environ Microbiol 81:6024-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Stelten A, Simpson JM, Chen Y, Scott VN, Whiting RC, Ross WH, Nightingale KK, 2011. Significant shift in median guinea pig infectious dose shown by an outbreak-associated Listeria monocytogenes epidemic clone strain and a strain carrying a premature stop codon mutation in inlA. Appl Environ Microbiol 77:2479-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Stelten A, Simpson JM, Ward TJ, Nightingale KK, 2010. Revelation by single-nucleotide polymorphism genotyping that mutations leading to a premature stop codon in inlA are common among Listeria monocytogenes isolates from ready-to-eat foods but not human listeriosis cases. Appl Environ Microbiol 76:2783-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Stelten A, Simpson JM, Ward TJ, Nightingale KK, 2010. Revelation by single-nucleotide polymorphism genotyping that mutations leading to a premature stop codon in inlA are common among Listeria monocytogenes isolates from ready-to-eat foods but not human listeriosis cases. Appl Environ Microbiol 76:2783-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Véghová A, Minarovičová J, Koreňová J, Drahovská H, Kaclíková E, 2017. Prevalence and tracing of persistent Listeria monocytogenes strains in meat processing facility production chain. J Food Safety 37:e12315. [Google Scholar]
- Véghová A, Minarovičová J, Koreňová J, Drahovská H, Kaclíková E, 2017. Prevalence and tracing of persistent Listeria monocytogenes strains in meat processing facility production chain. J Food Safety 37:e12315. [Google Scholar]